
Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development

Abstract
:1. Introduction
2. The Role of ROS and Gastric Carcinogenesis
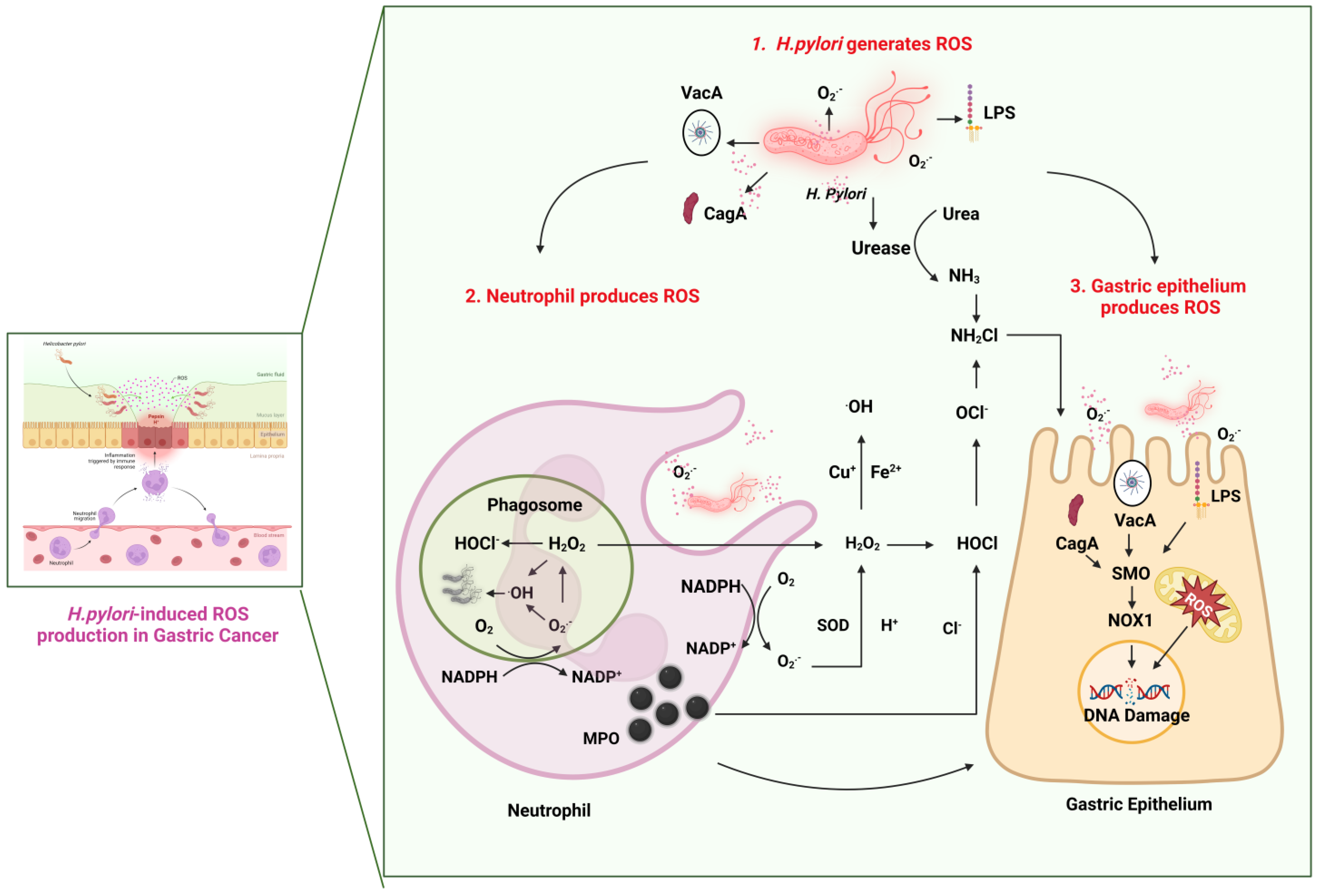
3. The Mechanisms Underlying H. pylori-Induced Oxidative Stress
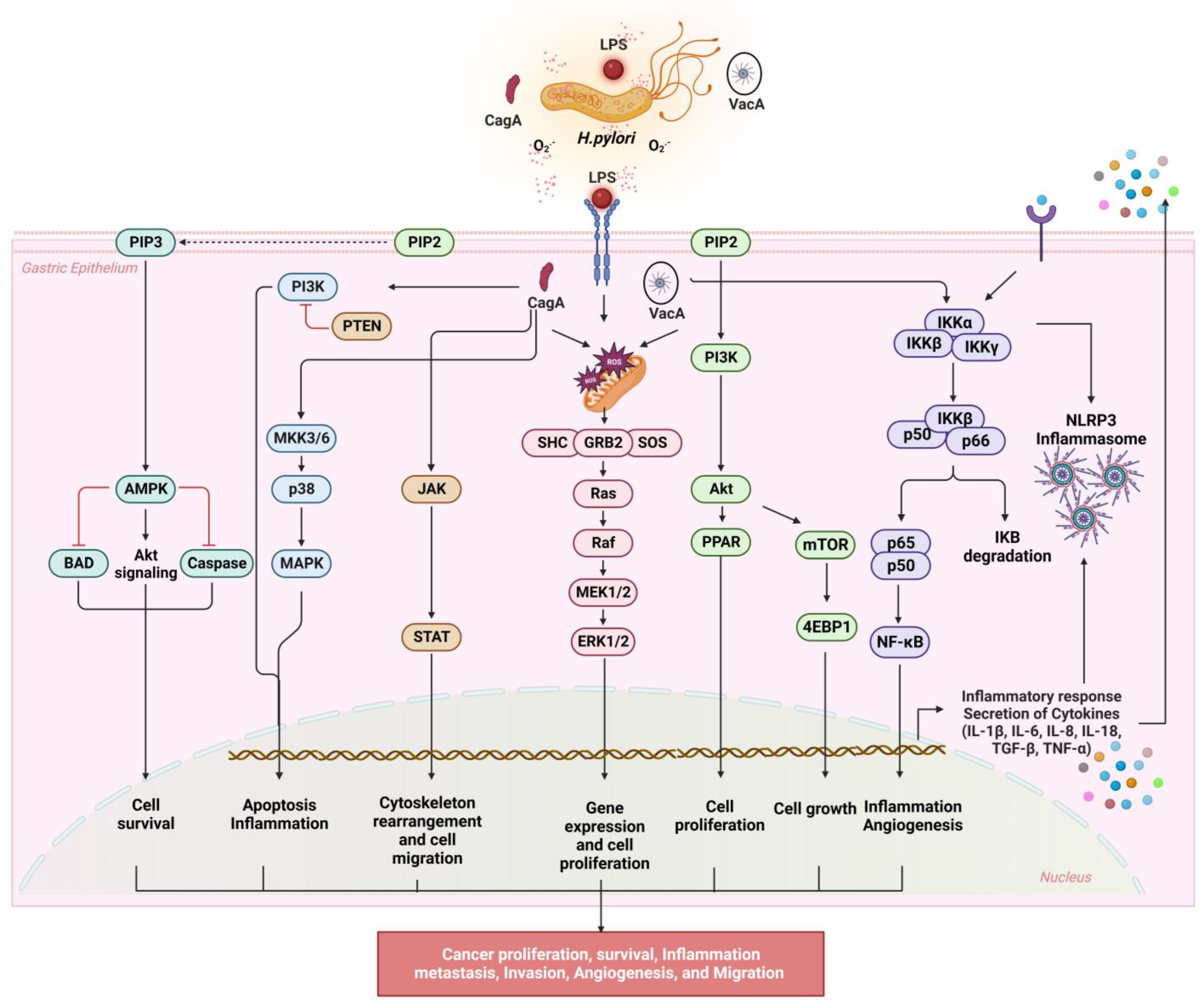
3.1. NF-κB Signaling
3.2. NLRP3 Inflammasome Activation
3.3. PI3K/AKT/mTOR Signaling
3.4. JAK/STAT Signaling
3.5. MAPK/ERK/JNK Signaling
3.6. ROS and Ferroptosis
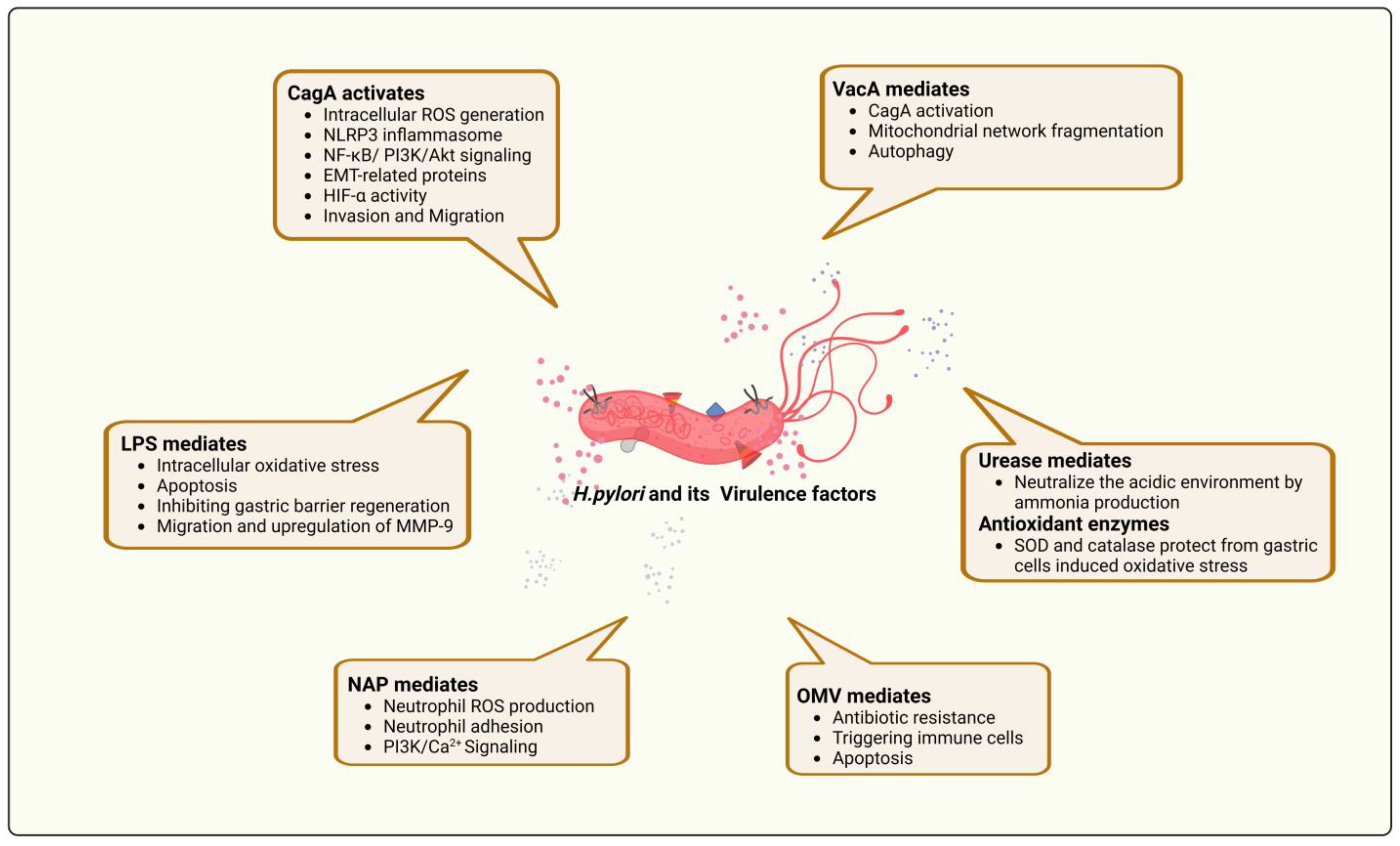
4. The Relationship between H. pylori Virulence Factors and ROS Production
4.1. Cytotoxin-Associated Gene A (CagA)
4.2. Vacuolating Cytotoxins (VacA)
4.3. Neutrophil-Activating Protein A
4.4. Other Virulence Factors
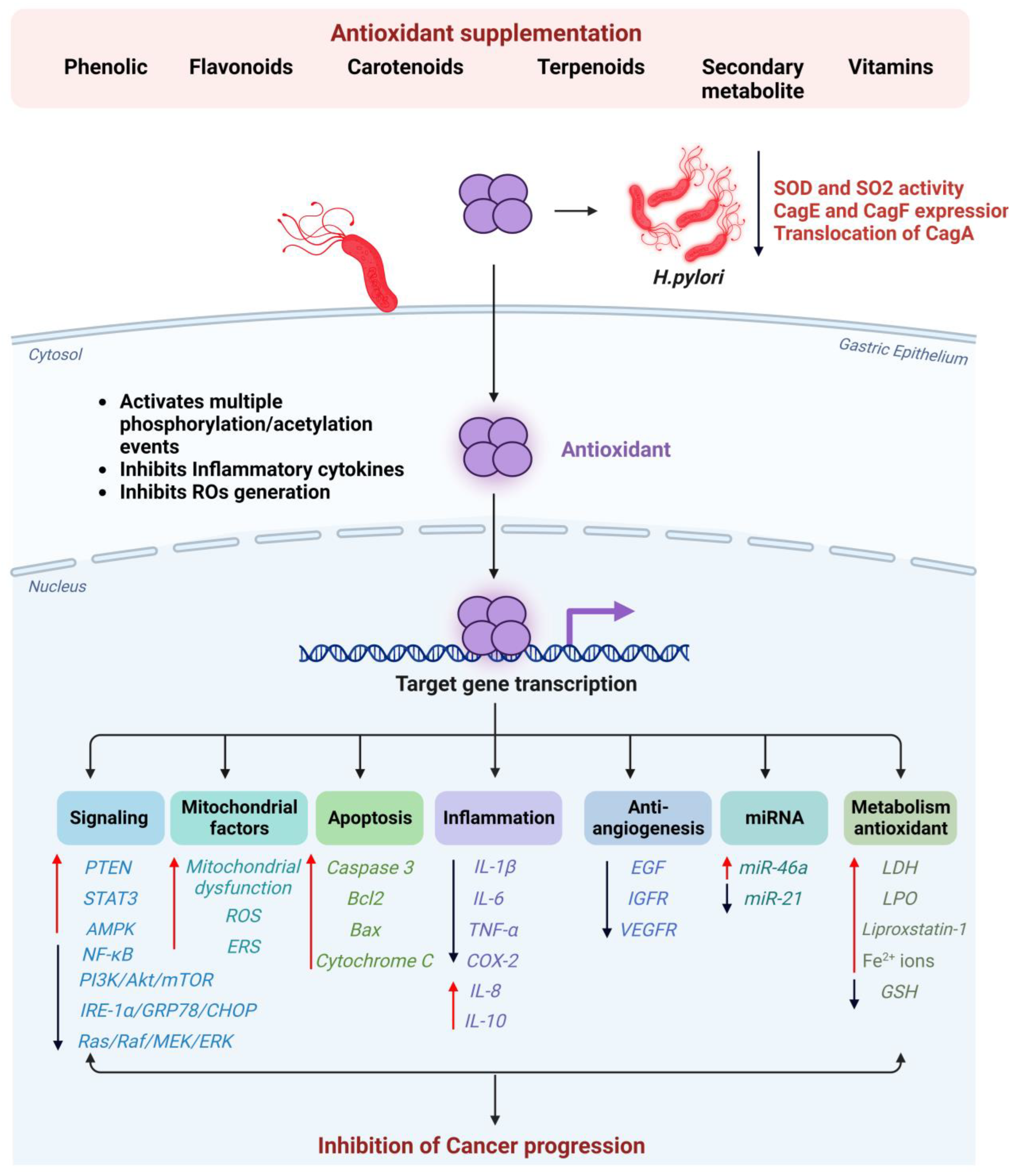
5. The Effectiveness of Antioxidant Supplementation in Preventing Gastric Cancer in H. pylori-Infected Individuals
5.1. Phenolics
5.2. Flavonoids
5.3. Terpenoids and Carotenoids
5.4. Other Phytochemical Compounds
5.5. Nanosystems in H. pylori-Induced Gastric Cancer
6. Future Directions for ROS and H. pylori-Related Gastric Cancer Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Röcken, C. Predictive biomarkers in gastric cancer. J. Cancer Res. Clin. Oncol. 2023, 149, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Gullo, I.; Carneiro, F.; Oliveira, C.; Almeida, G.M. Heterogeneity in gastric cancer: From pure morphology to molecular classifications. Pathobiology 2018, 85, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.-N.; Teng, Q.-X.; Tian, Q.; Chen, W.; Xie, Y.; Wu, K.; Zeng, Q.; Zeng, L.; Pan, Y.; Chen, Z.-S. Signaling pathways and therapeutic interventions in gastric cancer. Signal Transduct. Target. Ther. 2022, 7, 358. [Google Scholar] [CrossRef]
- Yang, L.; Kartsonaki, C.; Yao, P.; de Martel, C.; Plummer, M.; Chapman, D.; Guo, Y.; Clark, S.; Walters, R.G.; Chen, Y. The relative and attributable risks of cardia and non-cardia gastric cancer associated with Helicobacter pylori infection in China: A case-cohort study. Lancet Public Health 2021, 6, e888–e896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dai, J.; Hu, N.; Miao, X.; Abnet, C.C.; Yang, M.; Freedman, N.D.; Chen, J.; Burdette, L.; Zhu, X. Identification of new susceptibility loci for gastric non-cardia adenocarcinoma: Pooled results from two Chinese genome-wide association studies. Gut 2017, 66, 581–587. [Google Scholar] [CrossRef]
- Lee, K.E.; Khoi, P.N.; Xia, Y.; Park, J.S.; Joo, Y.E.; Kim, K.K.; Choi, S.Y.; Do Jung, Y. Helicobacter pylori and interleukin-8 in gastric cancer. World J. Gastroenterol. WJG 2013, 19, 8192. [Google Scholar] [CrossRef]
- Song, P.; Wu, L.; Guan, W. Dietary nitrates, nitrites, and nitrosamines intake and the risk of gastric cancer: A meta-analysis. Nutrients 2015, 7, 9872–9895. [Google Scholar] [CrossRef]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef]
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef]
- Moodley, Y.; Linz, B.; Bond, R.P.; Nieuwoudt, M.; Soodyall, H.; Schlebusch, C.M.; Bernhöft, S.; Hale, J.; Suerbaum, S.; Mugisha, L. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012, 8, e1002693. [Google Scholar] [CrossRef]
- Stefano, K.; Marco, M.; Federica, G.; Laura, B.; Barbara, B.; Gioacchino, L.; Gian, L.d.A. Helicobacter pylori, transmission routes and recurrence of infection: State of the art. Acta Bio Medica Atenei Parm. 2018, 89, 72. [Google Scholar]
- Joseph, A.O.; Tsai, Y.-J. Prevalence, Diversity and Public Health Implications of Helicobacter species in Pet and Stray Dogs. One Health 2022. [Google Scholar]
- Sah, D.K.; Arjunan, A.; Park, S.Y.; Do Jung, Y. Bile acids and microbes in metabolic disease. World J. Gastroenterol. 2022, 28, 6846. [Google Scholar] [CrossRef]
- Johnson, K.S.; Ottemann, K.M. Colonization, localization, and inflammation: The roles of H. pylori chemotaxis in vivo. Curr. Opin. Microbiol. 2018, 41, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Camargo, M.C.; El-Omar, E.; Liou, J.-M.; Peek, R.; Schulz, C.; Smith, S.I.; Suerbaum, S. Helicobacter pylori infection. Nat. Rev. Dis. Primers 2023, 9, 19. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, X.; Zeng, R.; Wu, Q.; Sun, H.; Wu, W.; Zhang, X.; Sun, G.; Yan, B.; Wu, L. Changes of the gastric mucosal microbiome associated with histological stages of gastric carcinogenesis. Front. Microbiol. 2020, 11, 997. [Google Scholar] [CrossRef]
- Salvatori, S.; Marafini, I.; Laudisi, F.; Monteleone, G.; Stolfi, C. Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms. Int. J. Mol. Sci. 2023, 24, 2895. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- de Almeida, A.J.P.O.; de Oliveira, J.C.P.L.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic concepts, sources, cellular signaling, and its implications in aging pathways. Oxidative Med. Cell. Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Ebik, B.; Aslan, N.; Ekin, N.; Bacaksiz, F.; Arpa, M.; Neselioglu, S.; Erel, O.; Ucmak, F. Oxidative stress and the importance of H. pylori eradication in patients with functional dyspepsia. Saudi J. Gastroenterol. 2022, 28, 434–440. [Google Scholar] [CrossRef]
- Hardbower, D.M.; de Sablet, T.; Chaturvedi, R.; Wilson, K.T. Chronic inflammation and oxidative stress: The smoking gun for Helicobacter pylori-induced gastric cancer? Gut Microbes 2013, 4, 475–481. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Redox biology and gastric carcinogenesis: The role of Helicobacter pylori. Redox Rep. 2011, 16, 1–7. [Google Scholar] [CrossRef]
- Díaz, P.; Valenzuela Valderrama, M.; Bravo, J.; Quest, A.F. Helicobacter pylori and gastric cancer: Adaptive cellular mechanisms involved in disease progression. Front. Microbiol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Sarem, M.; Corti, R. Role of Helicobacter pylori coccoid forms in infection and recrudescence. Gastroenterol. Y Hepatol. Engl. Ed. 2016, 39, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Jain, U.; Saxena, K.; Chauhan, N. Helicobacter pylori induced reactive oxygen Species: A new and developing platform for detection. Helicobacter 2021, 26, e12796. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Peri, S.; Versienti, G.; Fiorillo, C.; Becatti, M.; Magnelli, L.; Papucci, L. Gastric Cancer Vascularization and the Contribution of Reactive Oxygen Species. Biomolecules 2023, 13, 886. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.A.H. Phagocytosis and persistence of Helicobacter pylori. Cell. Microbiol. 2007, 9, 817–828. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.; Fieschi, F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Sultana, S.; Foti, A.; Dahl, J.-U. Bacterial defense systems against the neutrophilic oxidant hypochlorous acid. Infect. Immun. 2020, 88, e00964-19. [Google Scholar] [CrossRef]
