

Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
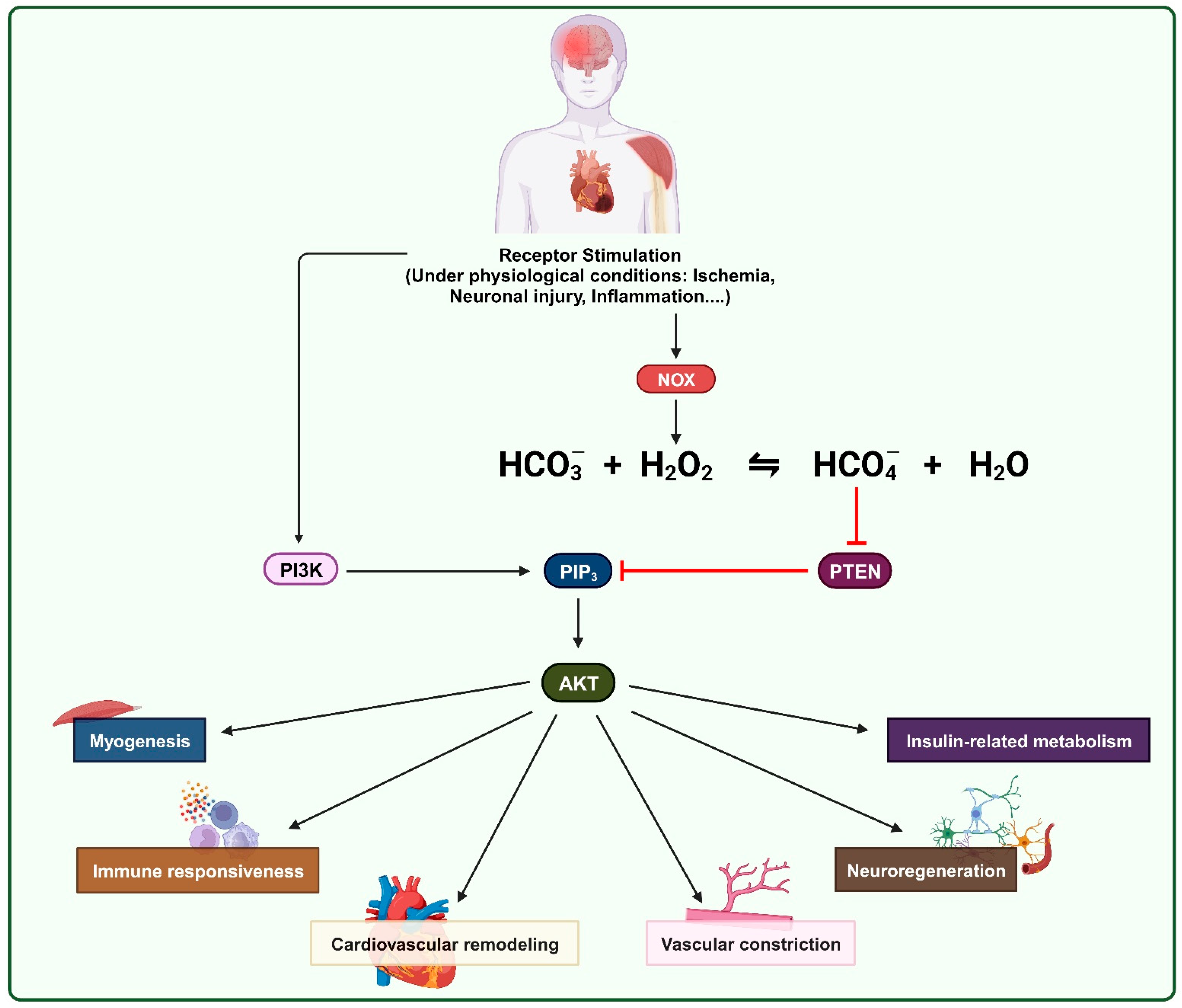
2. Oxidative Inhibition of PTEN by ROS in Physiological Processes
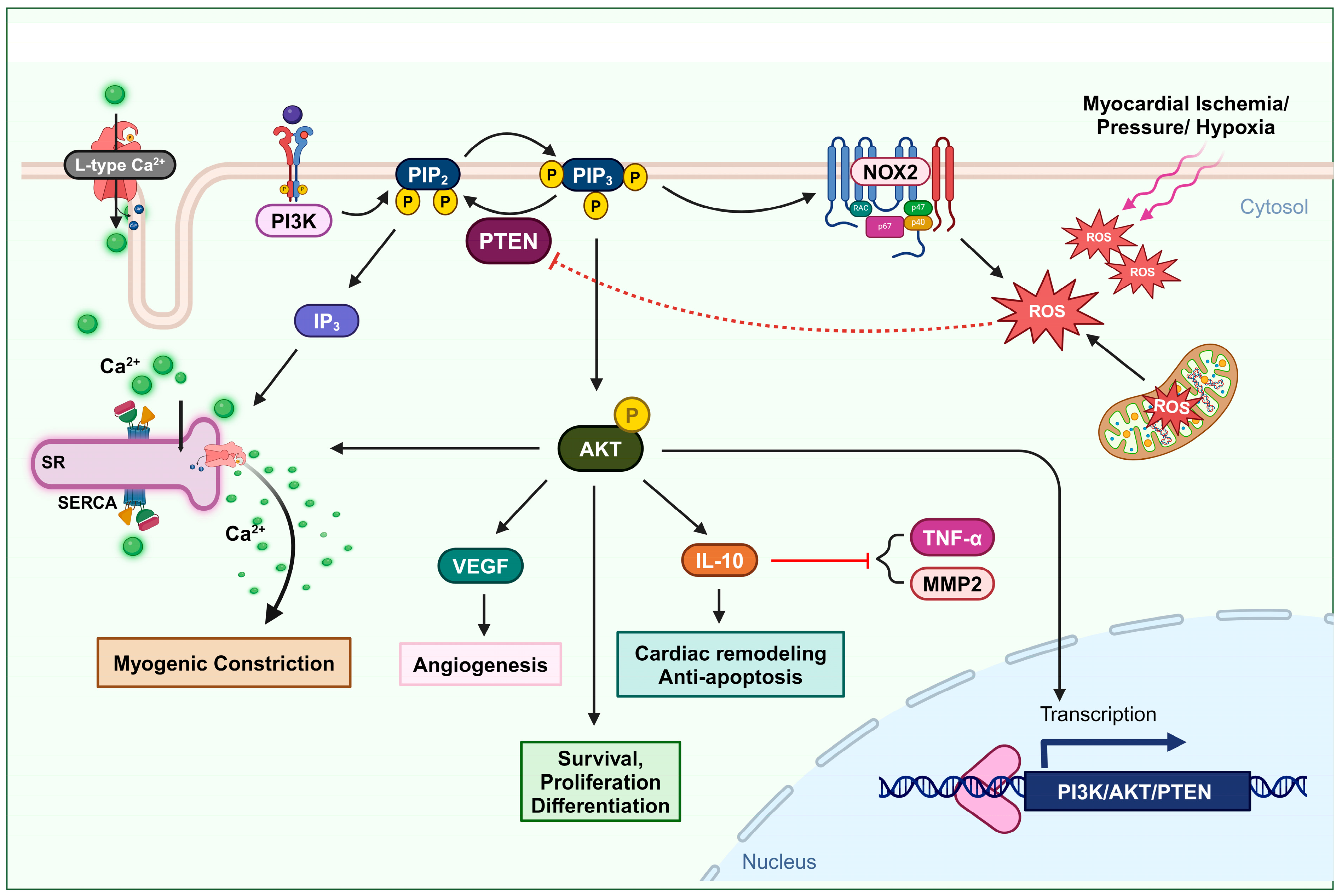
2.1. Cardiovascular Remodeling
2.2. Vascular Constriction
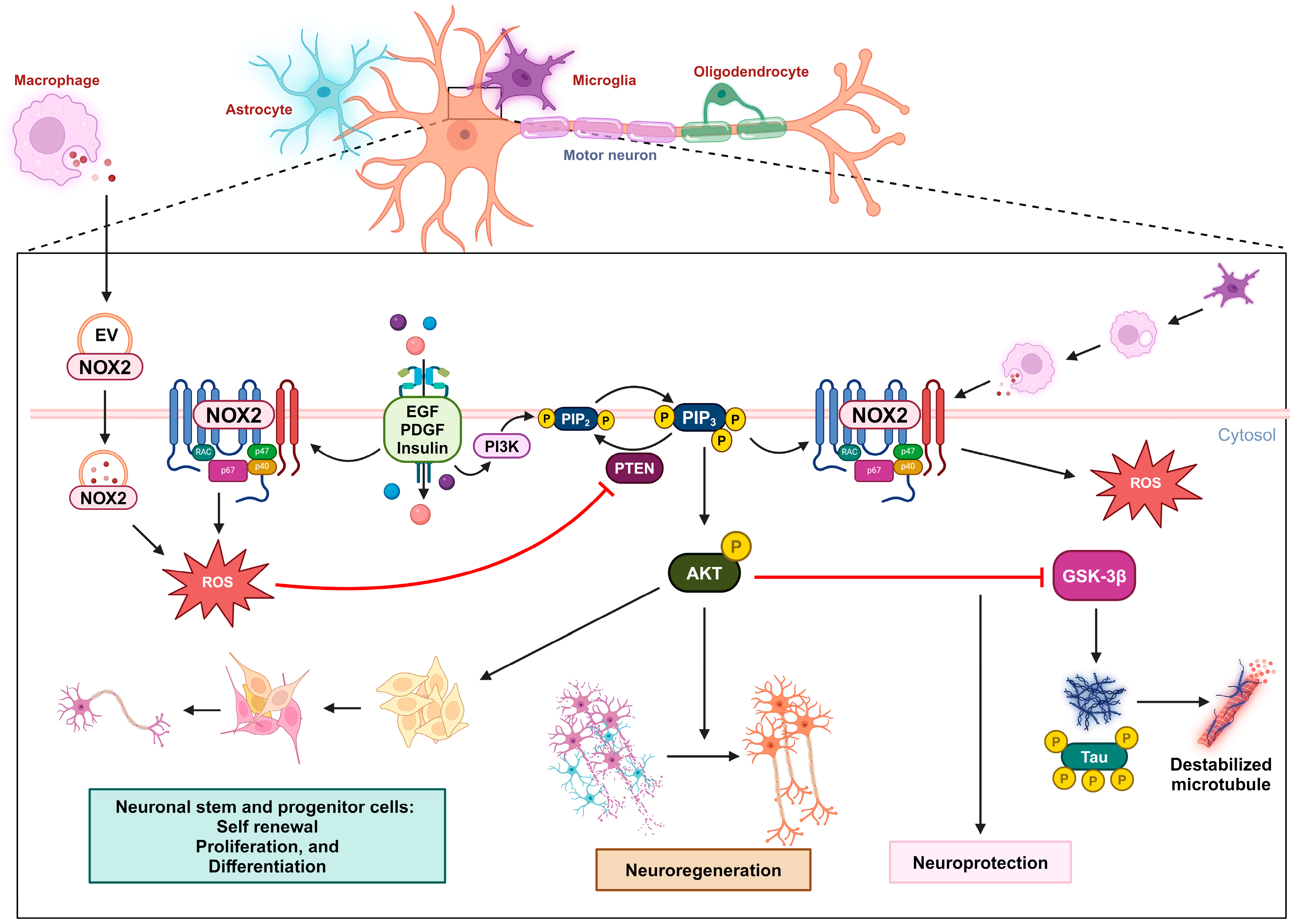
2.3. Neuro-Regeneration and Neuro-Survival
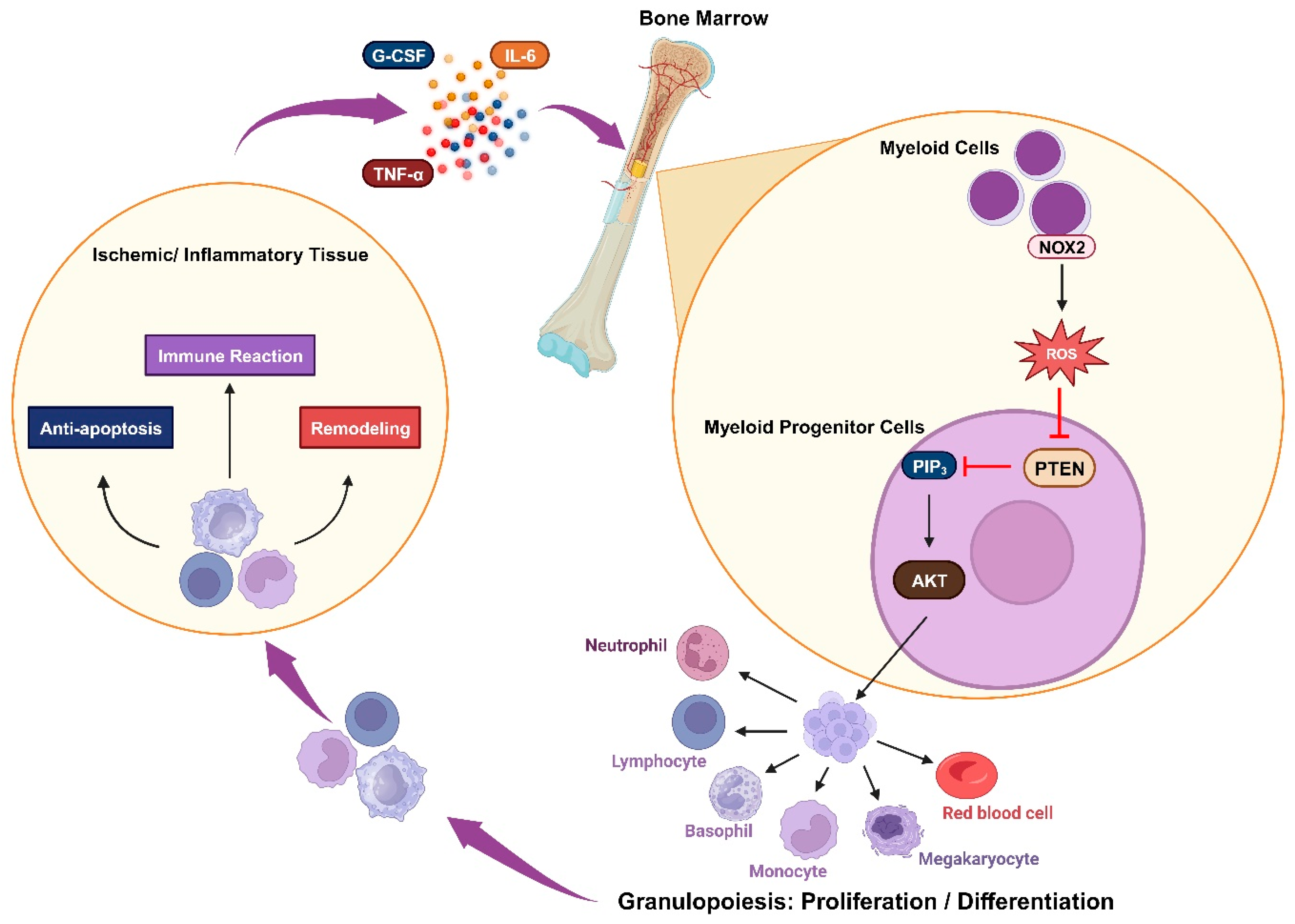
2.4. Immune Responsiveness
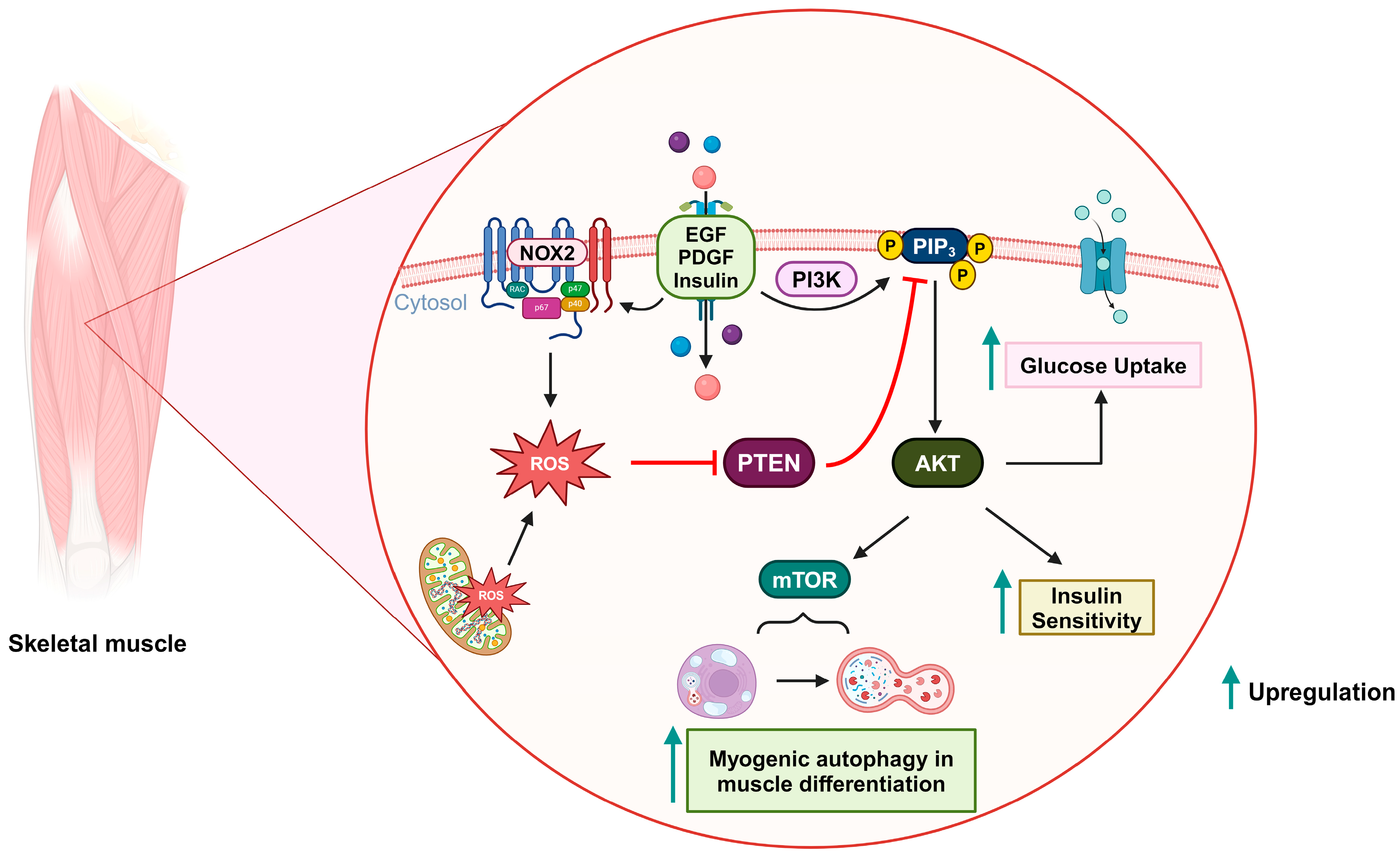
2.5. Insulin-Related Metabolism
2.6. Myogenic Autophagy in Muscle Differentiation
3. Role of Bicarbonate in the Oxidation of PTPs by H2O2
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, S.-J.; Park, I.; Kim, I.; Chay, K.-O.; Kim, S.M.; Jang, D.I.; Lee, T.-H.; Lee, S.-R. Redox regulation of the tumor suppressor PTEN by hydrogen peroxide and tert-butyl hydroperoxide. Int. J. Mol. Sci. 2017, 18, 982. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-J.; Zhang, Y.; Kim, I.; Chay, K.-O.; Yoon, H.J.; Jang, D.I.; Yang, S.Y.; Park, J.; Woo, H.A.; Park, I. Redox regulation of the tumor suppressor PTEN by the thioredoxin system and cumene hydroperoxide. Free Radic. Biol. Med. 2017, 112, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-J.; Ahn, Y.; Park, I.; Zhang, Y.; Kim, I.; Kim, H.W.; Ku, C.-S.; Chay, K.-O.; Yang, S.Y.; Ahn, B.W. Assay of the redox state of the tumor suppressor PTEN by mobility shift. Methods 2015, 77, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.S.; Gunasekar, P.; Agrawal, D.K. An update on PTEN modulators—A patent review. Expert Opin. Ther. Pat. 2019, 29, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, G.M.; Dennis, P.A. PTEN hamartoma tumor syndromes. Eur. J. Hum. Genet. 2008, 16, 1289–1300. [Google Scholar] [CrossRef]
- Baig, R.M.; Mahjabeen, I.; Sabir, M.; Masood, N.; Hafeez, S.; Malik, F.A.; Kayani, M.A. Genetic changes in the PTEN gene and their association with breast cancer in Pakistan. Asian Pac. J. Cancer Prev. 2011, 12, 2773–2778. [Google Scholar]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacoke, M. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Norimatsu, Y.; Moriya, T.; Kobayashi, T.K.; Sakurai, T.; Shimizu, K.; Tsukayama, C.; Ohno, E. Immunohistochemical expression of PTEN and β-catenin for endometrial intraepithelial neoplasia in Japanese women. Ann. Diagn. Pathol. 2007, 11, 103–108. [Google Scholar] [CrossRef]
- Patel, R.; Gao, M.; Ahmad, I.; Fleming, J.; Singh, L.B.; Rai, T.S.; McKie, A.B.; Seywright, M.; Barnetson, R.J.; Edwards, J. Sprouty2, PTEN, and PP2A interact to regulate prostate cancer progression. J. Clin. Investig. 2013, 123, 1157–1175. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Wang, J.; Chen, H.; Fang, J.-Y. Combined PTEN mutation and protein expression associate with overall and disease-free survival of glioblastoma patients. Transl. Oncol. 2014, 7, 196–205.e1. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.; Schepis, C. PTEN gene: A model for genetic diseases in dermatology. Sci. World J. 2012, 2012, 252457. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R. PTEN inhibition in human disease therapy. Molecules 2018, 23, 285. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez Brito, M.; Goulielmaki, E.; Papakonstanti, E.A. Focus on PTEN regulation. Front. Oncol. 2015, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y.; Han, L.; Piao, J. MicroRNA302-367-PI3K-PTEN-AKT-mTORC1 pathway promotes the development of cardiac hypertrophy through controlling autophagy. In Vitr. Cell. Dev. Biol.-Anim. 2020, 56, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Fan, J.; Li, H.; Yin, Z.; Zhao, Y.; Dai, B.; Dong, N.; Chen, C.; Wang, D.W. miR-217 promotes cardiac hypertrophy and dysfunction by targeting PTEN. Mol. Ther.-Nucleic Acids 2018, 12, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.y.; Chen, C.; Xu, X.; Lu, Q. miR-29a promotes pathological cardiac hypertrophy by targeting the PTEN/AKT/mTOR signalling pathway and suppressing autophagy. Acta Physiol. 2019, 227, e13323. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Song, X.W.; Li, Q.; Wang, G.K.; Jing, Q.; Qin, Y.W. Attenuation of microRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J. Cell. Physiol. 2012, 227, 1391–1398. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Sayad, A.; Taheri, M. Regulatory role of microRNAs on PTEN signaling. Biomed. Pharmacother. 2021, 133, 110986. [Google Scholar] [CrossRef]
- Denu, J.M.; Dixon, J.E. Protein tyrosine phosphatases: Mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 1998, 2, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Huu, T.; Park, J.; Zhang, Y.; Duong Thanh, H.; Park, I.; Choi, J.M.; Yoon, H.J.; Park, S.C.; Woo, H.A.; Lee, S.R. The Role of Oxidative Inactivation of Phosphatase PTEN and TCPTP in Fatty Liver Disease. Antioxidants 2023, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Oxidative stress and cardiac repair/remodeling following infarction. Am. J. Med. Sci. 2007, 334, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.-C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Jeong, W.; Chang, T.S.; Yang, K.S.; Woo, H.A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 2005, 17, 183–189. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chang, T.S.; Bae, Y.S.; Lee, S.R.; Kang, S.W. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol. 2003, 14, S211–S215. [Google Scholar] [CrossRef]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and oxidative stress in human diseases: From molecular mechanisms to novel treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Rhee, S.G. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Cheng, G.; Arnold, R.S.; Edens, W.A. Novel homologs of gp91phox. Trends Biochem. Sci. 2000, 25, 459–461. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Lee, S.-R.; Yang, K.-S.; Kwon, J.; Kang, S.W. Hydrogen peroxide as intracellular messenger: Identification of protein tyrosine phosphatases and PTEN as H2O2 target. In Signal Transduction by Reactive Oxygen and Nitrogen Species: Pathways and Chemical Principles; Springer: Dordrecht, The Netherlands, 2003; pp. 167–179. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Song, Y.B.; Kim, T.-Y.; Kim, I.; Han, S.-J.; Ahn, Y.; Cho, S.-H.; Choi, C.Y.; Chay, K.-O.; Yang, S.Y. Redox regulation of the tumor suppressor PTEN by glutathione. FEBS Lett. 2010, 584, 3550–3556. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chay, K.-O.; Kim, I.; Song, Y.B.; Kim, T.-Y.; Han, S.-J.; Ahn, Y.; Cho, S.-H.; Hoe, K.-L.; Ahn, B.W. Redox regulation of the tumor suppressor PTEN by glutaredoxin 5 and Ycp4. Biochem. Biophys. Res. Commun. 2011, 407, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, J.; Han, S.-J.; Lim, Y.; Park, I.; Kim, J.-S.; Woo, H.; Lee, S.-R. Peroxiredoxin III protects tumor suppressor PTEN from oxidation by 15-Hydroperoxy-eicosatetraenoic acid. Oxidative Med. Cell. Longev. 2019, 2019, 2828493. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Huu, T.; Park, J.; Zhang, Y.; Park, I.; Yoon, H.J.; Woo, H.A.; Lee, S.R. Redox Regulation of PTEN by Peroxiredoxins. Antioxidants 2021, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Takakura, K.; Beckman, J.S.; MacMillan-Crow, L.A.; Crow, J.P. Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1B, CD45, and LAR by peroxynitrite. Arch. Biochem. Biophys. 1999, 369, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R. The redox regulation of PI 3-kinase-dependent signaling. Antioxid. Redox Signal. 2006, 8, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Downes, C.P.; Ross, S.; Maccario, H.; Perera, N.; Davidson, L.; Leslie, N.R. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv. Enzym. Regul. 2007, 47, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Peskin, A.V.; Kleffmann, T.; Radi, R.; Pace, P.E. Carbon dioxide/bicarbonate is required for sensitive inactivation of mammalian glyceraldehyde-3-phosphate dehydrogenase by hydrogen peroxide. Proc. Natl. Acad. Sci. USA 2023, 120, e2221047120. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Interplay of carbon dioxide and peroxide metabolism in mammalian cells. J. Biol. Chem. 2022, 298, 102358. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ. Res. 2004, 94, 7–16. [Google Scholar] [CrossRef]
- Jonassen, A.K.; Sack, M.N.; Mjøs, O.D.; Yellon, D.M. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 2001, 89, 1191–1198. [Google Scholar] [CrossRef]
- Matsui, T.; Li, L.; del Monte, F.; Fukui, Y.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation 1999, 100, 2373–2379. [Google Scholar] [CrossRef]
- Uchiyama, T.; Engelman, R.M.; Maulik, N.; Das, D.K. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 2004, 109, 3042–3049. [Google Scholar] [CrossRef]
- Tong, H.; Chen, W.; Steenbergen, C.; Murphy, E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 2000, 87, 309–315. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Bell, R.M.; Yellon, D.M. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J. Mol. Cell. Cardiol. 2002, 34, 661–668. [Google Scholar] [CrossRef]
- Mocanu, M.; Yellon, D. PTEN, the Achilles’ heel of myocardial ischaemia/reperfusion injury? Br. J. Pharmacol. 2007, 150, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, N.; Yuan, Y.; Zheng, X.; Bedja, D.; Cai, Z.P. Phosphatase PTEN is critically involved in post-myocardial infarction remodeling through the Akt/interleukin-10 signaling pathway. Basic Res. Cardiol. 2012, 107, 248. [Google Scholar] [CrossRef]
