1. Introduction
Necrotizing enterocolitis (NEC) is a devastating gastrointestinal emergency that affects 4–7% of newborns admitted to the Neonatal Intensive Care Unit (NICU) [
1]. NEC preferentially occurs in premature and very low birth weight infants (less than 1.5 kg), although it can also affect full-term patients. Both treatment of the active disease and management of the associated long-term complications (i.e., short bowel syndrome) convey a significant clinical task for the patient, family, and involved medical care providers. As compared to other disorders of prematurity (i.e., bronchopulmonary dysplasia), the morbidity associated with NEC has not shown any major improvements with advances in neonatal care [
2]. Additionally, the estimated cost of NEC management in the United States ranges between USD 500 million and USD 1 billion annually [
3]. Despite years of advancements and resource allocation, NEC remains responsible for 1 in 10 of all neonatal deaths [
4].
Research focused on the underlying pathophysiology of NEC has identified several mechanisms that are involved in disease susceptibility. Introduction of formula feeding and increased caloric density of the latter, subsequent changes to the gastrointestinal microbiome, a leaky and immature intestinal barrier, and a dysregulated inflammatory and immune response to host–pathogen interactions in the gut have all been implicated as inciting factors for NEC. It has also been established that there is a role for oxidative distress in NEC. Previous data has shown that nitrosative stress in NEC promotes intestinal injury, impairs cellular repair mechanisms, and favors apoptosis [