- Ulfig, A.; Leichert, L.I. The effects of neutrophil-generated hypochlorous acid and other hypohalous acids on host and pathogens. Cell. Mol. Life Sci. 2021, 78, 385–414. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Fiske, C.; Wilson, K.T. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 2010, 90, 831–858. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Nishizawa, T. Oxidative stress and stomach cancer. In Cancer; Elsevier: Amsterdam, The Netherlands, 2014; pp. 33–40. [Google Scholar]
- Cheok, Y.Y.; Tan, G.M.Y.; Lee, C.Y.Q.; Abdullah, S.; Looi, C.Y.; Wong, W.F. Innate immunity crosstalk with Helicobacter pylori: Pattern recognition receptors and cellular responses. Int. J. Mol. Sci. 2022, 23, 7561. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Butcher, L.D.; den Hartog, G.; Ernst, P.B.; Crowe, S.E. Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Gobert, A.P.; Wilson, K.T. Polyamine-and NADPH-dependent generation of ROS during Helicobacter pylori infection: A blessing in disguise. Free Radic. Biol. Med. 2017, 105, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, R.L.; Bayat-Sarmadi, J.; Leben, R.; Niesner, R.; Hauser, A.E. NAD (P) H oxidase activity in the small intestine is predominantly found in enterocytes, not professional phagocytes. Int. J. Mol. Sci. 2018, 19, 1365. [Google Scholar] [CrossRef] [PubMed]
- Karkhah, A.; Ebrahimpour, S.; Rostamtabar, M.; Koppolu, V.; Darvish, S.; Vasigala, V.K.R.; Validi, M.; Nouri, H.R. Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases. Microbiol. Res. 2019, 218, 49–57. [Google Scholar] [CrossRef]
- Uno, K.; Kato, K.; Shimosegawa, T. Novel role of toll-like receptors in Helicobacter pylori-induced gastric malignancy. World J. Gastroenterol. WJG 2014, 20, 5244. [Google Scholar] [CrossRef]
- Yahiro, K.; Akazawa, Y.; Nakano, M.; Suzuki, H.; Hisatune, J.; Isomoto, H.; Sap, J.; Noda, M.; Moss, J.; Hirayama, T. Helicobacter pylori VacA induces apoptosis by accumulation of connexin 43 in autophagic vesicles via a Rac1/ERK-dependent pathway. Cell Death Discov. 2015, 1, 15035. [Google Scholar] [CrossRef]
- Chaithongyot, S.; Naumann, M. Helicobacter pylori-induced reactive oxygen species direct turnover of CSN-associated STAMBPL1 and augment apoptotic cell death. Cell. Mol. Life Sci. 2022, 79, 86. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and oxidative stress are elevated in mitosis during asynchronous cell cycle progression and are exacerbated by mitotic arrest. Cell Syst. 2019, 8, 163–167.e162. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Sah, D.K.; Khoi, P.N.; Li, S.; Arjunan, A.; Jeong, J.-U.; Jung, Y.D. (-)-Epigallocatechin-3-Gallate Prevents IL-1β-Induced uPAR Expression and Invasiveness via the Suppression of NF-κB and AP-1 in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2022, 23, 14008. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Ouyang, Y.; Lu, N.; Li, N. The NF-κB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: Recent advances. Front. Immunol. 2020, 11, 1387. [Google Scholar] [CrossRef]
- Sokolova, O.; Naumann, M. NF-κB signaling in gastric cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef]
- Sharma, S.A.; Tummuru, M.K.; Blaser, M.J.; Kerr, L.D. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-κB in gastric epithelial cells. J. Immunol. 1998, 160, 2401–2407. [Google Scholar] [CrossRef]
- Glocker, E.; Lange, C.; Covacci, A.; Bereswill, S.; Kist, M.; Pahl, H.L. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-κB activation. Infect. Immun. 1998, 66, 2346–2348. [Google Scholar] [CrossRef]
- Naumann, M.; Wessler, S.; Bartsch, C.; Wieland, B.r.; Covacci, A.; Haas, R.; Meyer, T.F. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. J. Biol. Chem. 1999, 274, 31655–31662. [Google Scholar] [CrossRef]
- Backert, S.; Tegtmeyer, N.; Fischer, W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015, 10, 955–965. [Google Scholar] [CrossRef]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef]
- Watanabe, T.; Asano, N.; Fichtner-Feigl, S.; Gorelick, P.L.; Tsuji, Y.; Matsumoto, Y.; Chiba, T.; Fuss, I.J.; Kitani, A.; Strober, W. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Investig. 2010, 120, 1645–1662. [Google Scholar] [CrossRef] [PubMed]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11, 77. [Google Scholar] [CrossRef]
- Belogolova, E.; Bauer, B.; Pompaiah, M.; Asakura, H.; Brinkman, V.; Ertl, C.; Bartfeld, S.; Nechitaylo, T.Y.; Haas, R.; Machuy, N. H elicobacter pylori outer membrane protein HopQ identified as a novel T4SS-associated virulence factor. Cell. Microbiol. 2013, 15, 1896–1912. [Google Scholar]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejías-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2, 16189. [Google Scholar] [CrossRef]
- Königer, V.; Holsten, L.; Harrison, U.; Busch, B.; Loell, E.; Zhao, Q.; Bonsor, D.A.; Roth, A.; Kengmo-Tchoupa, A.; Smith, S.I. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2016, 2, 16188. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kudo, T.; Lu, H.; Casola, A.; Brasier, A.R.; Graham, D.Y. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology 2004, 126, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Cho, S.O.; Kim, H. α-Lipoic acid inhibits expression of IL-8 by suppressing activation of MAPK, Jak/Stat, and NF-κB in H. pylori-infected gastric epithelial AGS cells. Yonsei Med. J. 2016, 57, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lim, J.W.; Kim, H. Astaxanthin inhibits matrix metalloproteinase expression by suppressing PI3K/AKT/mTOR activation in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2022, 14, 3427. [Google Scholar] [CrossRef]
- Cha, B.; Lim, J.W.; Kim, H. Jak1/Stat3 is an upstream signaling of NF-κB activation in Helicobacter pylori-induced IL-8 production in gastric epithelial AGS cells. Yonsei Med. J. 2015, 56, 862–866. [Google Scholar] [CrossRef]
- Tang, C.-L.; Hao, B.; Zhang, G.-X.; Shi, R.-H.; Cheng, W.-F. Helicobacter pylori tumor necrosis factor-α inducing protein promotes cytokine expression via nuclear factor-κB. World J. Gastroenterol. WJG 2013, 19, 399. [Google Scholar] [CrossRef]
- Maubach, G.; Vieth, M.; Boccellato, F.; Naumann, M. Helicobacter pylori-induced NF-κB: Trailblazer for gastric pathophysiology. Trends Mol. Med. 2022, 28, 210–222. [Google Scholar] [CrossRef]