- Burchfield, J.S.; Iwasaki, M.; Koyanagi, M.; Urbich, C.; Rosenthal, N.; Zeiher, A.M.; Dimmeler, S. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circ. Res. 2008, 103, 203–211. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.; Seybold, K.; Petzi, S.; Wasmeier, G.; Raaz, D.; Yilmaz, A.; Anger, T.; Daniel, W.G.; Garlichs, C.D. Interleukin-10 improves left ventricular function in rats with heart failure subsequent to myocardial infarction. Eur. J. Heart Fail. 2008, 10, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zingarelli, B.; Szabó, C. Crucial role of endogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation 2000, 101, 1019–1026. [Google Scholar] [CrossRef]
- Keyes, K.T.; Xu, J.; Long, B.; Zhang, C.; Hu, Z.; Ye, Y. Pharmacological inhibition of PTEN limits myocardial infarct size and improves left ventricular function postinfarction. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H1198–H1208. [Google Scholar] [CrossRef]
- Ruan, H.; Li, J.; Ren, S.; Gao, J.; Li, G.; Kim, R.; Wu, H.; Wang, Y. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2009, 46, 193–200. [Google Scholar] [CrossRef]
- Fukui, T.; Yoshiyama, M.; Hanatani, A.; Omura, T.; Yoshikawa, J.; Abe, Y. Expression of p22-phox and gp91-phox, essential components of NADPH oxidase, increases after myocardial infarction. Biochem. Biophys. Res. Commun. 2001, 281, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Krijnen, P.; Meischl, C.; Hack, C.; Meijer, C.; Visser, C.; Roos, D.; Niessen, H. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 2003, 56, 194–199. [Google Scholar] [CrossRef]
- Sirker, A.; Murdoch, C.E.; Protti, A.; Sawyer, G.J.; Santos, C.X.; Martin, D.; Zhang, X.; Brewer, A.C.; Zhang, M.; Shah, A.M. Cell-specific effects of Nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell. Cardiol. 2016, 98, 11–17. [Google Scholar] [CrossRef]
- Cai, Z.; Semenza, G.L. PTEN activity is modulated during ischemia and reperfusion: Involvement in the induction and decay of preconditioning. Circ. Res. 2005, 97, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Lu, Y.; Xin, L.; Gao, J.; Shang, C.; Jiang, Z.; Lin, H.; Fang, X.; Qu, Y.; Wang, Y. Role of oxidative stress in reperfusion following myocardial ischemia and its treatments. Oxidative Med. Cell. Longev. 2021, 2021, 6614009. [Google Scholar] [CrossRef]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef]
- Kanazawa, M.; Takahashi, T.; Ishikawa, M.; Onodera, O.; Shimohata, T.; Del Zoppo, G.J. Angiogenesis in the ischemic core: A potential treatment target? J. Cereb. Blood Flow Metab. 2019, 39, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Abo-Gresha, N.M. Rosuvastatin promotes angiogenesis and reverses isoproterenol-induced acute myocardial infarction in rats: Role of iNOS and VEGF. Eur. J. Pharmacol. 2012, 691, 134–142. [Google Scholar] [CrossRef]
- Connor, K.M.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.-T.; Aguirre-Ghiso, J.; Flores, S.C. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef]
- Latronico, M.V.; Costinean, S.; Lavitrano, M.L.; Peschle, C.; Condorelli, G. Regulation of cell size and contractile function by AKT in cardiomyocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 250–260. [Google Scholar] [CrossRef]