5]. Most of these studies have focused on rat and cell line models, and they have demonstrated that a rise in inducible nitric oxide synthase (iNOS) expression is a potential driver for the increased inflammatory response and apoptosis in NEC [
6].
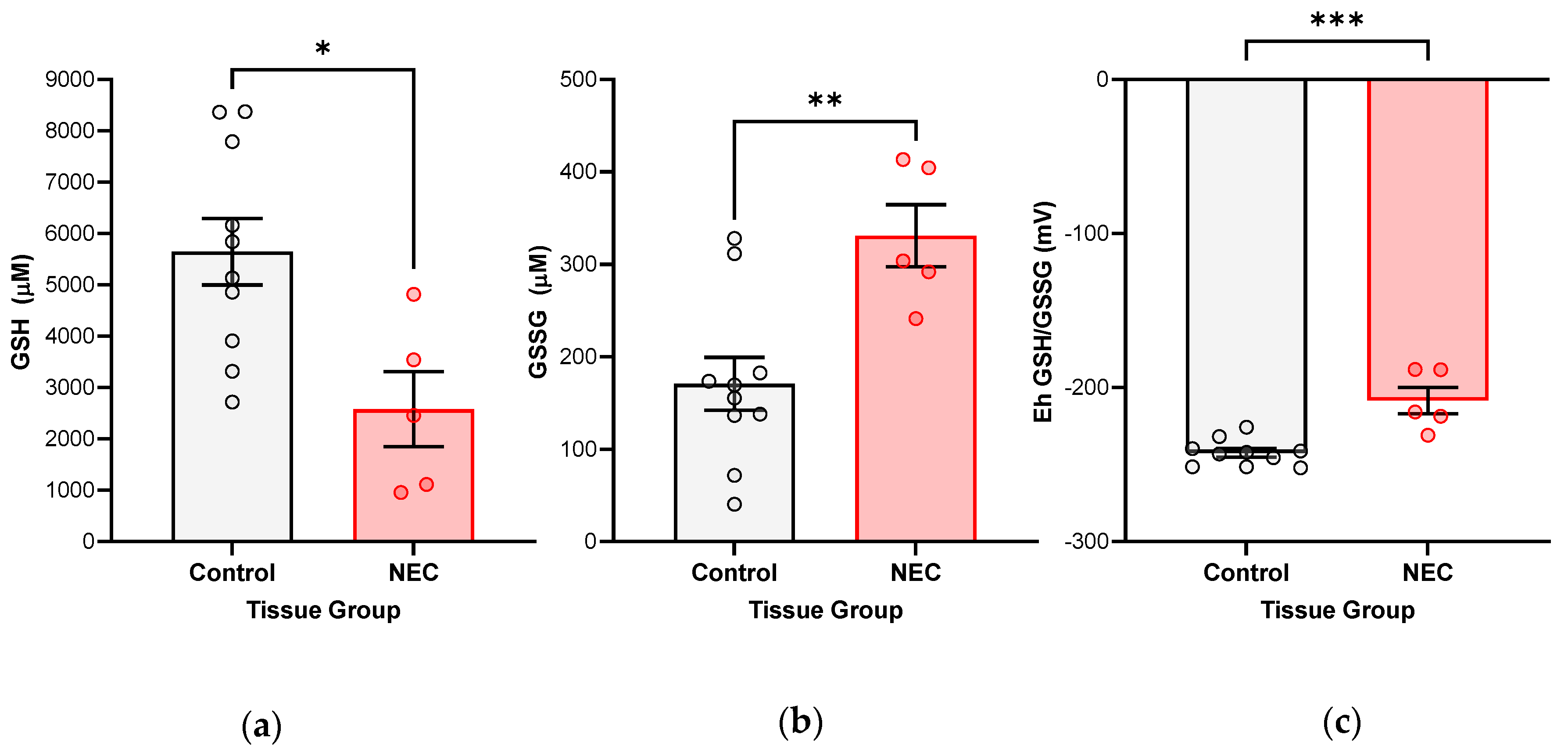
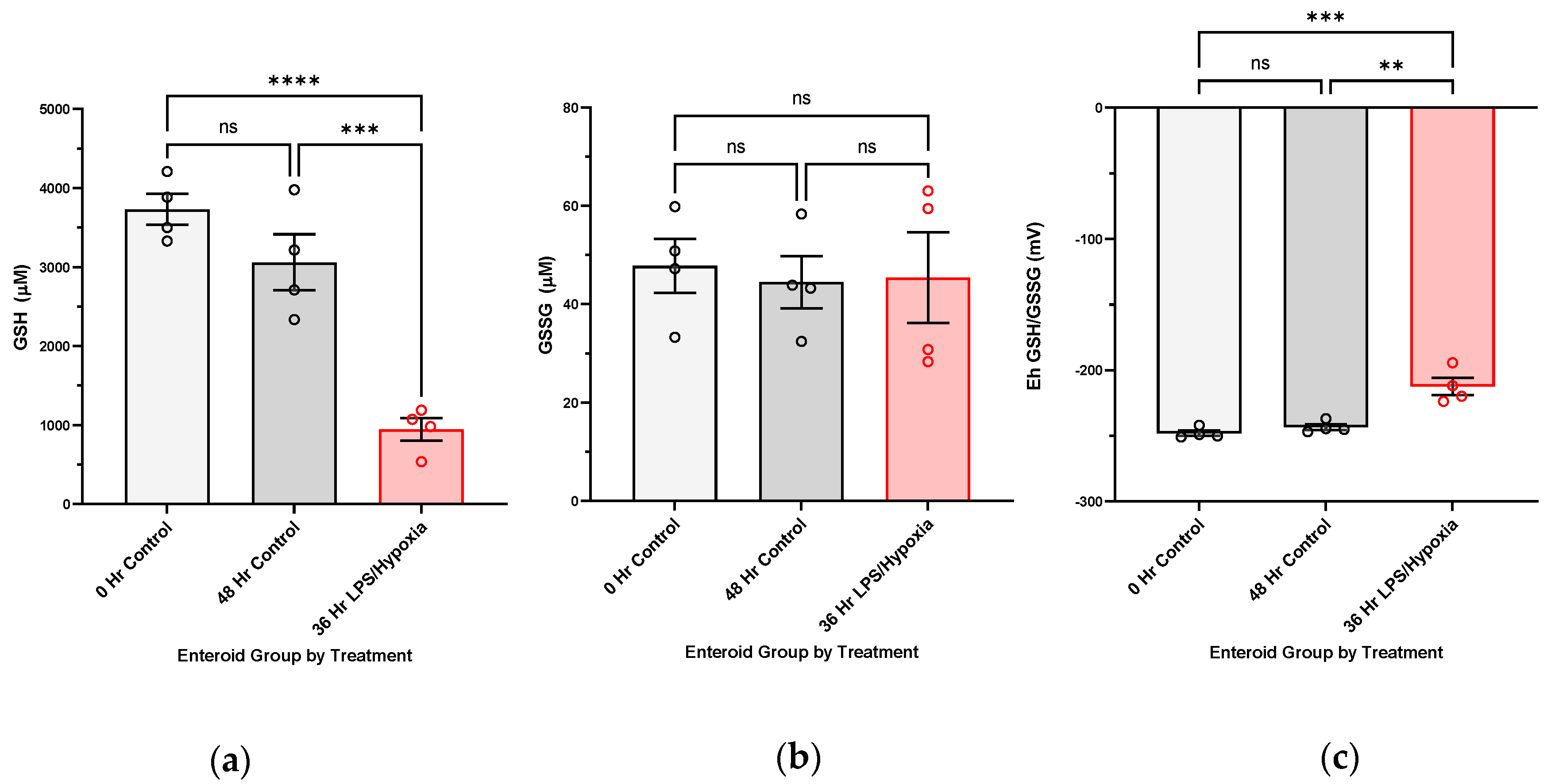
Unbalanced cellular redox aberrations are associated with disease, including NEC, and it may promote hyperinflammation, impair restitution, and favor cellular death. However, there are also intrinsic defense mechanisms in place to counteract oxidative stress and diminish the buildup of toxic radicals. Within the antioxidant network, the glutathione-dependent (GSH) defense system is of particular interest. GSH is considered one of the essential master antioxidants because of its multiple studied functions in the maintenance of intracellular redox homeostasis, growth and repair promotion, and maturation of tissue function. It is a recyclable molecule, abundant in the body in every organ, including intestinal tissue. GSH-dependent molecules (i.e., glutathione peroxidase family) make up one of the cell’s defense mechanisms against oxidative injury. GSH-dependent enzymes detoxify the cell from buildup of intracellular toxic radicals, utilizing glutathione’s reducing power in the process and converting it from reduced GSH to oxidized glutathione disulfide (GSSG). Earlier studies in rodent NEC models and IEC-6 cells (rat intestinal epithelial cells) have shown that several GSH-dependent antioxidants are upregulated with experimental disease induction, while chemical inhibition of GSH leads to increased cell death [
7].