- Ferrand, J.; Lehours, P.; Schmid-Alliana, A.; Mégraud, F.; Varon, C. Helicobacter pylori infection of gastrointestinal epithelial cells in vitro induces mesenchymal stem cell migration through an NF-κB-dependent pathway. PLoS ONE 2011, 6, e29007. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef]
- Kinoshita, T.; Imamura, R.; Kushiyama, H.; Suda, T. NLRP3 mediates NF-κB activation and cytokine induction in microbially induced and sterile inflammation. PLoS ONE 2015, 10, e0119179. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Israel, D.A.; Peek, R.M., Jr. Mechanisms of Helicobacter pylori-induced gastric inflammation. In Physiology of the Gastrointestinal Tract; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1517–1545. [Google Scholar]
- Pachathundikandi, S.K.; Blaser, N.; Backert, S. Mechanisms of inflammasome signaling, microRNA induction and resolution of inflammation by Helicobacter pylori. In Molecular Mechanisms of Inflammation: Induction, Resolution and Escape by Helicobacter pylori; Springer: Cham, Switzerland, 2019; pp. 267–302. [Google Scholar]
- Li, X.; Liu, S.; Luo, J.; Liu, A.; Tang, S.; Liu, S.; Yu, M.; Zhang, Y. Helicobacter pylori induces IL-1β and IL-18 production in human monocytic cell line through activation of NLRP3 inflammasome via ROS signaling pathway. Pathog. Dis. 2015, 73, ftu024. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Bae, S.Y.; Kim, H.R.; Kim, Y.S.; Kim, D.J.; Cho, B.J.; Yang, H.-K.; Hwang, Y.-I.; Kim, K.J.; Park, H.S. Interleukin-18 increases metastasis and immune escape of stomach cancer via the downregulation of CD70 and maintenance of CD44. Carcinogenesis 2009, 30, 1987–1996. [Google Scholar] [CrossRef]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-κB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An intracellular degradation pathway regulating plant survival and stress response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef]
- Ito, N.; Tsujimoto, H.; Ueno, H.; Xie, Q.; Shinomiya, N. Helicobacter pylori-mediated immunity and signaling transduction in gastric cancer. J. Clin. Med. 2020, 9, 3699. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Hisatsune, J.; Yamasaki, E.; Isomoto, H.; Kurazono, H.; Hatakeyama, M.; Azuma, T.; Yamaoka, Y.; Yahiro, K.; Moss, J. Helicobacter pylori VacA-induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J. Biol. Chem. 2009, 284, 1612–1619. [Google Scholar] [CrossRef]
- Ouyang, Y.; Liu, G.; Xu, W.; Yang, Z.; Li, N.; Xie, C.; Zhou, C.; Chen, J.; Zhu, Y.; Hong, J. Helicobacter pylori induces epithelial-mesenchymal transition in gastric carcinogenesis via the AKT/GSK3β signaling pathway. Oncol. Lett. 2021, 21, 165. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Yashiro, M. The role of PI3K/Akt/mTOR signaling in gastric carcinoma. Cancers 2014, 6, 1441–1463. [Google Scholar] [CrossRef]
- Shi, M.; Liu, D.; Duan, H.; Han, C.; Wei, B.; Qian, L.; Chen, C.; Guo, L.; Hu, M.; Yu, M. Catecholamine up-regulates MMP-7 expression by activating AP-1 and STAT3 in gastric cancer. Mol. Cancer 2010, 9, 269. [Google Scholar] [CrossRef]
- Aung, P.; Oue, N.; Mitani, Y.; Nakayama, H.; Yoshida, K.; Noguchi, T.; Bosserhoff, A.; Yasui, W. Systematic search for gastric cancer-specific genes based on SAGE data: Melanoma inhibitory activity and matrix metalloproteinase-10 are novel prognostic factors in patients with gastric cancer. Oncogene 2006, 25, 2546–2557. [Google Scholar] [CrossRef]
- Zhang, G.; Miyake, M.; Lawton, A.; Goodison, S.; Rosser, C.J. Matrix metalloproteinase-10 promotes tumor progression through regulation of angiogenic and apoptotic pathways in cervical tumors. BMC Cancer 2014, 14, 310. [Google Scholar] [CrossRef]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef]
- Li, M.; Zhao, L.; Liu, J.; Liu, A.; Jia, C.; Ma, D.; Jiang, Y.; Bai, X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell. Signal. 2010, 22, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. mTOR activity and autophagy in senescent cells, a complex partnership. Int. J. Mol. Sci. 2021, 22, 8149. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. PTEN/PTENP1:‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ueda, M. Mutual inhibition between PTEN and PIP3 generates bistability for polarity in motile cells. Nat. Commun. 2018, 9, 4481. [Google Scholar] [CrossRef]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Basseres, D.S.; Baldwin, A.S. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinase α (IKKα). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Miao, L.; Liang, F.; Huang, H.; Teng, X.; Li, S.; Nuriddinov, J.; Selzer, M.E.; Hu, Y. The mTORC1 effectors S6K1 and 4E-BP play different roles in CNS axon regeneration. Nat. Commun. 2014, 5, 5416. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.O.; Lim, J.W.; Kim, H. Red ginseng extract inhibits the expression of MCP-1 and iNOS in Helicobacter pylori-infected gastric epithelial cells by suppressing the activation of NADPH oxidase and Jak2/Stat3. J. Ethnopharmacol. 2013, 150, 761–764. [Google Scholar] [CrossRef]
- Woo, J.; Lim, J.W.; Kim, H. Astaxanthin inhibits integrin α5 expression by suppressing activation of JAK1/STAT3 in Helicobacter pylori-stimulated gastric epithelial cells. Mol. Med. Rep. 2023, 27, 127. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.Y.; Lee, H.G.; Kim, S.J.; Kim, D.H.; Han, H.j.; Ngo, H.K.C.; Park, S.A.; Woo, J.H.; Lee, J.S.; Na, H.K. Helicobacter pylori activates IL-6-STAT3 signaling in human gastric cancer cells: Potential roles for reactive oxygen species. Helicobacter 2016, 21, 405–416. [Google Scholar] [CrossRef]
- Yu, B.; Xiang, L.; Peppelenbosch, M.P.; Fuhler, G.M. Overlapping cytokines in H. pylori infection and gastric cancer: A tandem meta-analysis. Front. Immunol. 2023, 14, 1125658. [Google Scholar] [CrossRef]
- Nakagawa, H.; Tamura, T.; Mitsuda, Y.; Goto, Y.; Kamiya, Y.; Kondo, T.; Wakai, K.; Hamajima, N. Significant association between serum interleukin-6 and Helicobacter pylori antibody levels among H. pylori-positive Japanese adults. Mediat. Inflamm. 2013, 2013, 142358. [Google Scholar] [CrossRef]
- Omar, S.; Hassan, M.F.; Hasan, B.B. Impact of Serum Interleukin 6 among Helicobacter pylori-Positive Adult Patients in Relation to upper gastrointestinal endoscopy findings. Suez Canal Univ. Med. J. 2019, 22, 117–121. [Google Scholar] [CrossRef]
