The Role of Heme Oxygenase-1 in Remote Ischemic and Anesthetic Organ Conditioning


{kind=link}
{kind=link}
{kind=link}
Abstract
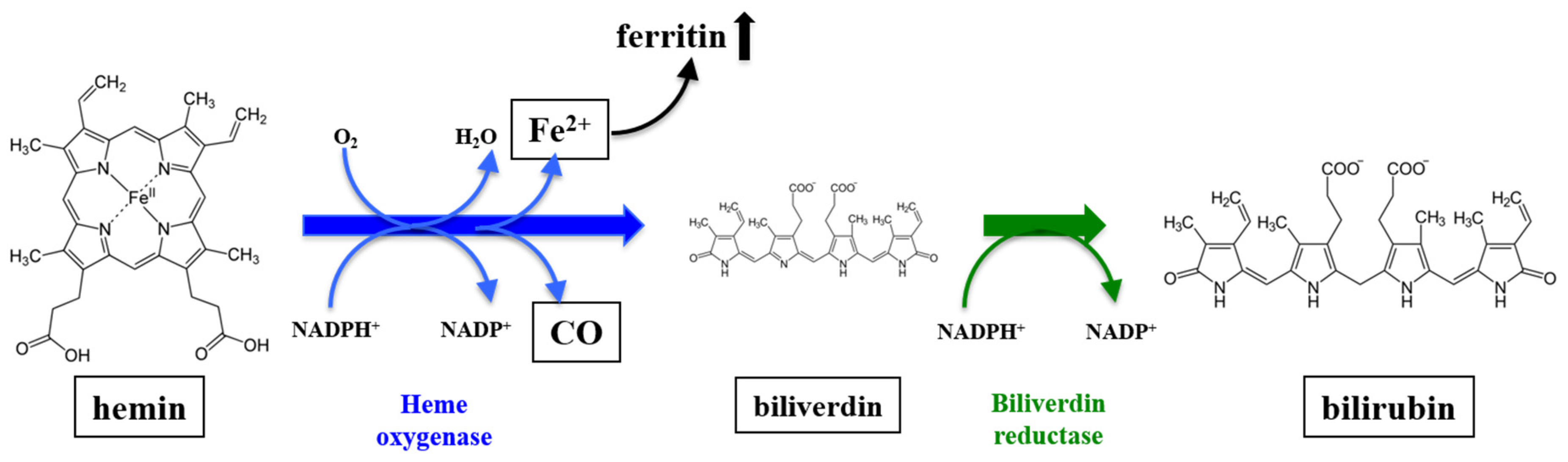
:1. Introduction
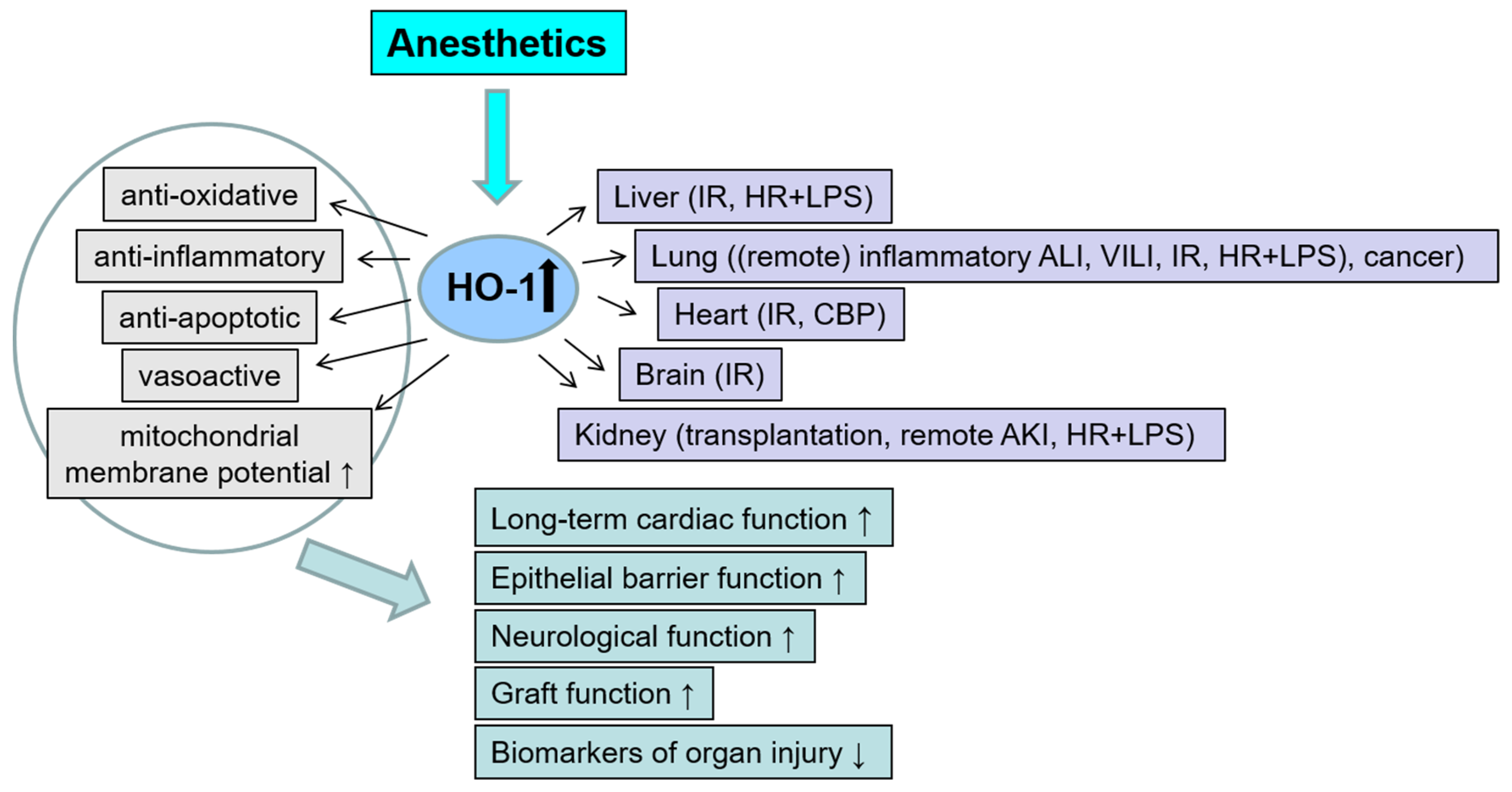
2. The Role of Anesthetic Agents on HO-1 Modulation
2.1. Isoflurane
2.1.1. Isoflurane: Preconditioning
2.1.2. Isoflurane: Postconditioning
2.1.3. Isoflurane: Perioperative Application
2.2. Sevoflurane
2.2.1. Sevoflurane: Preconditioning
2.2.2. Sevoflurane: Postconditioning
2.2.3. Sevoflurane: Continuous Application
2.3. Xenon
2.4. Morphine/Opioids
2.4.1. Morphine: Preconditioning
2.4.2. Morphine/Opioid: Postconditioning
2.5. Propofol
2.5.1. Propofol: Preconditioning
2.5.2. Propofol: Postconditioning
2.5.3. Propofol: Perioperative Setting
2.6. Ketamine
2.7. Dexmedetomidine
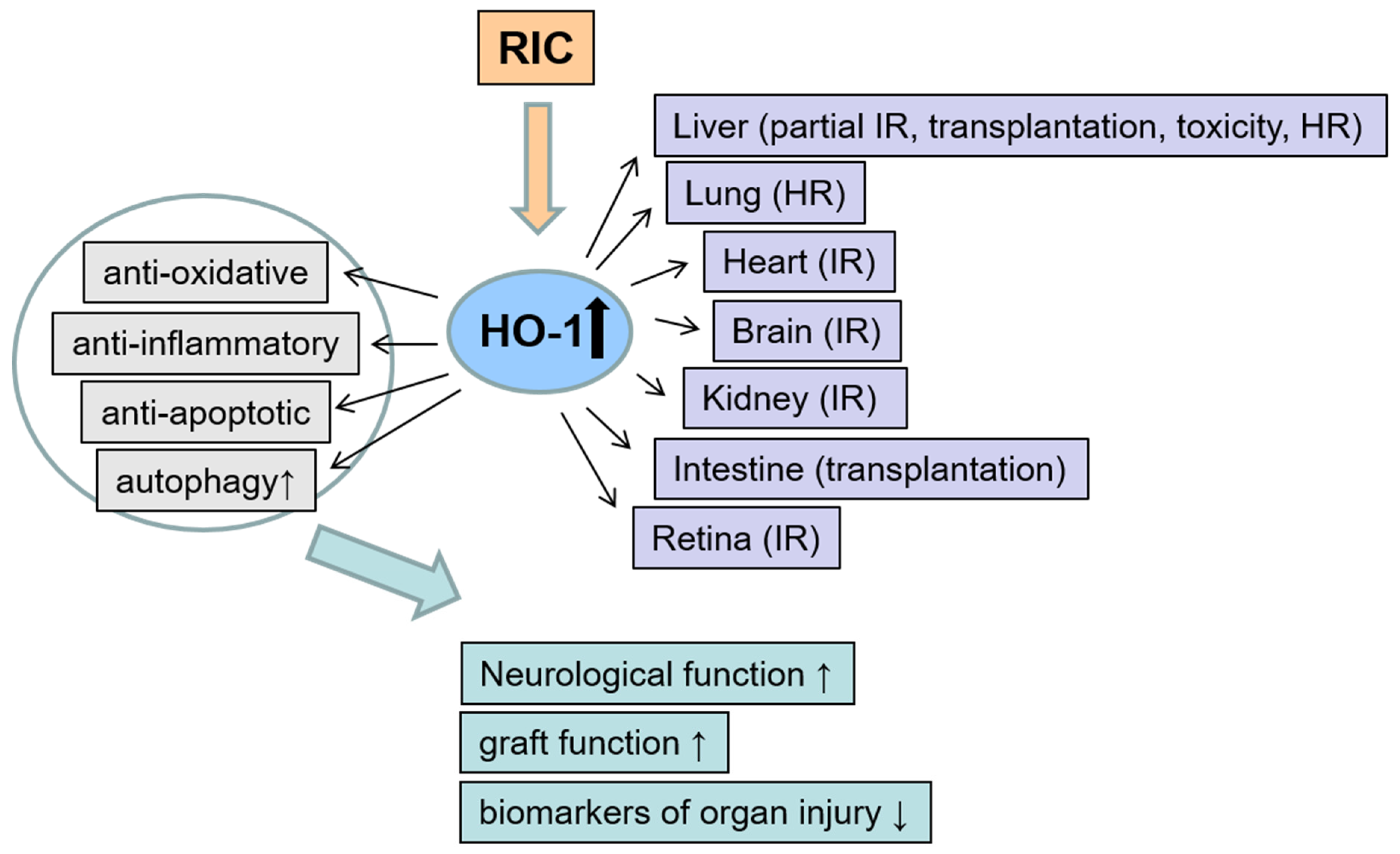
3. The Role of Remote Ischemic Conditioning on HO-1 Modulation
3.1. Remote Ischemic Preconditioning (Pre-RIC)
3.1.1. Liver
3.1.2. Lung
3.1.3. Heart
3.1.4. Kidney
3.1.5. Intestine
3.1.6. Various Organs
3.2. Remote Ischemic Postconditioning (Post-RIC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Lin, J.H.; Schwartzman, M.L.; Levere, R.D.; Shibahara, S. The physiological significance of heme oxygenase. Int. J. Biochem. 1988, 20, 543–558. [Google Scholar] [CrossRef]
- Bauer, I.; Wanner, G.A.; Rensing, H.; Alte, C.; Miescher, E.A.; Wolf, B.; Pannen, B.H.; Clemens, M.G.; Bauer, M. Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology 1998, 27, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Bauer, I.; Vollmar, B.; Jaeschke, H.; Rensing, H.; Kraemer, T.; Larsen, R.; Bauer, M. Transcriptional activation of heme oxygenase-1 and its functional significance in acetaminophen-induced hepatitis and hepatocellular injury in the rat. J. Hepatol. 2000, 33, 395–406. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Choi, A.M. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1029–L1037. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Hoetzel, A.; Schmidt, R. Regulatory role of anesthetics on heme oxygenase-1. Curr. Drug Targets 2010, 11, 1495–1503. [Google Scholar] [CrossRef]
- Berger, M.M.; Huhn, R.; Oei, G.T.; Heinen, A.; Winzer, A.; Bauer, I.; Preckel, B.; Weber, N.C.; Schlack, W.; Hollmann, M.W. Hypoxia induces late preconditioning in the rat heart in vivo. Anesthesiology 2010, 113, 1351–1360. [Google Scholar] [CrossRef]
- Rensing, H.; Bauer, I.; Datene, V.; Pätau, C.; Pannen, B.H.; Bauer, M. Differential expression pattern of heme oxygenase-1/heat shock protein 32 and nitric oxide synthase-II and their impact on liver injury in a rat model of hemorrhage and resuscitation. Crit. Care Med. 1999, 27, 2766–2775. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; Volk, H.-D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Gibbs, P.E.M. 30 some years of heme oxygenase: From a “molecular wrecking ball” to a “mesmerizing” trigger of cellular events. Biochem. Biophys. Res. Commun. 2005, 338, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M.K. Targeting heme oxygenase-1/carbon monoxide for therapeutic modulation of inflammation. Transl. Res. J. Lab. Clin. Med. 2016, 167, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Vogt, B.A.; Alam, J.; Croatt, A.J.; Vercellotti, G.M.; Nath, K.A. Acquired resistance to acute oxidative stress. Possible role of heme oxygenase and ferritin. Lab. Investig. J. Tech. Methods Pathol. 1995, 72, 474–483. [Google Scholar]
- Müllebner, A.; Moldzio, R.; Redl, H.; Kozlov, A.V.; Duvigneau, J.C. Heme degradation by heme oxygenase protects mitochondria but induces ER stress via formed bilirubin. Biomolecules 2015, 5, 679–701. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Pannen, B.H. Bench-to-bedside review: Carbon monoxide—From mitochondrial poisoning to therapeutic use. Crit. Care 2009, 13, 220. [Google Scholar] [CrossRef]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Drummond, H.A.; Mitchell, Z.L.; Abraham, N.G.; Stec, D.E. Targeting heme oxygenase-1 in cardiovascular and kidney disease. Antioxidants 2019, 8, 181. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote ischemic conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Chen, H.; Zhu, Q.; Cai, J.; Chen, C.; Yuan, D.; Jin, Y.; Yao, W.; Hei, Z. Propofol post-conditioning alleviates hepatic ischaemia reperfusion injury via BRG1-mediated Nrf2/HO-1 transcriptional activation in human and mice. J. Cell. Mol. Med. 2017, 21, 3693–3704. [Google Scholar] [CrossRef] [PubMed]
