
T3 Intratracheal Therapy Alleviates Pulmonary Pathology in an Elastase-Induced Emphysema-Dominant COPD Mouse Model
 , ,
, ,
Abstract
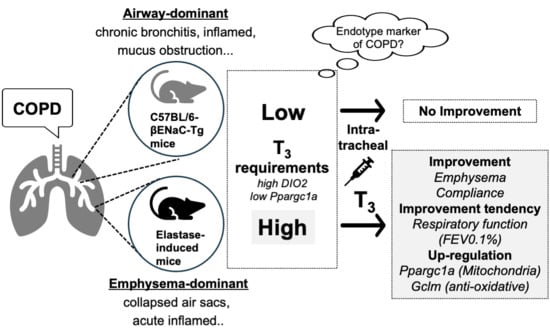
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Elastase-Induced COPD Model Mice
2.3. Administration of 3,3′,5-Triiodo-L-Thyronine (T3)
2.4. Measurement of Pulmonary Mechanics and Function by flexiVent
2.5. Sample Collection
2.6. Morphological Staining of Lung Tissue Section and Assessment of Emphysema
2.7. Image-Based Automatic Measurement of Alveolar Morphological Information
2.8. RNA Isolation, cDNA Synthesis, and Real-Time PCR
2.9. Immunoblotting
2.10. Measurement of Oxidative Stress and Plasma Antioxidant Capacity
2.11. Immunohistochemistry (IHC) and Quantification of Infiltration of Immune Cells
2.12. Statistical Analysis
3. Results
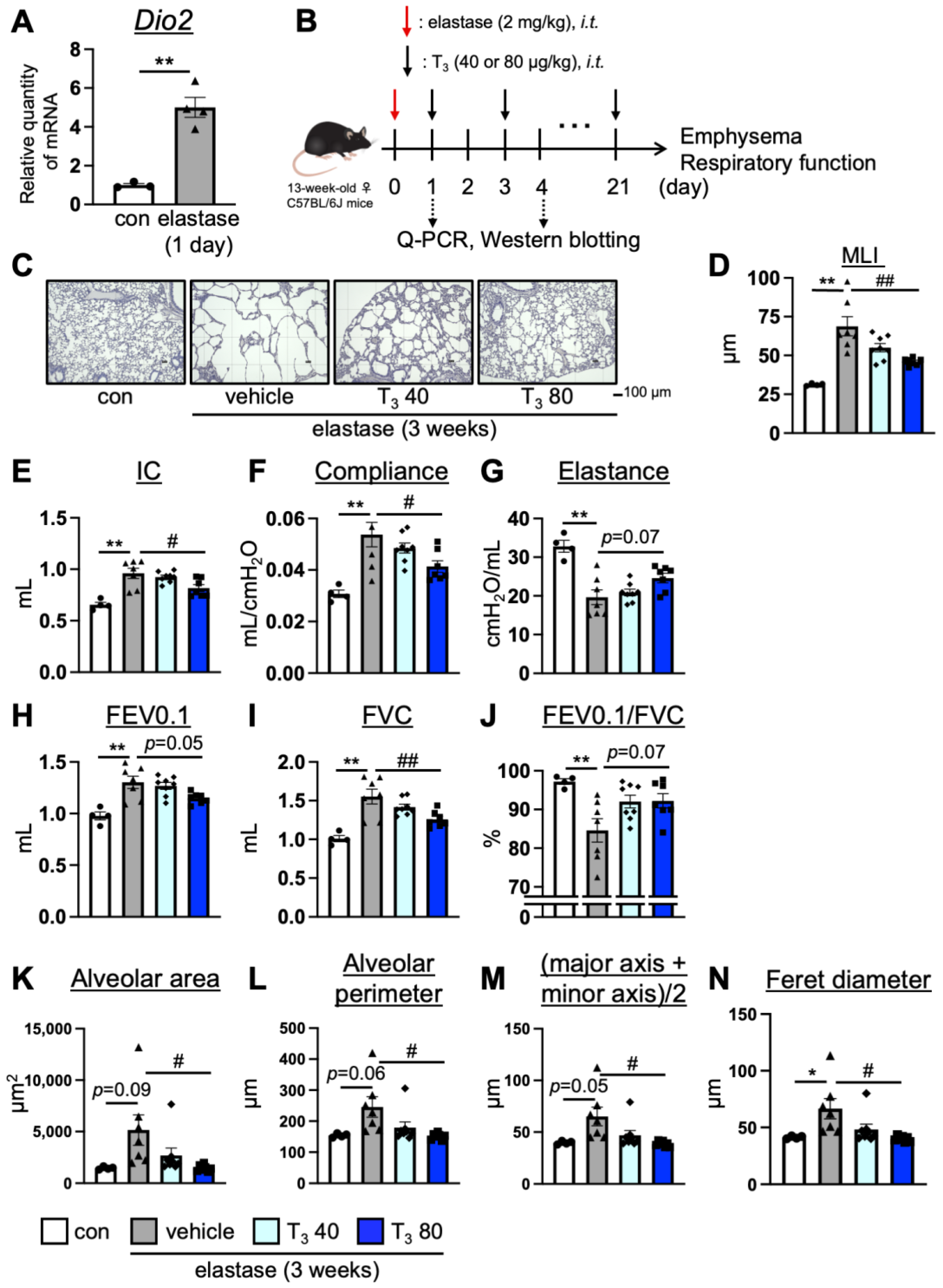
3.1. Intratracheal Administration of T3 Improves Pulmonary Pathology in Elastase-Induced COPD Mouse Model
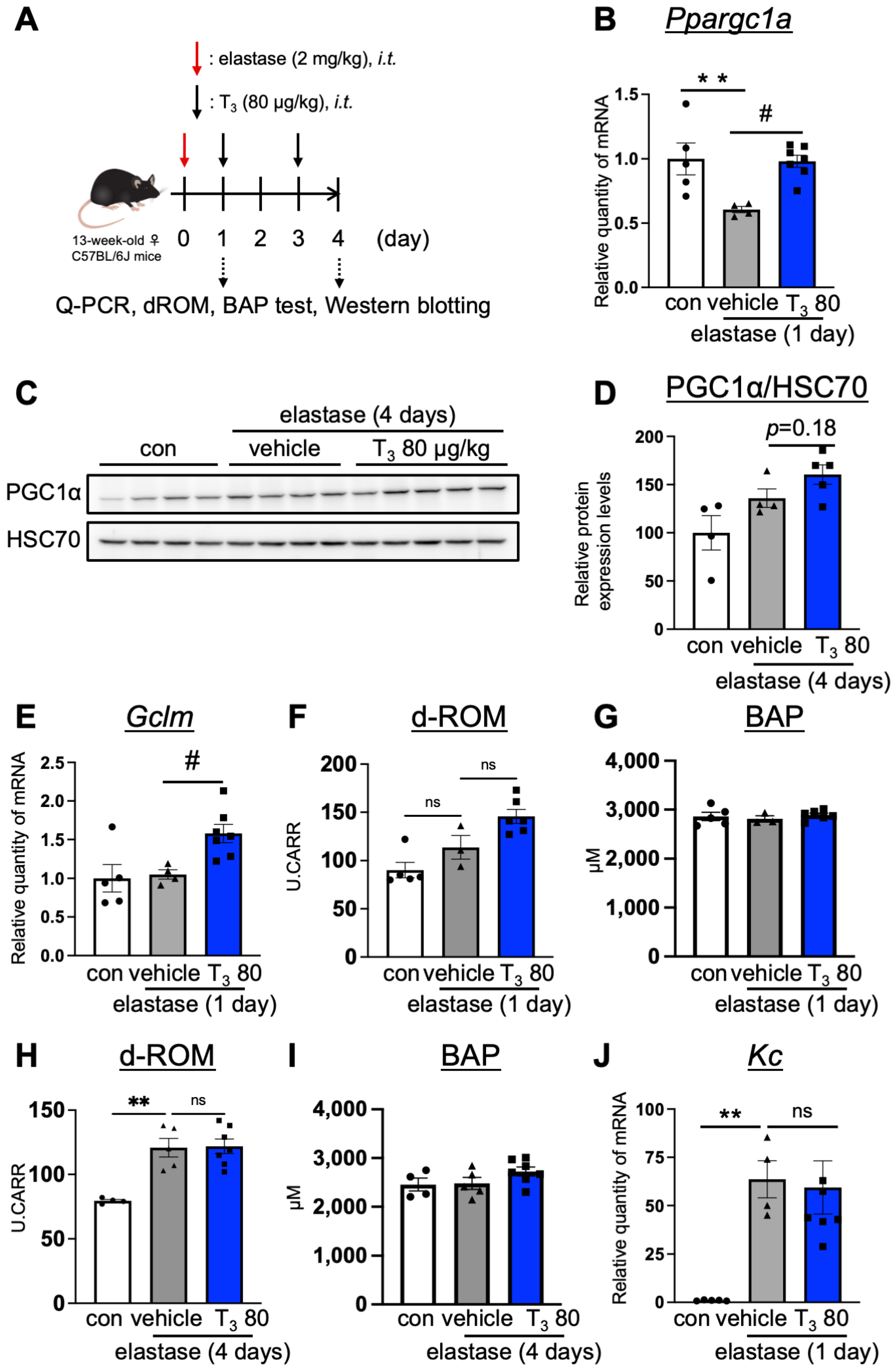
3.2. T3 Improves Pulmonary Pathology via the Ppargc1a-Gclm Pathway in the Lungs of the Elastase-Induced COPD Mouse Model
3.3. The Intratracheal Administration of T3 for 12 Days Did Not Improve COPD Pathology in C57BL/6J-βENaC-Tg Mice
3.4. The Intratracheal Administration of T3 for 2 Months Did Not Improve COPD Pathology in C57BL/6J-βENaC-Tg Mice
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Agustí, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Small Airways in COPD. N. Engl. J. Med. 2004, 350, 2635–2637. [Google Scholar] [CrossRef] [PubMed]
- Hansel, N.N.; McCormack, M.C.; Kim, V. The Effects of Air Pollution and Temperature on COPD. COPD J. Chronic Obstr. Pulm. Dis. 2016, 13, 372–379. [Google Scholar] [CrossRef]