- Jan, I.; Rather, R.A.; Mushtaq, I.; Malik, A.A.; Besina, S.; Baba, A.B.; Farooq, M.; Yousuf, T.; Rah, B.; Afroze, D. Helicobacter pylori subdues cytokine signaling to alter mucosal inflammation via hypermethylation of suppressor of cytokine signaling 1 gene during gastric carcinogenesis. Front. Oncol. 2021, 10, 604747. [Google Scholar] [CrossRef]
- Qi, H.; Yang, Z.; Dai, C.; Wang, R.; Ke, X.; Zhang, S.; Xiang, X.; Chen, K.; Li, C.; Luo, J. STAT3 activates MSK1-mediated histone H3 phosphorylation to promote NFAT signaling in gastric carcinogenesis. Oncogenesis 2020, 9, 15. [Google Scholar] [CrossRef]
- Xu, S.; Wu, X.; Zhang, X.; Chen, C.; Chen, H.; She, F. CagA orchestrates eEF1A1 and PKCδ to induce interleukin-6 expression in Helicobacter pylori-infected gastric epithelial cells. Gut Pathog. 2020, 12, 31. [Google Scholar] [CrossRef]
- Guo, Y.; Ding, S. Helicobacter pylori Thioredoxin1 May Play a Highly Pathogenic Role via the IL6/STAT3 Pathway. Gastroenterol. Res. Pract. 2022, 2022, 3175935. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, L.; Zhang, T.; Shen, L.; Liu, L.; Zhang, J.; Zhang, Y.; Wang, X.; Yang, S.; Lu, F. The involvement of Helicobacter pylori thioredoxin-1 in gastric carcinogenesis. J. Med. Microbiol. 2013, 62, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.O.; Lim, J.W.; Kim, K.H.; Kim, H. Diphenyleneiodonium inhibits the activation of mitogen-activated protein kinases and the expression of monocyte chemoattractant protein-1 in Helicobacter pylori-infected gastric epithelial AGS cells. Inflamm. Res. 2011, 60, 501–507. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Lu, C.-L.; Sheu, B.-S. Differential H. pylori-induced MAPK responses regulate Lewis antigen expression and colonization density on gastric epithelial cells between children and adults. Front. Immunol. 2022, 13, 849512. [Google Scholar] [CrossRef]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, X.; Chen, L.; Li, W.; Sun, Y.; Zeng, J.; Yu, H.; Chen, C.; Jia, J. Helicobacter pylori CagA inhibits the expression of Runx3 via Src/MEK/ERK and p38 MAPK pathways in gastric epithelial cell. J. Cell. Biochem. 2012, 113, 1080–1086. [Google Scholar] [CrossRef]
- Bae, S.; Lim, J.W.; Kim, H. β-carotene inhibits expression of matrix metalloproteinase-10 and invasion in Helicobacter pylori-infected gastric epithelial cells. Molecules 2021, 26, 1567. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin inhibits mitochondrial dysfunction and interleukin-8 expression in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2018, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Wu, D.; Wu, X.; Wang, F.; Yu, H.; Liu, P.; Cui, G.; Chen, Z. Phycocyanin inhibits Helicobacter pylori-induced hyper-proliferation in AGS cells via activation of the ROS/MAPK signaling pathway. Ann. Transl. Med. 2022, 10, 176. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Hirata, Y.; Kinoshita, H.; Sakitani, K.; Nakagawa, H.; Nakata, W.; Takahashi, R.; Sakamoto, K.; Maeda, S.; Koike, K. Differential roles of ASK1 and TAK1 in Helicobacter pylori-induced cellular responses. Infect. Immun. 2013, 81, 4551–4560. [Google Scholar] [CrossRef]
- Da Conceicao, V.N.; Sun, Y.; Ramachandran, K.; Chauhan, A.; Raveendran, A.; Venkatesan, M.; DeKumar, B.; Maity, S.; Vishnu, N.; Kotsakis, G.A. Resolving macrophage polarization through distinct Ca2+ entry channel that maintains intracellular signaling and mitochondrial bioenergetics. iScience 2021, 24, 103339. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lim, J.W.; Kim, H. Korean red ginseng extract inhibits IL-8 expression via Nrf2 activation in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2022, 14, 1044. [Google Scholar] [CrossRef]
- Gu, R.; Xia, Y.; Li, P.; Zou, D.; Lu, K.; Ren, L.; Zhang, H.; Sun, Z. Ferroptosis and its role in gastric cancer. Front. Cell Dev. Biol. 2022, 10, 860344. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e353. [Google Scholar] [CrossRef] [PubMed]
- Saint-Germain, E.; Mignacca, L.; Vernier, M.; Bobbala, D.; Ilangumaran, S.; Ferbeyre, G. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging 2017, 9, 2137. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, D.; Lu, Y.; Sun, J.; Zhu, J.; Xing, Y.; Ma, X.; Wang, Y.; Ji, M.; Jia, Y. PHKG2 regulates RSL3-induced ferroptosis in Helicobacter pylori related gastric cancer. Arch. Biochem. Biophys. 2023, 740, 109560. [Google Scholar] [CrossRef]
- Sicinschi, L.A.; Correa, P.; Bravo, L.E.; Peek, R.M., Jr.; Wilson, K.T.; Loh, J.T.; Yepez, M.C.; Gold, B.D.; Thompson, D.T.; Cover, T.L. Non-invasive genotyping of Helicobacter pylori cagA, vacA, and hopQ from asymptomatic children. Helicobacter 2012, 17, 96–106. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori virulence factors—Mechanisms of bacterial pathogenicity in the gastric microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Raghwan; Chowdhury, R. Host Cell Contact Induces Fur-dependent Expression of Virulence Factors CagA and VacA in H elicobacter pylori. Helicobacter 2014, 19, 17–25. [Google Scholar] [CrossRef]
- Sharndama, H.C.; Mba, I.E. Helicobacter pylori: An up-to-date overview on the virulence and pathogenesis mechanisms. Braz. J. Microbiol. 2022, 53, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, C.; Chen, D.; He, X.; Zhao, Y.; Bao, L.; Wang, Q.; Zhou, J.; Xie, Y. H. pylori CagA activates the NLRP3 inflammasome to promote gastric cancer cell migration and invasion. Inflamm. Res. 2022, 71, 141–155. [Google Scholar] [CrossRef]
- Yoon, J.H.; Seo, H.S.; Choi, S.S.; Chae, H.S.; Choi, W.S.; Kim, O.; Ashktorab, H.; Smoot, D.T.; Nam, S.W.; Lee, J.Y. Gastrokine 1 inhibits the carcinogenic potentials of Helicobacter pylori CagA. Carcinogenesis 2014, 35, 2619–2629. [Google Scholar] [CrossRef]
- Jung, D.E.; Yu, S.S.; Lee, Y.S.; Choi, B.K.; Lee, Y.C. Regulation of SIRT3 signal related metabolic reprogramming in gastric cancer by Helicobacter pylori oncoprotein CagA. Oncotarget 2017, 8, 78365. [Google Scholar]
- Winter, J.A.; Letley, D.P.; Cook, K.W.; Rhead, J.L.; Zaitoun, A.A.; Ingram, R.J.; Amilon, K.R.; Croxall, N.J.; Kaye, P.V.; Robinson, K. A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori–induced metaplasia in the stomach. J. Infect. Dis. 2014, 210, 954–963. [Google Scholar] [CrossRef]