- Saward Peter Zahradka, L. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ. Res. 1997, 81, 249–257. [Google Scholar] [CrossRef]
- Sugden, P.H. Ras, Akt, and mechanotransduction in the cardiac myocyte. Circ. Res. 2003, 93, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- McDowell, S.A.; McCall, E.; Matter, W.F.; Estridge, T.B.; Vlahos, C.J. Phosphoinositide 3-kinase regulates excitation-contraction coupling in neonatal cardiomyocytes. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H796–H805. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.A.; Ammit, A.J.; Irani, C.; Carroll, R.G.; Eszterhas, A.J.; Panettieri, R.A.; Krymskaya, V.P. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2002, 283, L354–L363. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Schroder, J.N.; Lima, B.; Villamizar, N.; Nienaber, J.J.; Milano, C.A.; Naga Prasad, S.V. Dynamic regulation of phosphoinositide 3-kinase-γ activity and β-adrenergic receptor trafficking in end-stage human heart failure. Circulation 2007, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Namgaladze, D.; Brüne, B. Phospholipase A2–modified low-density lipoprotein activates the phosphatidylinositol 3-kinase-akt pathway and increases cell survival in monocytic cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Northcott, C.A.; Hayflick, J.S.; Watts, S.W. PI3-Kinase upregulation and involvement in spontaneous tone in arteries from DOCA-salt rats: Is p110δ the culprit? Hypertension 2004, 43, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Wu, C.-A.; Wu, C.-W.; Chan, S.H.; Chang, A.Y.; Chan, J.Y. Redox-sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxid. Redox Signal. 2013, 18, 36–50. [Google Scholar] [CrossRef]
- Gebremedhin, D.; Terashvili, M.; Wickramasekera, N.; Zhang, D.X.; Rau, N.; Miura, H.; Harder, D.R. Redox signaling via oxidative inactivation of PTEN modulates pressure-dependent myogenic tone in rat middle cerebral arteries. PLoS ONE 2013, 8, e68498. [Google Scholar] [CrossRef]
- Carnevale, D.; Vecchione, C.; Mascio, G.; Esposito, G.; Cifelli, G.; Martinello, K.; Landolfi, A.; Selvetella, G.; Grieco, P.; Damato, A. PI3Kγ inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc. Res. 2012, 93, 200–209. [Google Scholar] [CrossRef]
- Ning, K.; Pei, L.; Liao, M.; Liu, B.; Zhang, Y.; Jiang, W.; Mielke, J.G.; Li, L.; Chen, Y.; El-Hayek, Y.H.; et al. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J. Neurosci. 2004, 24, 4052–4060. [Google Scholar] [CrossRef]
- Mao, L.; Jia, J.; Zhou, X.; Xiao, Y.; Wang, Y.; Mao, X.; Zhen, X.; Guan, Y.; Alkayed, N.J.; Cheng, J. Delayed administration of a PTEN inhibitor BPV improves functional recovery after experimental stroke. Neuroscience 2013, 231, 272–281. [Google Scholar] [CrossRef]