- Savran Karadeniz, M.; Senturk Ciftci, H.; Tefik, T.; Oktar, T.; Nane, I.; Turkmen, A.; Oguz, F.; Tugrul, K.M. Effects of different volatile anesthetics on cytokine and chemokine production after ischemia-reperfusion injury in patients undergoing living-donor kidney transplant. Middle East Soc. Organ Transplant. 2019, 17, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Beck-Schimmer, B.; Bonvini, J.M.; Schadde, E.; Dutkowski, P.; Oberkofler, C.E.; Lesurtel, M.; DeOliveira, M.L.; Figueira, E.R.R.; Rocha Filho, J.A.; Auler, J.O.C.; et al. Conditioning with sevoflurane in liver transplantation: Results of a multicenter randomized controlled trial. Transplantation 2015, 99, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Bettex, D.A.; Wanner, P.M.; Bosshart, M.; Balmer, C.; Knirsch, W.; Dave, H.; Dillier, C.; Bürki, C.; Hug, M.; Seifert, B.; et al. Role of sevoflurane in organ protection during cardiac surgery in children: A randomized controlled trial. Interact. Cardiovasc. Thorac. Surg. 2015, 20, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef]
- Zhou, D.; Ding, J.; Ya, J.; Pan, L.; Wang, Y.; Ji, X.; Meng, R. Remote ischemic conditioning: A promising therapeutic intervention for multi-organ protection. Aging 2018, 10, 1825–1855. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Yoon, D.-W.; Kim, J.-H.; Lee, J.-H.; Lim, C.-H. Effect of remote ischemic post-conditioning on systemic inflammatory response and survival rate in lipopolysaccharide-induced systemic inflammation model. J. Inflamm. 2014, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Cour, M.; Buisson, M.; Klouche, K.; Bouzgarrou, R.; Schwebel, C.; Quenot, J.-P.; Zeni, F.; Beuret, P.; Ovize, M.; Argaud, L. Remote ischemic conditioning in septic shock (RECO-Sepsis): Study protocol for a randomized controlled trial. Trials 2019, 20, 281. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote ischemic preconditioning and outcomes of cardiac surgery. N. Engl. J. Med. 2015, 373, 1408–1417. [Google Scholar] [CrossRef]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Böning, A.; Niemann, B.; et al. A Multicenter trial of remote ischemic preconditioning for heart surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef]
- Preckel, B.; Müllenheim, J.; Moloschavij, A.; Thämer, V.; Schlack, W. Xenon administration during early reperfusion reduces infarct size after regional ischemia in the rabbit heart in vivo. Anesth. Analg. 2000, 91, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Ma, D.; Maze, M.; Franks, N.P. Effects of xenon on in vitro and in vivo models of neuronal injury. Anesthesiology 2002, 96, 1485–1491. [Google Scholar] [CrossRef]
- De Hert, S.G.; Turani, F.; Mathur, S.; Stowe, D.F. Cardioprotection with volatile anesthetics: Mechanisms and clinical implications. Anesth. Analg. 2005, 100, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Toma, O.; Wolter, J.I.; Obal, D.; Müllenheim, J.; Preckel, B.; Schlack, W. The noble gas xenon induces pharmacological preconditioning in the rat heart in vivo via induction of PKC-epsilon and p38 MAPK. Br. J. Pharmacol. 2005, 144, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hossain, M.; Pettet, G.K.J.; Luo, Y.; Lim, T.; Akimov, S.; Sanders, R.D.; Franks, N.P.; Maze, M. Xenon preconditioning reduces brain damage from neonatal asphyxia in rats. J. Cereb. Blood Flow Metab. 2006, 26, 199–208. [Google Scholar] [CrossRef]
- Bein, B.; Höcker, J.; Scholz, J. Xenon—The ideal anaesthetic agent? Anasth. Intensivmed. Notfallmed. Schmerzther. AINS 2007, 42, 784–791. [Google Scholar] [CrossRef]
- Ma, D.; Lim, T.; Xu, J.; Tang, H.; Wan, Y.; Zhao, H.; Hossain, M.; Maxwell, P.H.; Maze, M. Xenon preconditioning protects against renal ischemic-reperfusion injury via HIF-1alpha activation. J. Am. Soc. Nephrol. JASN 2009, 20, 713–720. [Google Scholar] [CrossRef]
- Pagel, P.S. Cardioprotection by noble gases. J. Cardiothorac. Vasc. Anesth. 2010, 24, 143–163. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, H.; Wang, T.; Pac-Soo, C.; Ma, D. Cellular signaling pathways and molecular mechanisms involving inhalational anesthetics-induced organoprotection. J. Anesth. 2014, 28, 740–758. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-M.; Song, B.C.; Yeum, K.-J. Impact of volatile anesthetics on oxidative stress and inflammation. BioMed Res. Int. 2015, 2015, 242709. [Google Scholar] [CrossRef] [PubMed]