- Nakano, M.; Yahiro, K.; Yamasaki, E.; Kurazono, H.; Akada, J.; Yamaoka, Y.; Niidome, T.; Hatakeyama, M.; Suzuki, H.; Yamamoto, T. Helicobacter pylori VacA, acting through receptor protein tyrosine phosphatase α, is crucial for CagA phosphorylation in human duodenum carcinoma cell line AZ-521. Dis. Models Mech. 2016, 9, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Murata-Kamiya, N.; Hatakeyama, M. Helicobacter pylori CagA oncoprotein interacts with SHIP2 to increase its delivery into gastric epithelial cells. Cancer Sci. 2020, 111, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Luo, Z.-Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 16032–16037. [Google Scholar] [CrossRef]
- Zhu, P.; Xue, J.; Zhang, Z.-J.; Jia, Y.-P.; Tong, Y.-N.; Han, D.; Li, Q.; Xiang, Y.; Mao, X.-H.; Tang, B. Helicobacter pylori VacA induces autophagic cell death in gastric epithelial cells via the endoplasmic reticulum stress pathway. Cell Death Dis. 2017, 8, 3207. [Google Scholar] [CrossRef]
- Luo, J.; Bai, L.; Tao, J.; Wen, Y.; Li, M.; Zhu, Y.; Luo, S.; Pu, G.; Ma, L. Autophagy induced by H. pylori VacA regulated the survival mechanism of the SGC7901 human gastric cancer cell line. Genes Genom. 2021, 43, 1223–1230. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, W.; Yan, S.; Li, J.; Wei, H.; Zhao, W. CagA and VacA inhibit gastric mucosal epithelial cell autophagy and promote the progression of gastric precancerous lesions. Zhong Nan Da Xue Xue Bao Yi Xue Ban J. Cent. South Univ. Med. Sci. 2022, 47, 942–951. [Google Scholar]
- Fu, H.-W. Helicobacter pylori neutrophil-activating protein: From molecular pathogenesis to clinical applications. World J. Gastroenterol. WJG 2014, 20, 5294. [Google Scholar] [CrossRef]
- Zhao, Y.; Cai, Y.; Chen, Z.; Li, H.; Xu, Z.; Li, W.; Jia, J.; Sun, Y. SpoT-mediated NapA upregulation promotes oxidative stress-induced Helicobacter pylori biofilm formation and confers multidrug resistance. Antimicrob. Agents Chemother. 2021, 65, e00152-21. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Coletta, S.; D’Elios, M.M.; de Bernard, M. HP-NAP of Helicobacter pylori: The power of the immunomodulation. Front. Immunol. 2022, 3272, 944139. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.-W.; Lai, Y.-C. The Role of Helicobacter pylori Neutrophil-Activating Protein in the Pathogenesis of H. pylori and Beyond: From a Virulence Factor to Therapeutic Targets and Therapeutic Agents. Int. J. Mol. Sci. 2023, 24, 91. [Google Scholar] [CrossRef] [PubMed]
- Gonciarz, W.; Krupa, A.; Moran, A.P.; Tomaszewska, A.; Chmiela, M. Interference of LPS H. pylori with IL-33-Driven Regeneration of Caviae porcellus Primary Gastric Epithelial Cells and Fibroblasts. Cells 2021, 10, 1385. [Google Scholar] [CrossRef]
- Gonciarz, W.; Krupa, A.; Hinc, K.; Obuchowski, M.; Moran, A.P.; Gajewski, A.; Chmiela, M. The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts. PLoS ONE 2019, 14, e0220636. [Google Scholar] [CrossRef]
- Uddin, M.J.; Dawan, J.; Jeon, G.; Yu, T.; He, X.; Ahn, J. The role of bacterial membrane vesicles in the dissemination of antibiotic resistance and as promising carriers for therapeutic agent delivery. Microorganisms 2020, 8, 670. [Google Scholar] [CrossRef]
- Chmiela, M.; Walczak, N.; Rudnicka, K. Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. J. Biomed. Sci. 2018, 25, 78. [Google Scholar] [CrossRef]
- Xia, Q.; Casas-Martinez, J.C.; Zarzuela, E.; Muñoz, J.; Miranda-Vizuete, A.; Goljanek-Whysall, K.; McDonagh, B. Peroxiredoxin 2 is required for the redox mediated adaptation to exercise. Redox Biol. 2023, 60, 102631. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Z.; Zhu, S.; Lu, H.; Peng, D.; Soutto, M.; Naz, H.; Peek, R., Jr.; Xu, H.; Zaika, A. PRDX2 protects against oxidative stress induced by H. pylori and promotes resistance to cisplatin in gastric cancer. Redox Biol. 2020, 28, 101319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, X.; Chen, W.; Guo, X.; Chen, L.; Wei, Z.; Liu, D.; Liu, Z. Scutellarin Suppressed Proliferation and Induced Apoptosis in Gastric Cancer via Wnt/β-catenin Signaling Pathway. Curr. Pharm. Des. 2023, 29, 368–378. [Google Scholar] [PubMed]
- Zhuang, K.; Tang, H.; Guo, H.; Yuan, S. Geraniol prevents Helicobacterium pylori-induced human gastric cancer signalling by enhancing peroxiredoxin-1 expression in GES-1 cells. Microb. Pathog. 2023, 174, 105937. [Google Scholar] [CrossRef]
- Gunes-Bayir, A.; Guler, E.M.; Bilgin, M.G.; Ergun, I.S.; Kocyigit, A.; Dadak, A. Anti-Inflammatory and Antioxidant Effects of Carvacrol on N-Methyl-N′-Nitro-N-Nitrosoguanidine (MNNG) Induced Gastric Carcinogenesis in Wistar Rats. Nutrients 2022, 14, 2848. [Google Scholar] [CrossRef]
- Sun, J.; Meng, M. Chemoprotective Effect of Scutellarin against Gastric Cancer in Rats: An in vitro and in vivo Study. J. Oleo Sci. 2022, 71, 1003–1012. [Google Scholar] [CrossRef]
- Chaturvedi, M.; Mishra, M.; Pandey, A.; Gupta, J.; Pandey, J.; Gupta, S.; Malik, M.Z.; Somvanshi, P.; Chaturvedi, R. Oxidative Products of Curcumin Rather Than Curcumin Bind to Helicobacter pylori Virulence Factor VacA and Are Required to Inhibit Its Vacuolation Activity. Molecules 2022, 27, 6727. [Google Scholar] [CrossRef]
- Li, F.; Wang, S.; Niu, M. Scutellarin inhibits the growth and EMT of gastric cancer cells through regulating PTEN/PI3K pathway. Biol. Pharm. Bull. 2021, 44, 780–788. [Google Scholar] [CrossRef]
- Cho, K.; Lee, H.G.; Piao, J.-Y.; Kim, S.-J.; Na, H.-K.; Surh, Y.-J. Protective effects of silibinin on Helicobacter pylori-induced gastritis: NF-κB and STAT3 as potential targets. J. Cancer Prev. 2021, 26, 118. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, J.; Dong, Y.; Shen, X.; Zhang, Z. Vicenin-2 inhibits the Helicobacterium pylori infection associated gastric carcinogenic events through modulation of PI3K/AKT and Nrf2 signaling in GES-1 cells. J. Biochem. Mol. Toxicol. 2021, 35, e22680. [Google Scholar] [CrossRef]
- Radziejewska, I.; Borzym-Κluczyk, M.; Leszczyńska, K. Luteolin alters MUC1 extracellular domain, sT antigen, ADAM-17, IL-8, IL-10 and NF-κB expression in Helicobacter pylori-infected gastric cancer CRL-1739 cells: A preliminary study. Biomed. Rep. 2021, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lv, J.; Li, X.; Lin, Q. The flavonoid Astragalin shows anti-tumor activity and inhibits PI3K/AKT signaling in gastric cancer. Chem. Biol. Drug Des. 2021, 98, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Das, S.; Rahaman, A.; Talukdar, A.D.; Bhattacharjee, S.; Mandal, D.P. Eugenol and capsaicin exhibit anti-metastatic activity via modulating TGF-β signaling in gastric carcinoma. Food Funct. 2020, 11, 9020–9034. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, Y.; Luo, W.; Chen, S.; Lin, F.; Zhang, X.; Fan, S.; Shen, X.; Wang, Y.; Liang, G. Celastrol induces ROS-mediated apoptosis via directly targeting peroxiredoxin-2 in gastric cancer cells. Theranostics 2020, 10, 10290. [Google Scholar] [CrossRef]