Redox tone plays an important role in both inter- and intracellular signaling, promoting repair and growth as opposed to pathologic processes (i.e., ischemia/reperfusion, infection, immune response, cancer, and aging) [
8]. In redox homeostasis, a dynamic balance between oxidant production and the antioxidant response is imperative. Redox tone exists on a spectrum between oxidative eustress (physiologic balance of oxidants/antioxidants), which is beneficial for cellular health, and oxidative stress or distress, where pathologic production of oxidant species overwhelms intrinsic antioxidant machinery that should be providing balance and regulation [
9,
10]. Consequently, the redox environment can shift from a more reduced state to an oxidized state. It has previously been demonstrated that the cellular redox environment is crucial in the regulation of cellular proliferation and maturation, favoring growth in absence of oxidative stress [
11]. Intracellular redox tone is measured by comparing the fractions of reduced GSH to oxidized GSSG, from which GSH redox potentials (Eh GSH/GSSG) are derived. More reduced or negative glutathione redox potentials favor cellular proliferation, whereas the opposite is true in a more oxidative state, which can be inferred in the oxidative stress of NEC. A complete understanding of the capacity of the GSH antioxidant system in regulating redox aberrations in human NEC is lacking. A previous clinical study compared total GSH levels in infants affected by NEC to healthy controls by measuring this in blood samples [
12]. No significance was found in GSH blood levels among the two groups. Given the primary site of injury in NEC is the intestine, the functional capacity and quantity of GSH likely cannot be evaluated only by looking at total circulating GSH. Current basic science literature remains limited in the characterization of the GSH defense system in its response to NEC.
Herein, we investigate the role of the intestinal specific GSH defense system. We utilize human intestinal tissue collected from infants with active NEC and primarily derived human enteroid NEC models to investigate the response of the GSH defense system in vivo and under experimental conditions. We hypothesized that NEC-affected tissue and enteroid lines will show evidence of oxidative injury and deregulation of specific GSH-dependent enzymes that are responsible for detoxifying affected cells, as measured by increasingly oxidized GSH redox potentials. This is a novel line of investigation in human tissue and NEC models. We further analyzed whether lipid peroxidation is one of the potential significant targets of oxidative injury. Finally, we quantified the response of glutathione peroxidase 4 (Gpx4), an important GSH-dependent enzyme responsible for neutralizing lipid radicals [
13]. We postulated that lipid radical accumulation may occur when cellular defense mechanisms targeted against their accumulation fail to function efficiently and clear the toxic products of lipid peroxidation (i.e., deregulation of Gpx4 in abundance, maturity, or oxidative-injury related modification). This has downstream implications for cell fate and may be a new pathologic pathway to further help describe what leads to overwhelming cell death in terminal NEC pathophysiology.
2. Materials and Methods
Human intestinal sample collection. Institutional Review Board approval (#11610-11611) was obtained prior to commencement of human tissue collection. After parental consent, a section of pathologic intestinal tissue was collected from infants undergoing clinically indicated bowel resections at the Oklahoma Children’s Hospital (Oklahoma City, OK, USA). Indications for resection included Stage 3 surgical NEC as well as resections for non-inflammatory reasons, such as congenital gastrointestinal malformation (i.e., gastroschisis-related stricture or atresia requiring repair). The latter were used as control tissue for comparison. Gestational age at birth, age at surgery, intestinal section (i.e., ileum), gender, and indications for surgical resection were noted for each sample included. Samples from 27 infants were analyzed in the comparative studies described below, including ten infants with active NEC and 17 infants with non-NEC resections. The majority of infants had a history of prematurity, (gestational age range from 23 to 37 weeks), except for two who were born full term (40 weeks) but required intestinal resections for atresia (part of control group). See
Appendix A Table A1 for a comparative summary of basic clinical characteristics for included cases. All intestinal samples were sectioned and snap frozen in liquid nitrogen prior to further processing.
Enteroid lines were propagated from two of the human ileal specimens, both resected for active surgical NEC. Both lines had history of prematurity, born at 34 and 23 weeks, and underwent resections at 42 and 34 weeks, respectively. Birth weights were 1.79 and 0.55 kg. Both patients passed away at index hospitalization due to total bowel necrosis and respiratory failure, respectively.
Harvest and culture of enteroids from human intestinal samples. Tissue collected for enteroid line derivation was immediately washed in cold Dulbecco’s phosphate-buffered saline (DPBS, Sigma Life Science, #D1408, St. Louis, MO, USA) and processed on the same day as collection. Stem cell crypts were isolated for enteroid culture generation using a previously described protocol for enteroid processing [
14]. Enteroid cultures were suspended in basement membrane matrix or Matrigel (Corning, #CB-40230C, Corning, NY, USA) domes and grown in 50% L-WRN conditioned media prepared in our lab, supplemented with 50 ng/mL epidermal growth factor (Millipore Sigma, #GF144, Burlington, MA, USA), 1 mM N-Acetylcysteine (Millipore Sigma, #A9165-5G, Burlington, MA, USA), 500 nM A-83-01 (R&D Systems, #2939/10, Minneapolis, MN, USA), 10 μM SB202190 (Millipore Sigma, #S7067-5 MG, Burlington, MA, USA), 10 mM Nicotinamide (Millipore Sigma, #N0636-100G, Burlington, MA, USA), and 10 nM [leu] 15-gastrin 1 (Millipore Sigma, #G9145-.1 MG, USA). Enteroids were passaged weekly and experimented on once they reached maturity. All studies were completed between passages 5–10 for all described experiments in this study.