- Zhao, J.; Qu, Y.; Wu, J.; Cao, M.; Ferriero, D.; Zhang, L.; Mu, D. PTEN inhibition prevents rat cortical neuron injury after hypoxia–ischemia. Neuroscience 2013, 238, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, J.; Hu, H.; Liu, P.; Fang, Y.; Wu, D. Rho-kinase inhibitor, fasudil, prevents neuronal apoptosis via the Akt activation and PTEN inactivation in the ischemic penumbra of rat brain. Cell. Mol. Neurobiol. 2012, 32, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-Y.; Ding, J.; Yuan, F.; Chen, H.; Chen, S.-W.; Tian, H.-L. Dose-dependent protective effect of bisperoxovanadium against acute cerebral ischemia in a rat model of ischemia/reperfusion injury. Int. J. Mol. Sci. 2013, 14, 12013–12022. [Google Scholar] [CrossRef]
- Wu, D.-N.; Pei, D.-S.; Wang, Q.; Zhang, G.-Y. Down-regulation of PTEN by sodium orthovanadate inhibits ASK1 activation via PI3-K/Akt during cerebral ischemia in rat hippocampus. Neurosci. Lett. 2006, 404, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006, 34, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Christie, K.J.; Zochodne, D. Peripheral axon regrowth: New molecular approaches. Neuroscience 2013, 240, 310–324. [Google Scholar] [CrossRef]
- Garcia-Junco-Clemente, P.; Golshani, P. PTEN: A master regulator of neuronal structure, function, and plasticity. Commun. Integr. Biol. 2014, 7, e28358. [Google Scholar] [CrossRef]
- Knafo, S.; Esteban, J.A. PTEN: Local and Global Modulation of Neuronal Function in Health and Disease. Trends Neurosci. 2017, 40, 83–91. [Google Scholar] [CrossRef]
- Ohtake, Y.; Hayat, U.; Li, S. PTEN inhibition and axon regeneration and neural repair. Neural Regen. Res. 2015, 10, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Kanter, J.L.; He, Z. PTEN/mTOR and axon regeneration. Exp. Neurol. 2010, 223, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, L.; Chen, Y.; Zhan, L.; Gao, Z. Therapeutic Targets for Cerebral Ischemia Based on the Signaling Pathways of the GluN2B C Terminus. Stroke 2015, 46, 2347–2353. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P.; Rabin, S.L.; Cantley, L.C.; Kaplan, D.R. Nerve growth factor promotes the activation of phosphatidylinositol 3-kinase and its association with the trk tyrosine kinase. J. Biol. Chem. 1992, 267, 17472–17477. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lu, Y.; Lee, J.K.; Samara, R.; Willenberg, R.; Sears-Kraxberger, I.; Tedeschi, A.; Park, K.K.; Jin, D.; Cai, B.; et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 2010, 13, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Christie, K.J.; Webber, C.A.; Martinez, J.A.; Singh, B.; Zochodne, D.W. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J. Neurosci. 2010, 30, 9306–9315. [Google Scholar] [CrossRef]
- Little, D.; Valori, C.F.; Mutsaers, C.A.; Bennett, E.J.; Wyles, M.; Sharrack, B.; Shaw, P.J.; Gillingwater, T.H.; Azzouz, M.; Ning, K. PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol. Ther. 2015, 23, 270–277. [Google Scholar] [CrossRef]
- Ning, K.; Drepper, C.; Valori, C.F.; Ahsan, M.; Wyles, M.; Higginbottom, A.; Herrmann, T.; Shaw, P.; Azzouz, M.; Sendtner, M. PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum. Mol. Genet. 2010, 19, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, S.S.; Caplan, S. MICAL-family proteins: Complex regulators of the actin cytoskeleton. Antioxid. Redox Signal. 2014, 20, 2059–2073. [Google Scholar] [CrossRef]