- Marsh, S.E.; Travers, J.; Weatherall, M.; Williams, M.V.; Aldington, S.; Shirtcliffe, P.M.; Hansell, A.L.; Nowitz, M.R.; McNaughton, A.A.; Soriano, J.B.; et al. Proportional classifications of COPD phenotypes. Thorax 2008, 63, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.P.; Agusti, A.; Bafadhel, M.; Christenson, S.A.; Bon, J.; Donaldson, G.C.; Sin, D.D.; Wedzicha, J.A.; Martinez, F.J. Phenotypes, Etiotypes, and Endotypes of Exacerbations of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2023, 208, 1026–1041. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, M.E.; Lee, F.E.H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Scicluna, B.P.; van Vught, L.A.; Zwinderman, A.H.; Wiewel, M.A.; Davenport, E.E.; Burnham, K.L.; Nürnberg, P.; Schultz, M.J.; Horn, J.; Cremer, O.L.; et al. Classification of patients with sepsis according to blood genomic endotype: A prospective cohort study. Lancet Respir. Med. 2017, 5, 816–826. [Google Scholar] [CrossRef]
- Subramanian, D.R.; Gupta, S.; Burggraf, D.; Vom Silberberg, S.J.; Heimbeck, I.; Heiss-Neumann, M.S.; Haeussinger, K.; Newby, C.; Hargadon, B.; Raj, V.; et al. Emphysema- and airway-dominant COPD phenotypes defined by standardised quantitative computed tomography. Eur. Respir. J. 2016, 48, 92–103. [Google Scholar] [CrossRef]
- Nakashima, R.; Kamei, S.; Nohara, H.; Fujikawa, H.; Maruta, K.; Kawakami, T.; Eto, Y.; Takahashi, N.; Suico, M.A.; Takeo, T.; et al. Auto-measure emphysematous parameters and pathophysiological gene expression profiles in experimental mouse models of acute and chronic obstructive pulmonary diseases. J. Pharmacol. Sci. 2019, 140, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Shuto, T.; Kamei, S.; Nohara, H.; Fujikawa, H.; Tasaki, Y.; Sugahara, T.; Ono, T.; Matsumoto, C.; Sakaguchi, Y.; Maruta, K.; et al. Pharmacological and genetic reappraisals of protease and oxidative stress pathways in a mouse model of obstructive lung diseases. Sci. Rep. 2016, 6, 39305. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, H.; Sakamoto, Y.; Masuda, N.; Oniki, K.; Kamei, S.; Nohara, H.; Nakashima, R.; Maruta, K.; Kawakami, T.; Eto, Y.; et al. Higher blood uric acid in female humans and mice as a protective factor against pathophysiological decline of lung function. Antioxidants 2020, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Nohara, H.; Nakashima, R.; Kamei, S.; Fujikawa, H.; Ueno-Shuto, K.; Kawakami, T.; Eto, Y.; Suico, M.A.; Li, J.D.; Kai, H.; et al. Intratracheal GLP-1 receptor agonist treatment up-regulates mucin via p38 and exacerbates emphysematous phenotype in mucus hypersecretory obstructive lung diseases. Biochem. Biophys. Res. Commun. 2020, 524, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Nohara, H.; Takahashi, N.; Nasu, A.; Hayashi, M.; Kishimoto, T.; Kamei, S.; Fujikawa, H.; Maruta, K.; Kawakami, T.; et al. Metformin suppresses epithelial sodium channel hyperactivation and its associated phenotypes in a mouse model of obstructive lung diseases. J. Pharmacol. Sci. 2022, 149, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.; Yang, M.; Fan, L.; Xie, L.; Xu, Y.; Wang, B.; Xu, T.; Yu, L.; Ma, J.; Chen, W. Triiodothyronine attenuates silica-induced oxidative stress, inflammation, and apoptosis via thyroid hormone receptor α in differentiated thp-1 macrophages. Chem. Res. Toxicol. 2020, 33, 1256–1265. [Google Scholar] [CrossRef]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef]
- Hitchcock, K.R. Hormones and the lung. I. Thyroid hormones and glucocorticoids in lung development. Anat. Rec. 1979, 194, 15–40. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; De Castro, J.P.W.; Deiuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef]
- Terzano, C.; Romani, S.; Paone, G.; Conti, V.; Oriolo, F. COPD and thyroid dysfunctions. Lung 2014, 192, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Gałecka, E.; Kumor-Kisielewska, A.; Górski, P. Association of serum deiodinase type 2 level with chronic obstructive pulmonary disease in the Polish population. Acta Biochim. Pol. 2019, 66, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.S.; Izumchenko, E.; Dasgupta, S.; Hoque, M.O. Mitochondria in chronic obstructive pulmonary disease and lung cancer: Where are we now? Biomark. Med. 2017, 11, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; Lawrence Erlbaum Associates: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Barca-Mayo, O.; Liao, X.H.; DiCosmo, C.; Dumitrescu, A.; Moreno-Vinasco, L.; Wade, M.S.; Sammani, S.; Mirzapoiazova, T.; Garcia, J.G.N.; Refetoff, S.; et al. Role of type 2 deiodinase in response to acute lung injury (ALI) in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, G.; Kaminski, N.; Lee, P.J. PINK1 mediates the protective effects of thyroid hormone T3 in hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L1118–L1125. [Google Scholar] [CrossRef]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, Q.; Tuccitto, A.; Chan, D.; Alqawlaq, S.; Won, G.J.; Sivak, J.M. The AMPK-PGC-1α signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol. Dis. 2018, 113, 59–69. [Google Scholar] [CrossRef]
- Kheradmand, F.; Folkesson, H.G.; Shum, L.; Derynk, R.; Pytela, R.; Matthay, M.A. Transforming growth factor-α enhances alveolar epithelial cell repair in a new in vitro model. Am. J. Physiol. Lung Cell. Mol. Physiol. 1994, 267, L728–L738. [Google Scholar] [CrossRef]
- Ulich, T.R.; Yi, E.S.; Longmuir, K.; Yin, S.; Biltz, R.; Morris, C.F.; Housley, R.M.; Pierce, G.F. Keratinocyte growth factor is a growth factor for type II pneumocytes in vivo. J. Clin. Investig. 1994, 93, 1298–1306. [Google Scholar] [CrossRef]
- Prince, L.S. FGF10 and Human Lung Disease Across the Life Spectrum. Front. Genet. 2018, 9, 517. [Google Scholar] [CrossRef]
- Guzy, R.D.; Stoilov, I.; Elton, T.J.; Mecham, R.P.; Ornitz, D.M. Fibroblast growth factor 2 is required for epithelial recovery, but not for pulmonary fibrosis, in response to bleomycin. Am. J. Respir. Cell Mol. Biol. 2015, 52, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Allan Panganiban, R.M.; Day, R.M. Hepatocyte growth factor in lung repair and pulmonary fibrosis. Acta Pharmacol. Sin. 2011, 32, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, W.; Guo, Q.; Wang, Y.; Ma, L.; Zhang, X. Insulin-Like Growth Factor-1 Signaling in Lung Development and Inflammatory Lung Diseases. Biomed. Res. Int. 2018, 2018, 6057589. [Google Scholar] [CrossRef] [PubMed]