NEC induction in human enteroids. Enteroid cultures were grown in Matrigel domes (Corning, #CB-40230C, Corning, NY, USA), as previously described, until maturity (around seven days from passage). For experimental NEC induction, enteroids were subjected to treatment with 100 ug/mL lipopolysaccharide added to the media and hypoxic conditions (1% Oxygen) provided by the modular incubator chamber from Billings-Rothenberg, Inc. (Del Mar, CA, USA). N-Acetylcysteine was not supplemented into experimental media for either the control or the treated cultures during experimentation, given its proposed benefits against oxidative injury. Treatment duration lasted from 24 to 48 h. Control enteroids were harvested at both commencement (0 h) and end of experiment (48 h) to follow baseline redox changes within the cultures. Each sample was considered to be three pooled domes of enteroids, with at least three biological replicates per treatment group.
HPLC analysis for measurement of GSH redox potentials and protein bound GSH in human tissue and enteroid cultures. Methodology for HPLC analysis was previously described and modified by Harris and Hansen [
15]. Snap frozen intestinal tissue and enteroid pellets harvested for GSH redox potential measurements were preserved in cold 5% perchloric acid buffer containing 10 μM γ-glutamyl-glutamate at −80 °C until processing. Samples were solubilized in 100 mM sodium hydroxide, and protein concentrations were quantified using BCA assay. GSH and GSSG concentrations were determined using reverse-phase high performance liquid chromatography (HPLC (Waters 2695 Alliance Separations Module with a Supercoil LC-NH
2 column). Peak detection was made using a Waters 2474 fluorescence detector (335 nm/518 nm). Molarity values of GSH and GSSG were determined through standardization with the internal standard (γ-glutamyl-glutamate) and sample protein concentrations. Redox potentials were derived using the Nernst equation.
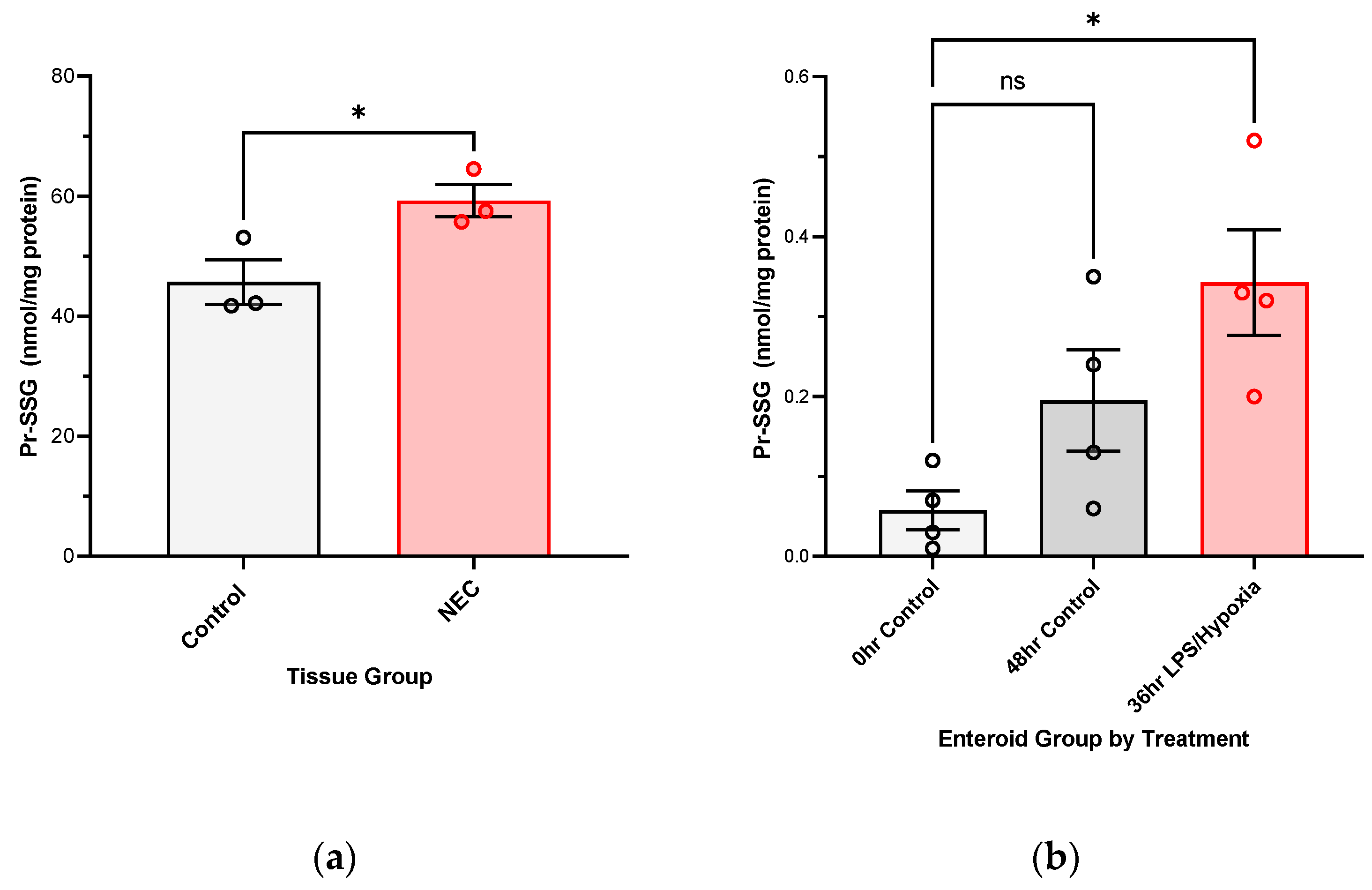
For protein bound GSH, S-glutathionylated proteins (PrSSG) pellets collected from samples during collection were washed with 5% perchloric acid and then re-pelleted, and the supernatants were removed. Pellets were treated with 0.5 mL 0.1 M NaOH to neutralize the perchloric acid, and the pellets were resuspended. An aliquot (0.25 mL) was then added to an equal volume of 0.1 M phosphate buffer (pH 6.0) containing 5 mM dithiothreitol, and the solution (final pH approximately 7.6) was incubated at room temperature for 30 min. Following the incubation, 0.5 mL of 10% perchloric acid/boric acid containing the internal standard was added. Samples were then processed and derivatized as described above via HPLC. Data is presented as GSH nmol/mg protein.