- Zhu, Y.; Hoell, P.; Ahlemeyer, B.; Sure, U.; Bertalanffy, H.; Krieglstein, J. Implication of PTEN in production of reactive oxygen species and neuronal death in in vitro models of stroke and Parkinson’s disease. Neurochem. Int. 2007, 50, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef]
- Trinczek, B.; Biernat, J.; Baumann, K.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol. Biol. Cell 1995, 6, 1887–1902. [Google Scholar] [CrossRef]
- Higuchi, M.; Lee, V.M.; Trojanowski, J.Q. Tau and axonopathy in neurodegenerative disorders. Neuromol. Med. 2002, 2, 131–150. [Google Scholar] [CrossRef]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases 2018, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Esteban, M.; Martin-Zanca, D.; Andres-Martin, L.; Almeida, A.; Bolaños, J.P. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signaling pathway. J. Neurochem. 2007, 102, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Walker, F.; Zhang, H.-H.; Matthews, V.; Weinstock, J.; Nice, E.C.; Ernst, M.; Rose-John, S.; Burgess, A.W. IL6/sIL6R complex contributes to emergency granulopoietic responses in G-CSF–and GM-CSF–deficient mice. Blood J. Am. Soc. Hematol. 2008, 111, 3978–3985. [Google Scholar] [CrossRef]
- Gwechenberger, M.; Mendoza, L.H.; Youker, K.A.; Frangogiannis, N.G.; Smith, C.W.; Michael, L.H.; Entman, M.L. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 1999, 99, 546–551. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFα in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The mechanistic basis of infarct healing. Antioxid. Redox Signal. 2006, 8, 1907–1939. [Google Scholar] [CrossRef]
- Kwak, H.-J.; Liu, P.; Bajrami, B.; Xu, Y.; Park, S.-Y.; Nombela-Arrieta, C.; Mondal, S.; Sun, Y.; Zhu, H.; Chai, L. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity 2015, 42, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Perlman, H.; Pagliari, L.J.; Pope, R.M. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: Role of Mcl-1, independent of nuclear factor (NF)-κB, Bad, or caspase activation. J. Exp. Med. 2001, 194, 113–126. [Google Scholar] [CrossRef]
- Tiganis, T. Reactive oxygen species and insulin resistance: The good, the bad and the ugly. Trends Pharmacol. Sci. 2011, 32, 82–89. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Seo, J.H.; Ahn, Y.; Lee, S.-R.; Yeo, C.Y.; Hur, K.C. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol. Biol. Cell 2005, 16, 348–357. [Google Scholar] [CrossRef]
- Li, Y.Z.; Di Cristofano, A.; Woo, M. Metabolic role of PTEN in insulin signaling and resistance. Cold Spring Harb. Perspect. Med. 2020, 10, a036137. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Fuentealba, C.; Contreras-Ferrat, A.E.; Altamirano, F.; Espinosa, A.; Li, Q.; Niu, W.; Lavandero, S.; Klip, A.; Jaimovich, E. Electrical stimuli release ATP to increase GLUT4 translocation and glucose uptake via PI3Kγ-Akt-AS160 in skeletal muscle cells. Diabetes 2013, 62, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.D.; Summers, S.A.; Birnbaum, M.J.; Roth, R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 1996, 271, 31372–31378. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.F.; Yang, H.J.; Gu, J.Q.; Cao, Y.L.; Meng, X.; Wang, X.L.; Lin, Y.C.; Gao, M. Suppression of phosphatase and tensin homolog protects insulin-resistant cells from apoptosis. Mol. Med. Rep. 2015, 12, 2695–2700. [Google Scholar] [CrossRef]