- Kwakkel, J.; Surovtseva, O.V.; De Vries, E.M.; Stap, J.; Fliers, E.; Boelen, A. A novel role for the thyroid hormone-activating enzyme type 2 deiodinase in the inflammatory response of macrophages. Endocrinology 2014, 155, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Nijampurkar, B.; Qureshi, F.; Jain, N.; Banerjee, T.; Kumar, A.; Parmar, H.S. Anti-Inflammatory Role of Thyroid Hormones on Rat Air Pouch Model of Inflammation. Inflamm. Allergy Drug Targets 2015, 14, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Breitzig, M.T.; Alleyn, M.D.; Lockey, R.F.; Kolliputi, N. Thyroid hormone: A resurgent treatment for an emergent concern. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L945–L950. [Google Scholar] [CrossRef]
- Agusti, A.; Gibson, P.G.; Mcdonald, V.M. Treatable Traits in Airway Disease: From Theory to Practice. J. Allergy Clin. Immunol. Pract. 2023, 11, 713–723. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Gene ID | Sequence |
|---|---|---|
| Dio2_QRT-FW | 13371 | 5′-TCAGGTAACAATTATGCCTCGGA-3′ |
| Dio2_QRT-RV | 5′-GCTGAACCAAAGTTGACCACC-3′ | |
| Ppargc1a_QRT-FW | 19017 | 5′-AAGTGGTGTAGCGACCAATCG-3′ |
| Ppargc1a_QRT-RV | 5′-AATGAGGGCAATCCGTCTTCA-3′ | |
| Il-8 (Kc)_QRT-FW | 14825 | 5′-TGTCAGTGCCTGCAGACCAT-3′ |
| Il-8 (Kc)_QRT-RV | 5′-CCTCGCGACCATTCTTGAGT-3′ | |
| Gclm_QRT-FW | 14630 | 5′-CTTCGCCTCCGATTGAAGATG-3′ |
| Gclm_QRT-RV | 5′-AAAGGCAGTCAAATCTGGTGG-3′ | |
| Nrf2_QRT-FW | 18024 | 5′-CACTCCAGCGAGCAGGCTAT-3′ |
| Nrf2_QRT-RV | 5′-CTGGGACTGTAGTCCTGGCG-3′ | |
| Nqo1_QRT-FW | 18104 | 5′-TTCTGTGGCTTCCAGGTCTT-3′ |
| Nqo1_QRT-RV | 5′-AGGCTGCTTGGAGCAAAATA-3′ | |
| Ho-1_QRT-FW | 15368 | 5′-GCCACCAAGGAGGTACACAT-3′ |
| Ho-1_QRT-RV | 5′-GCTTGTTGCGCTCTATCTCC-3′ | |
| Gsr_QRT-FW | 14782 | 5′-CACGGCTATGCAACATTCGC-3′ |