- Guan, Z.; Chen, J.; Li, X.; Dong, N. Tanshinone IIA induces ferroptosis in gastric cancer cells through p53-mediated SLC7A11 down-regulation. Biosci. Rep. 2020, 40, BSR20201807. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Li, L.; Ling, P. Nobiletin inhibits helicobacterium pylori infection-induced gastric carcinogenic signaling by blocking inflammation, apoptosis, and mitogen-activated protein kinase events in gastric epithelial-1 cells. J. Environ. Pathol. Toxicol. Oncol. 2020, 39, 77–88. [Google Scholar] [CrossRef]
- Sheng, Y.-N.; Luo, Y.-H.; Liu, S.-B.; Xu, W.-T.; Zhang, Y.; Zhang, T.; Xue, H.; Zuo, W.-B.; Li, Y.-N.; Wang, C.-Y. Zeaxanthin induces apoptosis via ROS-regulated MAPK and AKT signaling pathway in human gastric cancer cells. OncoTargets Ther. 2020, 13, 10995. [Google Scholar] [CrossRef]
- Yao, S.S.; Han, L.; Tian, Z.B.; Yu, Y.N.; Zhang, Q.; Li, X.Y.; Mao, T.; Yang, L. Celastrol inhibits growth and metastasis of human gastric cancer cell MKN45 by down-regulating microRNA-21. Phytother. Res. 2019, 33, 1706–1716. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, W.; Yang, H.; Bi, J.; Xie, Y.; Cheng, B.; Wang, Y.; Chen, S. Celastrol induces necroptosis and ameliorates inflammation via targeting biglycan in human gastric carcinoma. Int. J. Mol. Sci. 2019, 20, 5716. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin prevents decreases in superoxide dismutase 2 level and superoxide dismutase activity in Helicobacter pylori-infected gastric epithelial cells. J. Cancer Prev. 2019, 24, 54. [Google Scholar] [CrossRef]
- Xu, L.; Gong, C.; Li, G.; Wei, J.; Wang, T.; Meng, W.; Shi, M.; Wang, Y. Ebselen suppresses inflammation induced by Helicobacter pylori lipopolysaccharide via the p38 mitogen-activated protein kinase signaling pathway. Mol. Med. Rep. 2018, 17, 6847–6851. [Google Scholar] [PubMed]
- Jiang, J.; Cao, D.; Jia, Z.; You, L.; Tsukamoto, T.; Hou, Z.; Suo, Y.; Wang, S.; Cao, X. The green tea polyphenol epigallocatechin-3-gallate effectively inhibits Helicobacter pylori-induced gastritis in Mongolian gerbils. Int. J. Clin. Exp. Med. 2016, 9, 2479–2485. [Google Scholar]
- Jeong, M.; Park, J.M.; Han, Y.M.; Kangwan, N.; Kwon, S.O.; Kim, B.N.; Kim, W.H.; Hahm, K.B. Dietary intervention of artemisia and green tea extracts to rejuvenate Helicobacter pylori-associated chronic atrophic gastritis and to prevent tumorigenesis. Helicobacter 2016, 21, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Vetvicka, V.; Vetvickova, J.; Fernandez-Botran, R. Effects of curcumin on Helicobacter pylori infection. Ann. Transl. Med. 2016, 4, 479. [Google Scholar] [CrossRef]
- Moon, J.Y.; Cho, S.K. Nobiletin induces protective autophagy accompanied by ER-stress mediated apoptosis in human gastric cancer SNU-16 cells. Molecules 2016, 21, 914. [Google Scholar] [CrossRef]
- Sarkar, A.; Bhattacharjee, S.; Mandal, D.P. Induction of apoptosis by eugenol and capsaicin in human gastric cancer AGS cells-elucidating the role of p53. Asian Pac. J. Cancer Prev. 2015, 16, 6753–6759. [Google Scholar] [CrossRef]
- Byun, E.; Lim, J.W.; Kim, J.M.; Kim, H. α-Lipoic acid inhibits Helicobacter pylori-induced oncogene expression and hyperproliferation by suppressing the activation of NADPH oxidase in gastric epithelial cells. Mediat. Inflamm. 2014, 2014, 380830. [Google Scholar] [CrossRef]
- Lee, H.-W.; Jang, K.S.B.; Choi, H.J.; Jo, A.; Cheong, J.-H.; Chun, K.-H. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014, 47, 697. [Google Scholar] [CrossRef]
- Yang, J.-C.; Yang, H.-C.; Shun, C.-T.; Wang, T.-H.; Chien, C.-T.; Kao, J.Y. Catechins and sialic acid attenuate Helicobacter pylori-triggered epithelial caspase-1 activity and eradicate Helicobacter pylori infection. Evid. Based Complement. Altern. Med. 2013, 2013, 248585. [Google Scholar]
- Sha, M.; Ye, J.; Zhang, L.-X.; Luan, Z.-Y.; Chen, Y.-B. Celastrol induces apoptosis of gastric cancer cells by miR-146a inhibition of NF-κB activity. Cancer Cell Int. 2013, 13, 50. [Google Scholar] [CrossRef]
- Zhong, J.; Fang, L.; Chen, R.; Xu, J.; Guo, D.; Guo, C.; Guo, C.; Chen, J.; Chen, C.; Wang, X. Polysaccharides from sporoderm-removed spores of Ganoderma lucidum induce apoptosis in human gastric cancer cells via disruption of autophagic flux. Oncol. Lett. 2021, 21, 425. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Reis, F.S.; Sousa, D.; Tavares, C.; Lima, R.T.; Ferreira, I.C.; dos Santos, T.; Vasconcelos, M.H. A methanolic extract of Ganoderma lucidum fruiting body inhibits the growth of a gastric cancer cell line and affects cellular autophagy and cell cycle. Food Funct. 2014, 5, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, M. CARVACROL and human health: A comprehensive review. Phytother. Res. 2018, 32, 1675–1687. [Google Scholar] [CrossRef]
- Günes-Bayir, A.; Kiziltan, H.S.; Kocyigit, A.; Güler, E.M.; Karataş, E.; Toprak, A. Effects of natural phenolic compound carvacrol on the human gastric adenocarcinoma (AGS) cells in vitro. Anti-Cancer Drugs 2017, 28, 522–530. [Google Scholar] [CrossRef]
- Elbestawy, M.K.; El-Sherbiny, G.M.; Moghannem, S.A. Antibacterial, antibiofilm and anti-inflammatory activities of eugenol clove essential oil against resistant Helicobacter pylori. Molecules 2023, 28, 2448. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M.; Singh, S.K.; Gulati, M.; Chellappan, D.K.; Zacconi, F.; De Rubis, G.; Gupta, G.; Sharifi-Rad, J.; Cho, W.C. Luteolin: A flavonoid with a multifaceted anticancer potential. Cancer Cell Int. 2022, 22, 386. [Google Scholar] [CrossRef]
- Vajdi, M.; Farhangi, M.A. Citrus peel derived’Poly-Methoxylated Flavones (PMF). Int. J. Vitam. Nutr. Res. 2021, 93, 252–267. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, R.; Chen, Y.; Chen, X.; Li, Y.; Shen, J.; Yuan, M.; Chen, Y.; Wu, J.; Sun, Q. Nobiletin inhibits de novo FA synthesis to alleviate gastric cancer progression by regulating endoplasmic reticulum stress. Phytomedicine 2023, 116, 154902. [Google Scholar] [CrossRef]
- Omar, S.H.; Scott, C.J.; Hamlin, A.S.; Obied, H.K. Olive biophenols reduces alzheimer’s pathology in SH-SY5Y cells and APPswe mice. Int. J. Mol. Sci. 2018, 20, 125. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, C.; Li, J.; Lu, Y.; Li, H.; Zhuang, J.; Ren, X.; Wang, M.; Sun, C. Tanshinone IIA: New perspective on the anti-tumor mechanism of A traditional natural medicine. Am. J. Chin. Med. 2022, 50, 209–239. [Google Scholar] [CrossRef]