- Motayagheni, N.; Phan, S.; Eshraghi, C.; Nozari, A.; Atala, A. A review of anesthetic effects on renal function: Potential organ protection. Am. J. Nephrol. 2017, 46, 380–389. [Google Scholar] [CrossRef]
- Chen, S.; Lotz, C.; Roewer, N.; Broscheit, J.-A. Comparison of volatile anesthetic-induced preconditioning in cardiac and cerebral system: Molecular mechanisms and clinical aspects. Eur. J. Med. Res. 2018, 23, 10. [Google Scholar] [CrossRef]
- Krzych, L.J.; Szczepanska, A.J. Non-Anaesthetic effects of volatile anaesthetics: A short trip on the sea of translational medicine. Curr. Vasc. Pharmacol. 2018, 16, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Rylova, A.; Maze, M. Protecting the brain with xenon anesthesia for neurosurgical procedures. J. Neurosurg. Anesthesiol. 2019, 31, 18–29. [Google Scholar] [CrossRef]
- Headrick, J.P.; See Hoe, L.E.; Du Toit, E.F.; Peart, J.N. Opioid receptors and cardioprotection—“Opioidergic conditioning” of the heart. Br. J. Pharmacol. 2015, 172, 2026–2050. [Google Scholar] [CrossRef]
- Vasileiou, I.; Xanthos, T.; Koudouna, E.; Perrea, D.; Klonaris, C.; Katsargyris, A.; Papadimitriou, L. Propofol: A review of its non-anaesthetic effects. Eur. J. Pharmacol. 2009, 605, 1–8. [Google Scholar] [CrossRef]
- Schifilliti, D.; Grasso, G.; Conti, A.; Fodale, V. Anaesthetic-Related neuroprotection. CNS Drugs 2010, 24, 893–907. [Google Scholar] [CrossRef]
- Cai, Y.; Xu, H.; Yan, J.; Zhang, L.; Lu, Y. Molecular targets and mechanism of action of dexmedetomidine in treatment of ischemia/reperfusion injury. Mol. Med. Rep. 2014, 9, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Ding, W.; Jiang, W.; Chen, Y.; Hang, D.; Gu, D.; Jiang, G.; Tan, Y.; Ge, Z.; Ma, T. A comparison of the effects of midazolam, propofol and dexmedetomidine on the antioxidant system: A randomized trial. Exp. Ther. Med. 2015, 9, 2293–2298. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Hu, M.; Lu, Y.; Cao, Y.; Chang, Y.; Dai, Z. The protective effects of dexmedetomidine on ischemic brain injury: A meta-analysis. J. Clin. Anesth. 2017, 40, 25–32. [Google Scholar] [CrossRef]
- Dardalas, I.; Stamoula, E.; Rigopoulos, P.; Malliou, F.; Tsaousi, G.; Aidoni, Z.; Grosomanidis, V.; Milonas, A.; Papazisis, G.; Kouvelas, D.; et al. Dexmedetomidine effects in different experimental sepsis in vivo models. Eur. J. Pharmacol. 2019, 856, 172401. [Google Scholar] [CrossRef]
- Behmenburg, F.; Boekholt, Y.; van Caster, P.; Dorsch, M.; Heinen, A.; Hollmann, M.W.; Huhn, R. Extended Second Window of Protection of Sevoflurane-induced Preconditioning. J. Cardiovasc. Pharmacol. 2017, 70, 284–289. [Google Scholar] [CrossRef]
- Holak, E.J.; Mei, D.A.; Dunning, M.B.; Gundamraj, R.; Noseir, R.; Zhang, L.; Woehlck, H.J. Carbon monoxide production from sevoflurane breakdown: Modeling of exposures under clinical conditions. Anesth. Analg. 2003, 3, 757–764. [Google Scholar] [CrossRef]
- Levy, R.J. Anesthesia-Related carbon monoxide exposure: Toxicity and potential therapy. Anesth. Analg. 2016, 123, 670–681. [Google Scholar] [CrossRef]
- Levy, R.J. Carbon monoxide and anesthesia-induced neurotoxicity. Neurotoxicol. Teratol. 2017, 60, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Hopper, C.P.; Wollborn, J. Delivery of carbon monoxide via halogenated ether anesthetics. Nitric Oxide Biol. Chem. 2019, 89, 93–95. [Google Scholar] [CrossRef]
- Rudolph, U.; Antkowiak, B. Molecular and neuronal substrates for general anaesthetics. Nat. Rev. Neurosci. 2004, 5, 709–720. [Google Scholar] [CrossRef]
- Khan, K.S.; Hayes, I.; Buggy, D.J. Pharmacology of anaesthetic agents II: Inhalation anaesthetic agents. Contin. Educ. Anaesth. Crit. Care Pain 2014, 14, 106–111. [Google Scholar] [CrossRef]
- Schmidt, R.; Hoetzel, A.; Baechle, T.; Loop, T.; Humar, M.; Bauer, M.; Pahl, H.L.; Geiger, K.K.; Pannen, B.H.J. Isoflurane pretreatment lowers portal venous resistance by increasing hepatic heme oxygenase activity in the rat liver in vivo. J. Hepatol. 2004, 41, 706–713. [Google Scholar] [CrossRef]
- Schmidt, R.; Tritschler, E.; Hoetzel, A.; Loop, T.; Humar, M.; Halverscheid, L.; Geiger, K.K.; Pannen, B.H.J. Heme oxygenase-1 induction by the clinically used anesthetic isoflurane protects rat livers from ischemia/reperfusion injury. Ann. Surg. 2007, 245, 931–942. [Google Scholar] [CrossRef]