| Gsr_QRT-RV | 5′-GTGTGGAGCGGTAAACTTTTTC-3′ | |
| Gclc_QRT-FW | 14629 | 5′-GGACAAACCCCAACCATCC-3′ |
| Gclc_QRT-RV | 5′-GTTGAACTCAGACATCGTTCCT-3′ | |
| Fgf2_QRT-FW | 14173 | 5′-GCGACCCACACGTCAAACTA-3′ |
| Fgf2_QRT-RV | 5′-TCCCTTGATAGACACAACTCCTC-3′ | |
| Fgf7_QRT-FW | 14178 | 5′-ACCTGAGGATTGACAAACGAGG-3′ |
| Fgf7_QRT-RV | 5′-CCACGGTCCTGATTTCCATGA-3′ | |
| Fgf10_QRT-FW | 14165 | 5′-GCAGGCAAATGTATGTGGCAT-3′ |
| Fgf10_QRT-RV | 5′-ATGTTTGGATCGTCATGGGGA-3′ | |
| Igf1_QRT-FW | 16000 | 5′-CTACCAAAATGACCGCATCT-3′ |
| Igf1_QRT-RV | 5′-CAACACTCATCCACAATGCC-3′ | |
| Tgfa_QRT-FW | 21802 | 5′-CACTCTGGGTACGTGGGTG-3′ |
| Tgfa_QRT-RV | 5′-CACAGGTGATAATGAGGACAGC-3′ | |
| Hgf_QRT-FW | 15234 | 5′-AACAGGGGCTTTACGTTCACT-3′ |
| Hgf_QRT-RV | 5′-CGTCCCTTTATAGCTGCCTCC-3′ | |
| Gapdh_QRT-FW | 14433 | 5′-CCTGGAGAAACCTGCCAAGTATG-3′ |
| Gapdh_QRT-RV | 5′-GGTCCTCAGTGTAGCCCAAGATG-3′ | |
| 18s_QRT-FW | 19791 | 5′-GTAACCCGTTGAACCCCATT-3′ |
| 18s_QRT-RV | 5′-CCATCCAATCGGTAGTAGCG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, N.; Nakashima, R.; Nasu, A.; Hayashi, M.; Fujikawa, H.; Kawakami, T.; Eto, Y.; Kishimoto, T.; Fukuyama, A.; Ogasawara, C.; et al. T3 Intratracheal Therapy Alleviates Pulmonary Pathology in an Elastase-Induced Emphysema-Dominant COPD Mouse Model. Antioxidants 2024, 13, 30. https://doi.org/10.3390/antiox13010030
Takahashi N, Nakashima R, Nasu A, Hayashi M, Fujikawa H, Kawakami T, Eto Y, Kishimoto T, Fukuyama A, Ogasawara C, et al. T3 Intratracheal Therapy Alleviates Pulmonary Pathology in an Elastase-Induced Emphysema-Dominant COPD Mouse Model. Antioxidants. 2024; 13(1):30. https://doi.org/10.3390/antiox13010030
Chicago/Turabian StyleTakahashi, Noriki, Ryunosuke Nakashima, Aoi Nasu, Megumi Hayashi, Haruka Fujikawa, Taisei Kawakami, Yuka Eto, Tomoki Kishimoto, Ayami Fukuyama, Choyo Ogasawara, and et al. 2024. "T3 Intratracheal Therapy Alleviates Pulmonary Pathology in an Elastase-Induced Emphysema-Dominant COPD Mouse Model" Antioxidants 13, no. 1: 30. https://doi.org/10.3390/antiox13010030