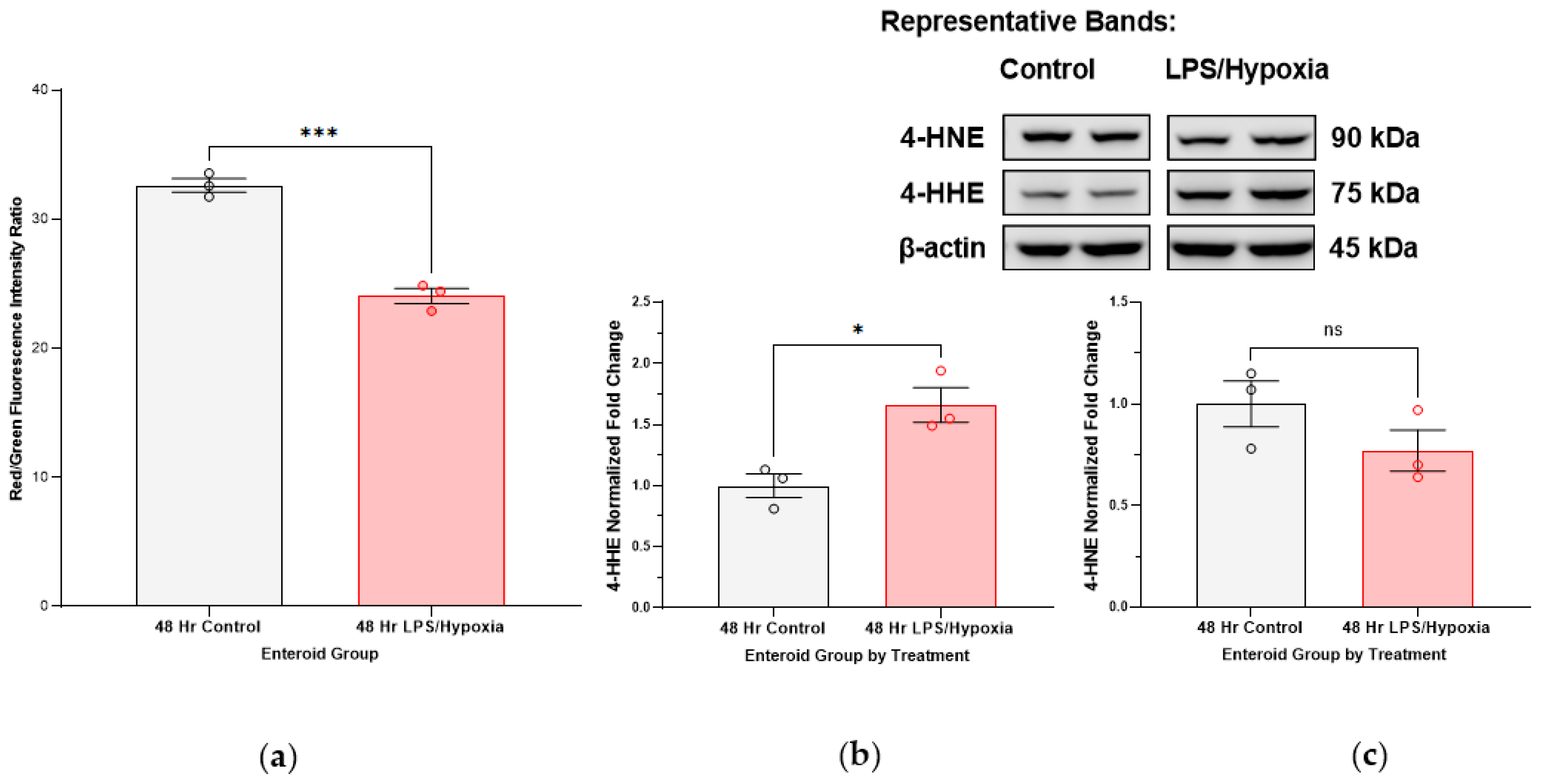
Lipid peroxidation measurements via flow cytometry in human enteroid NEC model. Enteroid cultures were subjected to our previously described experimental NEC conditions for 48 h. Matrigel domes were then dissolved and the enteroid cultures were suspended in growth media with the Image-It® Lipid Peroxidation Sensor (#C10445, Life Technologies, Carlsbad, CA, USA) at a manufacturer-recommended concentration of 10 µM. The Lipid Peroxidation Sensor is a fluorescent reporter based on BODIPY® 581/591 C11 reagent which, in live cells, shifts from red to green with increasing lipid peroxidation products, providing a ratiometric indication of lipid radical accumulation. Enteroids were incubated at 37 degrees Celsius for 30 min with the sensor, after which time the cultures were washed in 1× phosphate buffered saline (PBS) and further quickly processed into single cells using trypsin (ThermoFisher Scientific, #78840, Rockford, IL, USA) as the dissociation agent. Cells were analyzed with flow cytometry (Stratedigm S1200Ex flow cytometer platform, Stratedigm-3) at a total of 1000 events collected per sample, with a 488 nm laser excitation and fluorescence emission collected from 530/30 nm (green) and 488 nm laser excitation and fluorescence emission collected from 580/30 nm (red). Ratios of red/green fluorescence intensities were quantified and compared among control and NEC-induced enteroid groups (each run in triplicate). A decrease in red/green fluorescence intensity ratios corresponded with an increase in lipid peroxidation in the sample. A positive control was included, using enteroid cultures treated with 100 µM cumene hydroperoxide for 2 h prior to analysis.
Lipid profiling of fatty acid composition in intestinal tissue samples. Total lipid extraction was performed using a protocol described by Folch et al., in which samples were homogenized in methanol [
16]. Fifty nanomoles of pentadecanoic (15:0) and heptadecanoic (17:0) acids were added as internal standards. Lipid extracts underwent acid hydrolysis/methanolysis by heating at 100 degrees Celsius in 16%