- Faller, S.; Strosing, K.M.; Ryter, S.W.; Buerkle, H.; Loop, T.; Schmidt, R.; Hoetzel, A. The volatile anesthetic isoflurane prevents ventilator-induced lung injury via phosphoinositide 3-kinase/Akt signaling in mice. Anesth. Analg. 2012, 114, 747–756. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, Y.; Jiang, H.; Xu, H.; Liu, H. Up-regulation of heme oxygenase-1 by isoflurane preconditioning during tolerance against neuronal injury induced by oxygen glucose deprivation. Acta Biochim. Biophys. Sin. 2008, 40, 803–810. [Google Scholar] [CrossRef]
- Li, Q.-F.; Zhu, Y.-S.; Jiang, H.; Xu, H.; Sun, Y. Heme oxygenase-1 mediates the anti-inflammatory effect of isoflurane preconditioning in LPS-stimulated macrophages. Acta Pharmacol. Sin. 2009, 30, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Hu, R.; Sun, Y.; Li, Q.; Jiang, H. Isoflurane post-treatment improves pulmonary vascular permeability via upregulation of heme oxygenase-1. Exp. Lung Res. 2013, 39, 295–303. [Google Scholar] [CrossRef]
- Goris, R.J.; Boekholtz, W.K.; van Bebber, I.P.; Nuytinck, J.K.; Schillings, P.H. Multiple-organ failure and sepsis without bacteria. An experimental model. Arch. Surg. 1986, 121, 897–901. [Google Scholar] [CrossRef]
- Volman, T.J.H.; Hendriks, T.; Goris, R.J.A. Zymosan-induced generalized inflammation: Experimental studies into mechanisms leading to multiple organ dysfunction syndrome. Shock 2005, 23, 291–297. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Li, W.; Wang, L.; Hou, L.; Mu, J.; Liu, X.; Chen, H.; Xie, K.; Li, N.; et al. Anesthetic isoflurane posttreatment attenuates experimental lung injury by inhibiting inflammation and apoptosis. Mediators Inflamm. 2013, 2013, 16. [Google Scholar] [CrossRef]
- Konrad, F.M.; Braun, S.; Ngamsri, K.-C.; Vollmer, I.; Reutershan, J. Heme oxygenase-1 attenuates acute pulmonary inflammation by decreasing the release of segmented neutrophils from the bone marrow. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L707–L717. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, Y.-L.; Liu, N.-N.; Zhu, X.-S.; Liu, Q.-Q.; Mei, H.-Y.; Wang, L.-F.; Yang, A.-G.; Gao, C.-F.; Li, J.-T. Epithelial HO-1/STAT3 affords the protection of subanesthetic isoflurane against zymosan-induced lung injury in mice. Oncotarget 2017, 8, 54889–54903. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.-Y.; Lee, J.-S.; Roan, J.-N.; Tsai, Y.-C.; Lam, C.-F. Isoflurane impairs motor function recovery by increasing neuroapoptosis and degeneration during spinal ischemia-reperfusion injury in rats. Anesth. Analg. 2017, 124, 254–261. [Google Scholar] [CrossRef]
- Behne, M.; Wilke, H.J.; Harder, S. Clinical pharmacokinetics of sevoflurane. Clin. Pharmacokinet. 1999, 36, 13–26. [Google Scholar] [CrossRef]
- Zhang, S.-B.; Liu, T.-J.; Pu, G.-H.; Li, B.-Y.; Gao, X.-Z.; Han, X.-L. MicroRNA-374 exerts protective effects by inhibiting sp1 through activating the Pi3k/Akt pathway in rat models of myocardial ischemia-reperfusion after sevoflurane preconditioning. Cell. Physiol. Biochem. 2018, 46, 1455–1470. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lee, H.; Park, Y.H.; Jeon, Y.T.; Hwang, J.W.; Lim, Y.J.; Kim, E.; Park, S.Y.; Park, H.P. Sevoflurane post-conditioning increases nuclear factor erythroid 2-related factor and haemoxygenase-1 expression via protein kinase C pathway in a rat model of transient global cerebral ischaemia. Br. J. Anaesth. 2015, 114, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-L.; Kunsch, C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: A new therapeutic approach for the treatment of inflammatory diseases. Curr. Pharm. Des. 2004, 10, 879–891. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Ye, Z.; Guo, Q.; Xia, P.; Wang, N.; Wang, E.; Yuan, Y. Sevoflurane postconditioning involves an up-regulation of HIF-1α and HO-1 expression via PI3K/Akt pathway in a rat model of focal cerebral ischemia. Brain Res. 2012, 1463, 63–74. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Zhang, D.-X. The dual neuroprotective-neurotoxic effects of sevoflurane after hemorrhagic shock injury. J. Surg. Res. 2019, 235, 591–599. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, J.; Zhang, L.; Ai, Y. Post-conditioning with sevoflurane induces heme oxygenase-1 expression via the PI3K/Akt pathway in lipopolysaccharide-induced acute lung injury. Mol. Med. Rep. 2014, 9, 2435–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Yang, M.; Zhang, F.; Yu, J.; Wang, J.; Ma, L.; Zhong, Y.; Qian, L.; Chen, G.; Yu, L.; et al. Gender-related difference of sevoflurane postconditioning in isolated rat hearts: Focus on phosphatidylinositol-3-kinase/Akt signaling. J. Surg. Res. 2011, 170, e3–e9. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K. Efficacy of cardioprotective “conditioning” strategies in aging and diabetic cohorts: The co-morbidity conundrum. Drugs Aging 2011, 28, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Kleinbongard, P.; Skyschally, A.; Levkau, B.; Schulz, R.; Erbel, R. The coronary circulation in cardioprotection: More than just one confounder. Cardiovasc. Res. 2012, 94, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Huhn, R.; Heinen, A.; Weber, N.C.; Hollmann, M.W.; Schlack, W.; Preckel, B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: Cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br. J. Anaesth. 2008, 100, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Drenger, B.; Ostroversusky, I.A.; Barak, M.; Nechemia-Arbely, Y.; Ziv, E.; Axelrod, J.H. Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: Phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology 2011, 114, 1364–1372. [Google Scholar] [CrossRef]
- Tyagi, S.; Singh, N.; Virdi, J.K.; Jaggi, A.S. Diabetes abolish cardioprotective effects of remote ischemic conditioning: Evidences and possible mechanisms. J. Physiol. Biochem. 2019, 75, 19–28. [Google Scholar] [CrossRef]
- Grievink, H.; Kuzmina, N.; Chevion, M.; Drenger, B. Sevoflurane postconditioning is not mediated by ferritin accumulation and cannot be rescued by simvastatin in isolated streptozotocin-induced diabetic rat hearts. PLoS ONE 2019, 14, e0211238. [Google Scholar] [CrossRef]
- Goergens, J.I.; Heinen, N.M.; Zoller, J.; Preckel, B.; Bauer, I.; Huhn, R.; Ebel, D.; Raupach, A. Influence of hyperglycemia during different phases of ischemic preconditioning on cardioprotection—A focus on apoptosis and aggregation of granulocytes. Shock 2019, in press. [Google Scholar] [CrossRef]
- Gao, S.; Yang, Z.; Shi, R.; Xu, D.; Li, H.; Xia, Z.; Wu, Q.-P.; Yao, S.; Wang, T.; Yuan, S. Diabetes blocks the cardioprotective effects of sevoflurane postconditioning by impairing Nrf2/Brg1/HO-1 signaling. Eur. J. Pharmacol. 2016, 779, 111–121. [Google Scholar] [CrossRef]
- Shiraishi, S.; Cho, S.; Akiyama, D.; Ichinomiya, T.; Shibata, I.; Yoshitomi, O.; Maekawa, T.; Ozawa, E.; Miyaaki, H.; Hara, T. Sevoflurane has postconditioning as well as preconditioning properties against hepatic warm ischemia-reperfusion injury in rats. J. Anesth. 2019, 33, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Bein, B.; Renner, J.; Caliebe, D.; Hanss, R.; Bauer, M.; Fraund, S.; Scholz, J. The effects of interrupted or continuous administration of sevoflurane on preconditioning before cardio-pulmonary bypass in coronary artery surgery: Comparison with continuous propofol. Anaesthesia 2008, 63, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Frässdorf, J.; Borowski, A.; Ebel, D.; Feindt, P.; Hermes, M.; Meemann, T.; Weber, R.; Müllenheim, J.; Weber, N.C.; Preckel, B.; et al. Impact of preconditioning protocol on anesthetic-induced cardioprotection in patients having coronary artery bypass surgery. J. Thorac. Cardiovasc. Surg. 2009, 137, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Lin, L.; Wang, L.; Jin, L. Sevoflurane attenuates pulmonary inflammation and ventilator-induced lung injury by upregulation of HO-1 mRNA expression in mice. Int. J. Nanomed. 2013, 6, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, B.; Armbruster, S.; Schairer, W.; Landstra, M.; Trouwborst, A.; Van Daal, G.J.; Kusuma, A.; Erdmann, W. Safety and efficacy of xenon in routine use as an inhalational anaesthetic. Lancet 1990, 335, 1413–1415. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, R.; Franks, N.P. Bench-to-bedside review: Molecular pharmacology and clinical use of inert gases in anesthesia and neuroprotection. Crit. Care 2010, 14, 229. [Google Scholar] [CrossRef] [PubMed]
- Coburn, M.; Kunitz, O.; Baumert, J.-H.; Hecker, K.; Haaf, S.; Zühlsdorff, A.; Beeker, T.; Rossaint, R. Randomized controlled trial of the haemodynamic and recovery effects of xenon or propofol anaesthesia. Br. J. Anaesth. 2005, 94, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Neukirchen, M.; Schaefer, M.S.; Kern, C.; Brett, S.; Werdehausen, R.; Rellecke, P.; Reyle-Hahn, M.; Kienbaum, P. Xenon does not increase heart rate-corrected cardiac QT interval in volunteers and in patients free of cardiovascular disease. Anesthesiology 2015, 123, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Bein, B.; Turowski, P.; Renner, J.; Hanss, R.; Steinfath, M.; Scholz, J.; Tonner, P.H. Comparison of xenon-based anaesthesia compared with total intravenous anaesthesia in high risk surgical patients. Anaesthesia 2005, 60, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yoshida, A.; Xiao, W.; Ologunde, R.; O’Dea, K.P.; Takata, M.; Tralau-Stewart, C.; George, A.J.T.; Ma, D. Xenon treatment attenuates early renal allograft injury associated with prolonged hypothermic storage in rats. FASEB J. 2013, 27, 4076–4088. [Google Scholar] [CrossRef]