- Su, C.-C.; Chiu, T.-L. Tanshinone IIA decreases the protein expression of EGFR, and IGFR blocking the PI3K/Akt/mTOR pathway in gastric carcinoma AGS cells both in vitro and in vivo. Oncol. Rep. 2016, 36, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-C. Tanshinone IIA inhibits gastric carcinoma AGS cells by decreasing the protein expression of VEGFR and blocking Ras/Raf/MEK/ERK pathway. Int. J. Mol. Med. 2018, 41, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-I. Astaxanthin as a peroxisome proliferator-activated receptor (PPAR) modulator: Its therapeutic implications. Mar. Drugs 2019, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Wang, Y.; Zhang, Q.; Chen, Q.; Liang, C.-L.; Liu, H.; Qiu, F.; Chen, Y.; Huang, H.; Lu, W. Polyphyllin I suppresses the gastric cancer growth by promoting cancer cell ferroptosis. Front. Pharmacol. 2023, 14, 1145407. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Li, H.; Zhou, H.; Zhang, S.; Liu, Z.; Zhou, Q.; Sun, F. Recombinant Lz-8 from Ganoderma lucidum induces endoplasmic reticulum stress-mediated autophagic cell death in SGC-7901 human gastric cancer cells. Oncol. Rep. 2012, 27, 1079–1089. [Google Scholar] [CrossRef]
- Radziejewska, I.; Supruniuk, K.; Tomczyk, M.; Izdebska, W.; Borzym-Kluczyk, M.; Bielawska, A.; Bielawski, K.; Galicka, A. p-Coumaric acid, Kaempferol, Astragalin and Tiliroside Influence the Expression of Glycoforms in AGS Gastric Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8602. [Google Scholar] [CrossRef]
- Kwiecien, S.; Magierowski, M.; Majka, J.; Ptak-Belowska, A.; Wojcik, D.; Sliwowski, Z.; Magierowska, K.; Brzozowski, T. Curcumin: A potent protectant against esophageal and gastric disorders. Int. J. Mol. Sci. 2019, 20, 1477. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.K.; Luis, P.B.; Mishra, S.K.; Barry, D.P.; Asim, M.; Pandey, A.; Chaturvedi, M.; Gupta, J.; Gupta, S.; Mahant, S. Curcumin oxidation is required for inhibition of Helicobacter pylori growth, translocation and phosphorylation of CAG a. Front. Cell. Infect. Microbiol. 2021, 11, 1304. [Google Scholar] [CrossRef]
- Alam, J.; Dilnawaz, F.; Sahoo, S.K.; Singh, D.V.; Mukhopadhyay, A.K.; Hussain, T.; Pati, S. Curcumin encapsulated into biocompatible co-polymer PLGA nanoparticle enhanced anti-gastric cancer and anti-Helicobacter pylori effect. Asian Pac. J. Cancer Prev. APJCP 2022, 23, 61. [Google Scholar] [CrossRef]
- Srinivasan, K. Black pepper and its pungent principle-piperine: A review of diverse physiological effects. Crit. Rev. Food Sci. Nutr. 2007, 47, 735–748. [Google Scholar] [CrossRef]
- Li, S.; Nguyen, T.T.; Ung, T.T.; Sah, D.K.; Park, S.Y.; Lakshmanan, V.-K.; Jung, Y.D. Piperine attenuates lithocholic acid-stimulated interleukin-8 by suppressing Src/EGFR and reactive oxygen species in human colorectal cancer cells. Antioxidants 2022, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, N.; Kim, S.-H.; Park, M.; Woo, H.J.; Kim, H.W.; Yang, J.Y.; Rhee, K.-J.; Kim, J.B. Inhibitory effect of piperine on Helicobacter pylori growth and adhesion to gastric adenocarcinoma cells. Infect. Agents Cancer 2014, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Abou Baker, D.H.; Abbas, H.S. Helicobacter pylori: A real stomach bug and Future treatment prospects. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, W.; Zhang, J.; Xia, X. Eradication of Helicobacter pylori: The power of nanosized formulations. Nanomedicine 2020, 15, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Misba, L.; Khan, A.U. Nano-therapeutics: A revolution in infection control in post antibiotic era. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2281–2301. [Google Scholar] [CrossRef]
- Safarov, T.; Kiran, B.; Bagirova, M.; Allahverdiyev, A.M.; Abamor, E.S. An overview of nanotechnology-based treatment approaches against Helicobacter pylori. Expert Rev. Anti-Infect. Ther. 2019, 17, 829–840. [Google Scholar] [CrossRef]
- Arif, M. Complete life of cobalt nanoparticles loaded into cross-linked organic polymers: A review. RSC Adv. 2022, 12, 15447–15460. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Antioxidant Supplementation | Model | Inference | References |
|---|---|---|---|---|
| 1 | Scutellarin (20 and 80 μmol/L) | AGS, HGC-27, and GES-1 cell lines | SCU suppressed gastric cancer cell proliferation and increased apoptosis by inhibiting the Wnt/β-catenin pathway. | [146] |
| 2 | ASX (1 or 5 µM) for 3 h | H. pylori-infected AGS cells | ASX inhibited H. pylori-induced integrin α5-mediated cell adhesion and migration by decreasing ROS levels and suppressing JAK1/STAT3 activation. | [94] |
| 3 | Geraniol (30–100 μM) | H. pylori-infected GES-1 cells | Increased the expression of peroxiredoxin-1 (Prdx-1) in GES-1 cells. | [147] |
| 4 | Carvacrol (10, 25, 50, and 100 mg/kg BW) | MNNG-induced GC (200 mg/kg BW)/60 days/Wistar albino rats | High doses of carvacrol (50 and 100 mg/kg BW) increased oxidative stress, inflammation, and apoptosis. | [148] |
| 5 | SCU (10, 20, and 30 mg/kg) | MNNG-induced gastric carcinogenesis/AGS cell line and Wistar albino rats | A reduction in LDH activity, ulcer index, pH, mucus weight, and percentage inhibition of ulcers was observed after SC treatment. | [149] |
| 6 | Korean red ginseng extract (RGE) (0.01–1μg/mL) for 1 h | H. pylori-infected AGS cells | RGE treatment decreased IL-8 production, mitochondrial dysfunction, and ROS production by activating Nrf2, inducing SOD-1 and HO-1, and decreasing ROS levels. | [115] |
| 7 | ASX (1 or 10 µM) for 3 h | H. pylori-infected AGS cells | ASX suppressed MMP expression, cell invasion, and migration via inhibition of PI3K/AKT/mTOR/NF-κB signaling. | [60] |
| 8 | Curcumin (5 mM and 20 mM) | H. pylori-infected AGS cells | Curcumin treatment inhibited the vacuolation activity of H. pylori. | [150] |
| 9 | Phycocyanin (150 µM) | H. pylori-infected AGS cells | Phycocyanin inhibited AGS cell hyperproliferation by regulating ROS/MAPK signaling pathways and reducing c-myc and CyclinD1 expression. | [112] |
| 10 | SCU (0–80 µM) | MGC-803 and AGS | SCU inhibited GC growth and EMT by regulating the PTEN/PI3K pathway. | [151] |
| 11 | Silibinin (20 mg/kg or 200 mg/kg) for 4 or 8 weeks | H. pylori-infected C57BL/6 mice/ MKN-1 cell line | Silibinin suppressed H. pylori infection by inhibiting COX2 and inducing iNOS expression by suppressing NF-κB and STAT pathways. | [152] |