v/
v concentrated hydrochloric acid (HCl) in methanol to generate fatty acid methyl esters (FAMEs). FAMEs were extracted in hexane and purified via thin-layer chromatography (TLC) based on previously described protocol [
17]. FAMEs were identified using an Agilent Technologies 7890A gas chromatograph with a 5975C inert XL mass spectrometer detector (Agilent Technologies, Lexington, MA, USA) [
18]. FAMEs were quantified using an Agilent Technologies 6890N gas chromatograph with flame ionization detector [
18]. Sample concentrations were determined by comparison to internal standards (15:0 and 17:0). Data are represented as relative mole percent of each fatty acid.
SDS-PAGE and immunoblotting of protein extracted from intestinal tissue and treated enteroid cultures. Protein extraction was completed using a RIPA lysis cocktail (1×, Cell Signaling Technology, #9803, Danvers, MA, USA) with phosphatase and proteinase inhibitors (Phosphatase Inhibitor Cocktail 2, Sigma Aldrich, #P5726, St. Louis, MO, USA; Protease Inhibitor Cocktail, Sigma Aldrich, #P8340, St. Louis, MO, USA). Total protein concentrations were quantified using the PierceTM BCA Protein Assay kit (ThermoFisher Scientific, #23225 and 23227, Rockford, IL, USA). Gels with 10% hand-cast acrylamide (BioRad, #1610156, Hercules, CA, USA) were used for SDS-PAGE. Protein was loaded at 10 μg per well for both proteins derived from intestinal tissue and enteroid NEC studies. Gels were transferred on to nitrocellulose membranes and blocked in 5% milk. Membranes were incubated with primary antibodies overnight at 4 degrees Celsius and for 1–2 h at room temperature with secondary HRP-linked antibodies. Washes were completed with 1× tris-buffered saline, 0.1% Tween® 20 (TBS-T). The primary antibodies used were glutathione peroxidase 4 (Gpx4) at a dilution of 1:5000 (ab125066, abcam, Boston, MA, USA), 4-HNE at a dilution of 1:1000 (MA5-27570, Invitrogen, Danvers, MA, USA), and 4-HHE at a dilution of 1:5000 (MA5-27556, Invitrogen, Danvers, MA, USA). Β-actin served as a reference protein for normalization at a dilution of 1:1000 (#3700, Cell Signaling, Danvers, MA, USA). Secondary antibodies included horse anti-mouse HRP-linked antibody at 1:1000 dilution (#7076s, Cell Signaling, Danvers, MA, USA) and goat anti-rabbit HRP-linked antibody at 1:10,000 dilution (#31460, Invitrogen, Danvers, MA, USA).