- Patel, K.; Bhaskaran, M.; Dani, D.; Reddy, K.; Singhal, P.C. Role of heme oxygenase-1 in morphine-modulated apoptosis and migration of macrophages. J. Infect. Dis. 2003, 187, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wong, G.T.C.; Man, K.; Irwin, M.G. Pretreatment with intrathecal or intravenous morphine attenuates hepatic ischaemia-reperfusion injury in normal and cirrhotic rat liver. Br. J. Anaesth. 2012, 109, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Yun, N.; Eum, H.-A.; Lee, S.-M. Protective role of heme oxygenase-1 against liver damage caused by hepatic ischemia and reperfusion in rats. Antioxid. Redox Signal. 2010, 13, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Guo, H.; Li, Y.-C.; Hao, Z.-M. Heme oxygenase-1 induction by hemin protects liver cells from ischemia/reperfusion injury in cirrhotic rats. World J. Gastroenterol. 2007, 13, 5384–5390. [Google Scholar] [CrossRef] [Green Version]
- Godai, K.; Hasegawa-Moriyama, M.; Kurimoto, T.; Saito, T.; Yamada, T.; Sato, T.; Kojima, M.; Kanmura, Y. Peripheral administration of morphine attenuates postincisional pain by regulating macrophage polarization through COX-2-dependent pathway. Mol. Pain 2014, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Grochot-Przeczek, A.; Lach, R.; Mis, J.; Skrzypek, K.; Gozdecka, M.; Sroczynska, P.; Dubiel, M.; Rutkowski, A.; Kozakowska, M.; Zagorska, A.; et al. Heme oxygenase-1 accelerates cutaneous wound healing in mice. PLoS ONE 2009, 4, e5803. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.; Zhang, B.; Zhou, X.; Zhao, J.; Sun, Z.; Tao, Y.; Pei, J.; Zhang, W. Kappa-Opioid agonist U50,488H-mediated protection against heart failure following myocardial ischemia/reperfusion: Dual roles of heme oxygenase-1. Cell. Physiol. Biochem. 2016, 39, 2158–2172. [Google Scholar] [CrossRef] [PubMed]
- Trapani, G.; Altomare, C.; Liso, G.; Sanna, E.; Biggio, G. Propofol in anesthesia. Mechanism of action, structure-activity relationships, and drug delivery. Curr. Med. Chem. 2000, 7, 249–271. [Google Scholar] [CrossRef]
- Sahinovic, M.M.; Struys, M.M.R.F.; Absalom, A.R. Clinical pharmacokinetics and pharmacodynamics of propofol. Clin. Pharmacokinet. 2018, 57, 1539–1558. [Google Scholar] [CrossRef]
- Yao, W.; Luo, G.; Zhu, G.; Chi, X.; Zhang, A.; Xia, Z.; Hei, Z. Propofol activation of the Nrf2 pathway is associated with amelioration of acute lung injury in a rat liver transplantation model. Oxid. Med. Cell. Longev. 2014, 2014, 258567. [Google Scholar] [CrossRef]
- Ge, M.; Luo, G.; Yao, W.; Luo, C.; Zhou, S.; Yuan, D.; Chi, X.; Hei, Z. Propofol pretreatment attenuates remote kidney injury induced by orthotopic liver autotransplantation, which is correlated with the activation of Nrf2 in rats. Mol. Med. Rep. 2015, 11, 3962–3968. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, H.; Irwin, M.G.; Xia, Z.-Y.; Mao, X.; Lei, S.; Wong, G.T.; Hung, V.; Cheung, C.W.; Fang, X.; et al. Propofol ameliorates hyperglycemia-induced cardiac hypertrophy and dysfunction via heme oxygenase-1/signal transducer and activator of transcription 3 signaling pathway in rats. Crit. Care Med. 2014, 42, e583–e594. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Rezzani, R.; Rodella, L.; Kruger, A.; Taller, D.; Li Volti, G.; Goodman, A.I.; Kappas, A. Overexpression of human heme oxygenase-1 attenuates endothelial cell sloughing in experimental diabetes. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2468–H2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Volti, G.; Murabito, P. Pharmacologic induction of heme oxygenase-1: It is time to take it seriously. Crit. Care Med. 2014, 42, 1967–1968. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, T.; Tanaka, T.; Okuda, H.; Kawaguchi, A.T.; Oh-Hashi, K.; Terada, Y.; Isonishi, A.; Morita-Takemura, S.; Tatsumi, K.; Kawaguchi, M.; et al. Propofol induces nuclear localization of Nrf2 under conditions of oxidative stress in cardiac H9c2 cells. PLoS ONE 2018, 13, e0196191. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Xue, Z.; Wang, H.; Li, P. Propofol upregulates heme oxygenase-1 through activation of ERKs in human umbilical vein endothelial cells under oxidative stress conditions. J. Neurosurg. Anesthesiol. 2011, 23, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Chi, M.; Sun, X.; Wang, G.; Li, M.; Liu, L.; Li, X. Propofol-induced protection of SH-SY5Y cells against hydrogen peroxide is associated with the HO-1 via the ERK pathway. Int. J. Med. Sci. 2013, 10, 599–606. [Google Scholar] [CrossRef]