| 12 | β-carotene (0.1 or 0.2 µM) for 2 h | H. pylori-infected AGS cells | β-carotene suppressed MAPK-driven MMP-10 expression and cell invasion by promoting PPAR-γ-mediated catalase expression and inhibiting ROS levels. | [110] |
| 13 | Vicenin-2 (40 µM) | H. pylori-infected GES cells | Vicenin-2 enhanced Nrf2 and PTEN in GES cells. | [153] |
| 14 | Luteolin (30 µM) | H. pylori-infected CRL-1739 cells | Luteolin significantly induced IL-8, IL-10, and NF-κB expression and reduced ADAM-17 expression. | [154] |
| 15 | Astragalin (0–40 µM)—6 h | HGC-27, MGC-803, and MKN-28 cell lines | Astragalin-induced apoptosis inhibited the migration and invasion via inhibition of the PI3K/AKT signaling pathway. | [155] |
| 16 | Eugenol (0–240 ug/mL) | AGS cell lines | Eugenol inhibited the TGF-β/SMAD4 signaling pathway in GC. | [156] |
| 17 | Celastrol (5 mM for 1 h) | SGC-7901 and BGC-823 cell lines | Increased cellular ROS levels led to ROS-dependent endoplasmic reticulum stress, mitochondrial dysfunction, and apoptosis. | [157] |
| 18 | Tanshinone IIA (2 and 4 4 μM) | BGC-823 and NCI-H87 | Tan IIA upregulated p53 expression and lipid peroxidation (LPO); ferroptosis downregulated xCT expression, intracellular glutathione (GSH), and cysteine levels.. | [158] |
| 19 | Nobiletin | H. pylori-infected GES-1 cells. | Nobiletin significantly decreased the expression of TNF-α, IL-6, COX-2, PI3K, AKT, and MAPK molecules, including p38 and c-Jun amino-terminal expression in H. pylori-infected GES-1 cells. | [159] |
| 20 | Zeaxanthin (100 μM for 24 h) | AGS, KATO-3, MKN-28, MKN-45, NCI-N87, YCC-1, YCC-6, YCC-16, SUN-5, SUN-216, SUN-484, and SUN-668 cell lines | Zeaxanthin against GC by inhibiting the ROS-mediated MAPK, AKT, NF-κB, and STAT3 signaling pathways. | [160] |
| 21 | Celastrol | MKN45 cells | Celastrol inhibited proliferation, migration, and invasion and inactivated PTEN/PI3K/AKT and NF- κB signaling pathways in MKN45 cells by downregulating miR-21. | [161] |
| 22 | Celastrol (0 or 2 μM) | HGC27 and AGS cells | Celastrol activated RIP1/RIP3/MLKL pathways and suppressed the level of pro-inflammatory cytokines by downregulating biglycan (BGN) in HGC-27 and AGS cells. | [162] |
| 23 | ASX (1 or 5 μM) for 3 h | H. pylori-infected AGS cells | ASX inhibited the reduction in mitochondrial ROS caused by H. pylori and decreased SOD2 and SOD activity. | [163] |
| 24 | ASX (1 or 5 μM) for 3 h | H. pylori-infected AGS cells | Astaxanthin inhibited H. pylori-induced mitochondrial dysfunction and ROS-mediated IL-8 expression by activating PPAR-γ and catalase. | [111] |
| 25 | Ebselen (0 or 100 μM) | AGS and MGC-803 cells | Ebselen may inhibit ROS production triggered by H. pylori LPS treatment via GPX2/4 instead of TLR4 signaling and reduce phosphorylation of p38 MAPK, resulting in altered production of IL-8. | [164] |
| 26 | α-lipoic acid (10 and 20 µM for 2 h) | H. pylori-treated AGS cells | α-lipoic acid inhibited ROS levels, IL-8 expression, activation of MAPK, ERK1/2, JNK1/2, p38, JAK1/2, STAT3, and NF-κB signaling pathways. | [59] |
| 27 | Epigallocatechin Gallate (EGCG) (0.05% EGCG in drinking water) | H. pylori-infected Mongolian gerbils | EGCG inhibited the IL-1β, TNF-α, COX-2, and iNOS in the gerbil model of H. pylori-induced inflammation. | [165] |
| 28 | Artemisia and/or green tea extracts | H. pylori-infected and high-salt-diet-administered C57BL/6 mice | Artemisia and/or green tea extract treatment significantly decreased the expressions of COX-2, TNF-α, IL-6, lipid peroxide, and activated STAT3 relevant to H. pylori infection. | [166] |
| 29 | Curcumin | H. pylori-infected 8-week-old BALB/c mice | Curcumin reduced the LPO, MPO level, urease activity, the number of colonized bacteria, levels of anti-H. pylori antibodies, biofilm formation, IFN-γ, IL-4, gastrin and somatostatin levels in serum, and minimum inhibitory concentration. | [167] |
| 30 | Nobiletin (0–50 µM) | SNU-16 cells | Nobiletin-induced apoptosis in SNU-16 cells was mediated via intracellular ER stress-mediated protective autophagy. | [168] |
| 31 | Eugenol (0.1–1.7 mM) | AGS cells | Eugenol induced apoptosis (caspase 3 and caspase 8) in the presence of as well as in the absence of functional p53. | [169] |
| 32 | α-LA (10 μM and 20 μM) 2 h | H. pylori-infected AGS cells | α-LA inhibited NADPH oxidase and ROS production, inhibition of NF-κB and AP-1 activation, induction of oncogenes, β-catenin nuclear translocation, and hyperproliferation in AGS cells. | [170] |
| 33 | Celastrol | AGS and YCC-2 cells GC xenografted mice | Celastrol induced apoptosis and autophagy in gastric cancer cells. | [171] |
| 34 | RGE (at various concentrations) | H. pylori-infected AGS cells | RGE inhibited the expression of MCP-1 and iNOS by suppressing the activation of NADPH oxidase and Jak2/Stat3 signaling. | [93] |
| 35 | Catechins (CAs), sialic acid (SA) combination of CA and SA (CASA) | H. pylori-infected AGS cells and BALB/c mice | CASA attenuated the caspase-1-mediated epithelial damage. | [172] |
| 36 | Celastrol (0–5 µM) | BGC-823, MGC-803, and SGC-7901 cells | Celastrol induced apoptosis by inhibiting NF-κB activity by upregulating miR-146a expression. | [173] |
| 37 | Diphenyleneiodonium (DPI) (2.5 or 5.0 μM) 2 h | H. pylori-infected AGS cells | DPI inhibited H. pylori-induced activation of MAPKs and MCP-1 expression in AGS cells. | [106] |
| 38 | Sporoderm-removed spores of G. lucidum (RSGLP) | H. pylori-infected AGS cells | RSGLP is more effective at inhibiting gastric cancer cell viability and may serve as a promising autophagy inhibitor for gastric cancer. | [174] |
| 39 | Resveratrol (100 mg/kg body weight/day) orally for six weeks | Male Kunming mice | Resveratrol inhibited oxidative stress and inflammation in H. pylori-infected mucosa via the suppression of IL-8, iNOS, and NF-κB and the activation of the Nrf2/HO-1 pathway. | [175] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sah, D.K.; Arjunan, A.; Lee, B.; Jung, Y.D. Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development. Antioxidants 2023, 12, 1712. https://doi.org/10.3390/antiox12091712
Sah DK, Arjunan A, Lee B, Jung YD. Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development. Antioxidants. 2023; 12(9):1712. https://doi.org/10.3390/antiox12091712
Chicago/Turabian StyleSah, Dhiraj Kumar, Archana Arjunan, Bora Lee, and Young Do Jung. 2023. "Reactive Oxygen Species and H. pylori Infection: A Comprehensive Review of Their Roles in Gastric Cancer Development" Antioxidants 12, no. 9: 1712. https://doi.org/10.3390/antiox12091712