Statistical analysis. Results are represented as means with SEM as error bars, unless otherwise reported. Statistical analysis and figures were completed using GraphPad Prism 9.5.0 software. Comparative analyses were run using Student’s t-test or analysis of variance (ANOVA), as appropriate. All compared groups had at least three biological replicates and were run in triplicate to improve rigor of results.
4. Discussion
The role of oxidative injury in the pathogenesis of NEC has been proposed in the literature but not fully explained. Neither has the response of the innate glutathione antioxidant defense system been comprehensively characterized. It is established, though, that oxidative stress is partly responsible for cellular injury that is seen in NEC and other inflammatory bowel disease such as Crohn’s and experimental DSS-colitis [
19,
20,
21,
22]. Aberrations in the redox state of a cell are also one of the main regulatory checkpoints that may usher a cell towards death as opposed to repair and restitution [
7,
11,
22,
23]. Studies of human intestinal tissue have yet been able to confirm glutathione-dependent antioxidant system dysfunction in NEC, including changes in the GSH-controlled redox environment (i.e., GSH redox potentials). Furthermore, measures of oxidative stress in NEC-affected infants are mostly completed by blood analyses [
12,
20]. In this investigation, we aimed to investigate the role and function of the glutathione antioxidant defense as well as the potential targets of oxidative injury in the intestine, the target tissue affected by NEC.
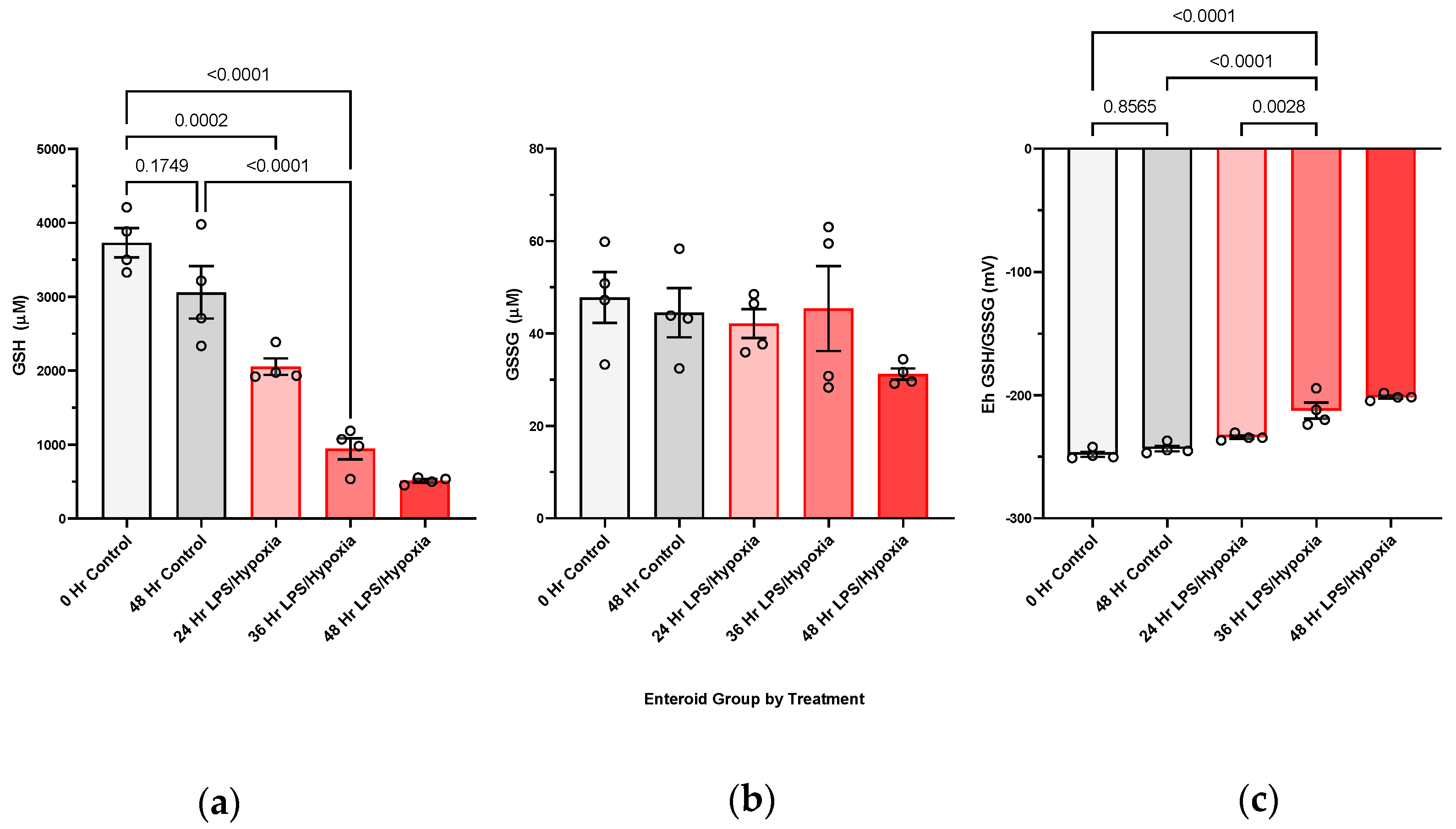
We have shown, for the first time, that NEC injury of the intestine is associated with increasingly oxidized GSH redox potentials in the target intestinal tissue in vivo. We have also been able to confirm this in an experimental human NEC enteroid model. A shift towards increasingly oxidized GSH redox potentials in both tissue and enteroids demonstrates that glutathione-dependent changes are occurring intracellularly in response to the disease. The implication of an oxidized redox intracellular environment is inhibition of cellular proliferation and increased regulatory cell death [
7,
11]. Consequently, the ability of NEC-affected intestinal tissue to repair itself may be impaired, leading to eventual necrosis and perforation as seen in the more severe, surgical NEC stages. In addition, with the derivation of protein glutathionylation, we have showed that both human intestinal tissues affected by NEC and experimental NEC enteroid model have increasing levels of glutathionylated protein. S-glutathionylation can lead to modification of target enzymes [
24].