- Liang, C.; Cang, J.; Wang, H.; Xue, Z. Propofol attenuates cerebral ischemia/reperfusion injury partially using heme oxygenase-1. J. Neurosurg. Anesthesiol. 2013, 25, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, Y.; Zhou, Q.; Tang, Q.; Zou, H. Comparison of the effects of propofol and midazolam on inflammation and oxidase stress in children with congenital heart disease undergoing cardiac surgery. Yonsei Med. J. 2011, 52, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Peltoniemi, M.A.; Hagelberg, N.M.; Olkkola, K.T.; Saari, T.I. Ketamine: A review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin. Pharmacokinet. 2016, 55, 1059–1077. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A.; et al. Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, Q.; She, Y.; Bi, X.; Zhao, B. Ketamine reduces LPS-induced HMGB1 via activation of the Nrf2/HO-1 pathway and NF-κB suppression. J. Trauma Acute Care Surg. 2015, 78, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Singh, P.M. Current role of dexmedetomidine in clinical anesthesia and intensive care. Anesth. Essays Res. 2011, 5, 128–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Wang, Y.; Zhao, J.; Su, A. Effects of dexmedetomidine pretreatment on heme oxygenase-1 expression and oxidative stress during one-lung ventilation. Int. J. Clin. Exp. Pathol. 2015, 8, 3144–3149. [Google Scholar] [PubMed]
- Feng, X.; Guan, W.; Zhao, Y.; Wang, C.; Song, M.; Yao, Y.; Yang, T.; Fan, H. Dexmedetomidine ameliorates lipopolysaccharide-induced acute kidney injury in rats by inhibiting inflammation and oxidative stress via the GSK-3β/Nrf2 signaling pathway. J. Cell. Physiol. 2019, 234, 18994–19009. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xia, M.; Huang, Q.; Ding, D.; Li, Y.; Zhang, Z.; Zhang, X. Protective effect of dexmedetomidine against organ dysfunction in a two-hit model of hemorrhage/resuscitation and endotoxemia in rats. Braz. J. Med. Biol. Res. 2019, 52. [Google Scholar] [CrossRef] [PubMed]
- Stokfisz, K.; Ledakowicz-Polak, A.; Zagorski, M.; Zielinska, M. Ischaemic preconditioning—Current knowledge and potential future applications after 30 years of experience. Adv. Med. Sci. 2017, 62, 307–316. [Google Scholar] [CrossRef] [PubMed]
- McClanahan, T.B.; Nao, B.S.; Wolke, L.J.; Martin, B.J.; Mertz, T.E.; Gallagher, K.P. Brief renal occlusion and reperfusion reduces myocardial infarct size in rabbits. Faseb J. 1993, 7, A118. [Google Scholar]
- Kleinbongard, P.; Skyschally, A.; Heusch, G. Cardioprotection by remote ischemic conditioning and its signal transduction. Eur. J. Physiol. 2017, 469, 159–181. [Google Scholar] [CrossRef]
- Zhou, G.; Li, M.H.; Tudor, G.; Lu, H.T.; Kadirvel, R.; Kallmes, D. Remote ischemic conditioning in cerebral diseases and neurointerventional procedures: Recent research progress. Front. Neurol. 2018, 9, 339. [Google Scholar] [CrossRef]
- Zhou, C.; Jeon, Y.; Meybohm, P.; Zarbock, A.; Young, P.J.; Li, L.; Hausenloy, D.J. Renoprotection by remote ischemic conditioning during elective coronary revascularization: A systematic review and meta-analysis of randomized controlled trials. Int. J. Cardiol. 2016, 222, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, I.-R.; Chang, K.-J.; Chen, C.-F.; Tsai, H.-W. Transient limb ischemia induces remote preconditioning in liver among rats: The protective role of heme oxygenase-1. Transplantation 2006, 81, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, J.; Xiong, X.; Xu, Y.; Zhang, H.; Huang, C.; Tian, Y.; Jiao, C.; Wang, X.; Li, X. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PLoS ONE 2014, 9, e98834. [Google Scholar] [CrossRef] [PubMed]