- Nakashima, N.; Sharma, P.M.; Imamura, T.; Bookstein, R.; Olefsky, J.M. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3-L1 adipocytes. J. Biol. Chem. 2000, 275, 12889–12895. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Yan Lu, S.; Nguyen, K.-T.T.; Schroer, S.A.; Suzuki, A.; Mak, T.W.; Gaisano, H.; Woo, M. Deletion of Pten in pancreatic β-cells protects against deficient β-cell mass and function in mouse models of type 2 diabetes. Diabetes 2010, 59, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Konrad, D.; Eweida, M.; Jefferies, C.; Liadis, N.; Giacca, A.; Crackower, M.; Suzuki, A.; Mak, T.W.; Kahn, C.R. Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol. Cell. Biol. 2005, 25, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Rosivatz, E. Inhibiting PTEN. Biochem. Soc. Trans. 2007, 35, 257–259. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, T.G.; Park, S.; Yun, H.R.; Nguyen, N.N.Y.; Jo, Y.H.; Jang, M.; Kim, J.; Kim, J.; Kang, I.; et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 2018, 25, 1921–1937. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-R.; Kwon, K.-S.; Kim, S.-R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.J.; Parsons, Z.D.; Cummings, A.H.; Zhou, H.; Gates, K.S. Redox regulation of protein tyrosine phosphatases: Structural and chemical aspects. Antioxid. Redox Signal. 2011, 15, 77–97. [Google Scholar] [CrossRef]
- Zhou, H.; Singh, H.; Parsons, Z.D.; Lewis, S.M.; Bhattacharya, S.; Seiner, D.R.; LaButti, J.N.; Reilly, T.J.; Tanner, J.J.; Gates, K.S. The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. J. Am. Chem. Soc. 2011, 133, 15803–15805. [Google Scholar] [CrossRef]
- Bakhmutova-Albert, E.V.; Yao, H.; Denevan, D.E.; Richardson, D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 2010, 49, 11287–11296. [Google Scholar] [CrossRef]
- Trindade, D.F.; Cerchiaro, G.; Augusto, O. A role for peroxymonocarbonate in the stimulation of biothiol peroxidation by the bicarbonate/carbon dioxide pair. Chem. Res. Toxicol. 2006, 19, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Aalkjaer, C.; Boedtkjer, E.; Choi, I.; Lee, S. Cation-coupled bicarbonate transporters. Compr. Physiol. 2014, 4, 1605–1637. [Google Scholar] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh, V.H.; Nguyen Huu, T.; Sah, D.K.; Choi, J.M.; Yoon, H.J.; Park, S.C.; Jung, Y.S.; Lee, S.-R. Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes. Antioxidants 2024, 13, 199. https://doi.org/10.3390/antiox13020199
Trinh VH, Nguyen Huu T, Sah DK, Choi JM, Yoon HJ, Park SC, Jung YS, Lee S-R. Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes. Antioxidants. 2024; 13(2):199. https://doi.org/10.3390/antiox13020199
Chicago/Turabian StyleTrinh, Vu Hoang, Thang Nguyen Huu, Dhiraj Kumar Sah, Jin Myung Choi, Hyun Joong Yoon, Sang Chul Park, Yu Seok Jung, and Seung-Rock Lee. 2024. "Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes" Antioxidants 13, no. 2: 199. https://doi.org/10.3390/antiox13020199