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 2019, 39, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Zhang, Z.; Liu, S.; Bi, J.; Zhang, J.; Du, L.; Ding, X.; Liu, C. Remote ischemic conditioning protects against acetaminophen-induced acute liver injury in mice. Hepatol. Res. 2017, 47, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Czigany, Z.; Bleilevens, C.; Beckers, C.; Stoppe, C.; Möhring, M.; Fülöp, A.; Szijarto, A.; Lurje, G.; Neumann, U.P.; Tolba, R.H. Limb remote ischemic conditioning of the recipient protects the liver in a rat model of arterialized orthotopic liver transplantation. PLoS ONE 2018, 13, e0195507. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Hata, K.; Tanaka, H.; Hirao, H.; Kubota, T.; Okamura, Y.; Iwaisako, K.; Takada, Y.; Uemoto, S. Intestinal ischemic preconditioning ameliorates hepatic ischemia/reperfusion injury in rats: Role of heme oxygenase 1 in the second window of protection. Liver Transplant. 2015, 21, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Caldarone, C.A.; Guan, R.; Wen, X.-Y.; Ailenberg, M.; Kapus, A.; Szaszi, K.; Rotstein, O.D. Nuclear factor (erythroid-derived 2)-like 2 regulates the hepatoprotective effects of remote ischemic conditioning in hemorrhagic shock. Antioxid. Redox Signal. 2019, 30, 1760–1773. [Google Scholar] [CrossRef] [PubMed]
- Jan, W.-C.; Chen, C.-H.; Tsai, P.-S.; Huang, C.-J. Limb ischemic preconditioning mitigates lung injury induced by haemorrhagic shock/resuscitation in rats. Resuscitation 2011, 82, 760–766. [Google Scholar] [CrossRef]
- Zhou, C.; Li, L.; Li, H.; Gong, J.; Fang, N. Delayed remote preconditioning induces cardioprotection: Role of heme oxygenase-1. J. Surg. Res. 2014, 191, 51–57. [Google Scholar] [CrossRef]
- Zhou, C.; Li, H.; Yao, Y.; Li, L. Delayed remote ischemic preconditioning produces an additive cardioprotection to sevoflurane postconditioning through an enhanced heme oxygenase 1 level partly via nuclear factor erythroid 2-related factor 2 nuclear translocation. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 558–566. [Google Scholar] [CrossRef]
- Hussein, A.M.; Harraz, A.M.; Awadalla, A.; Barakat, N.; Khater, S.; Shokeir, A.A. Remote limb ischemic preconditioning (rIPC) activates antioxidant and antiapoptotic genes and inhibits proinflammatory cytokine genes in renal ischemia/reperfusion injury. Gen. Physiol. Biophys. 2016, 35, 77–86. [Google Scholar] [PubMed]
- Saeki, I.; Matsuura, T.; Hayashida, M.; Taguchi, T. Ischemic preconditioning and remote ischemic preconditioning have protective effect against cold ischemia-reperfusion injury of rat small intestine. Pediatr. Surg. Int. 2011, 27, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Cremers, N.A.J.; Wever, K.E.; Wong, R.J.; van Rheden, R.E.M.; Vermeij, E.A.; van Dam, G.M.; Carels, C.E.; Lundvig, D.M.S.; Wagener, F.A. Effects of remote ischemic preconditioning on heme oxygenase-1 expression and cutaneous wound repair. Int. J. Mol. Sci. 2017, 18, 438. [Google Scholar] [CrossRef] [PubMed]
- Wever, K.E.; Warlé, M.C.; Wagener, F.A.; van der Hoorn, J.W.; Masereeuw, R.; van der Vliet, J.A.; Rongen, G.A. Remote ischaemic preconditioning by brief hind limb ischaemia protects against renal ischaemia-reperfusion injury: The role of adenosine. Nephrol. Dial. Transplant. 2011, 26, 3108–3117. [Google Scholar] [CrossRef]
- Zhang, X.; Jizhang, Y.; Xu, X.; Kwiecien, T.D.; Li, N.; Zhang, Y.; Ji, X.; Ren, C.; Ding, Y. Protective effects of remote ischemic conditioning against ischemia/reperfusion-induced retinal injury in rats. Vis. Neurosci. 2014, 31, 245–252. [Google Scholar] [CrossRef]
- Ramagiri, S.; Taliyan, R. Protective effect of remote limb post conditioning via upregulation of heme oxygenase-1/BDNF pathway in rat model of cerebral ischemic reperfusion injury. Brain Res. 2017, 1669, 44–54. [Google Scholar] [CrossRef]
- Gao, S.; Zhan, L.; Yang, Z.; Shi, R.; Li, H.; Xia, Z.; Yuan, S.; Wu, Q.-P.; Wang, T.; Yao, S. Remote limb ischaemic postconditioning protects against myocardial ischaemia/reperfusion injury in mice: Activation of JAK/STAT3-mediated Nrf2-antioxidant signalling. Cell. Physiol. Biochem. 2017, 43, 1140–1151. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, I.; Raupach, A. The Role of Heme Oxygenase-1 in Remote Ischemic and Anesthetic Organ Conditioning. Antioxidants 2019, 8, 403. https://doi.org/10.3390/antiox8090403
Bauer I, Raupach A. The Role of Heme Oxygenase-1 in Remote Ischemic and Anesthetic Organ Conditioning. Antioxidants. 2019; 8(9):403. https://doi.org/10.3390/antiox8090403
Chicago/Turabian StyleBauer, Inge, and Annika Raupach. 2019. "The Role of Heme Oxygenase-1 in Remote Ischemic and Anesthetic Organ Conditioning" Antioxidants 8, no. 9: 403. https://doi.org/10.3390/antiox8090403