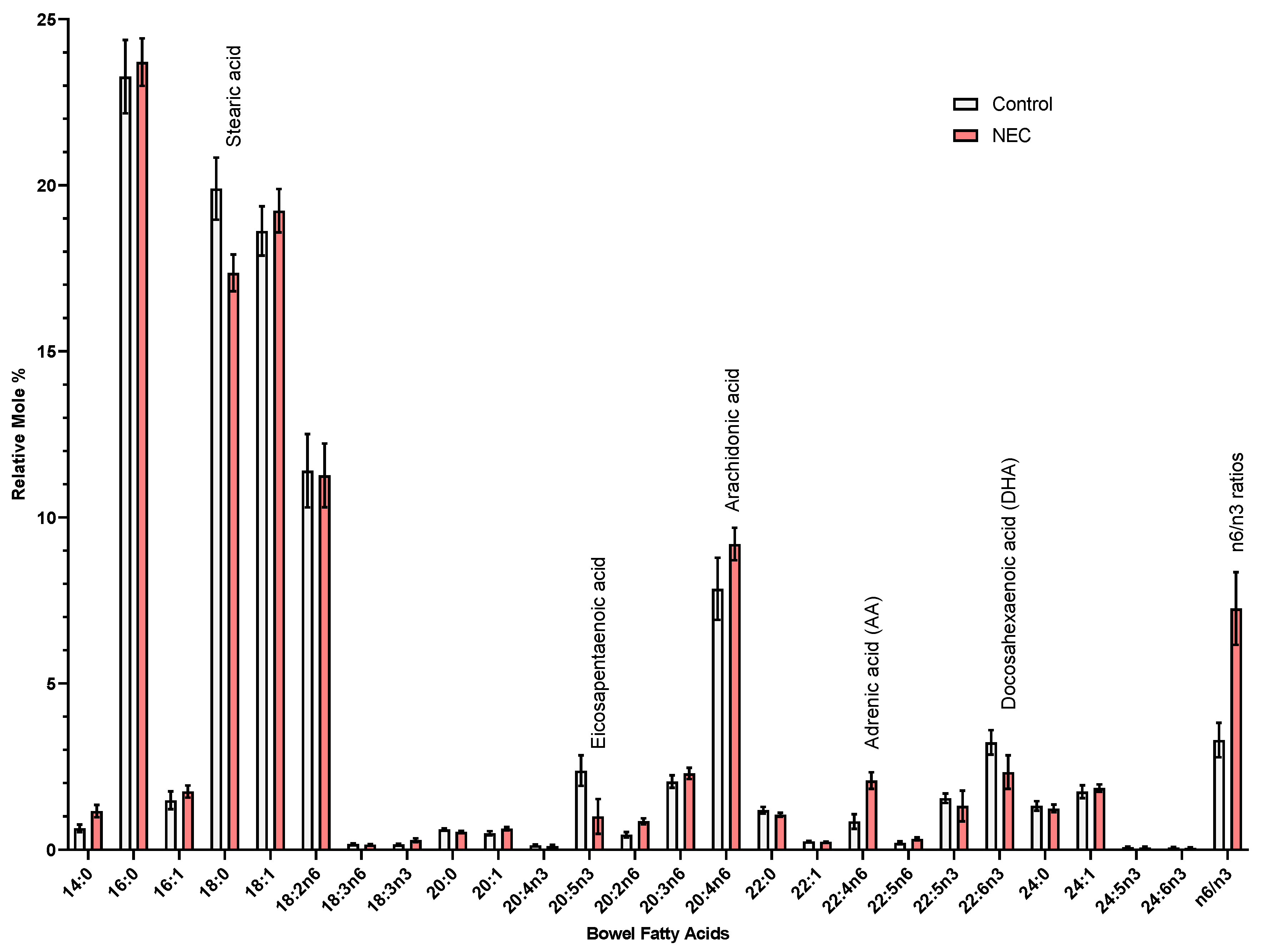
Furthermore, we show that lipids are targets of oxidative injury in NEC. Polyunsaturated fatty acids (PUFAs) are abundant in the cell (e.g., lipid bilayer of the cell membrane as well as fatty acids part of inflammatory signaling cascades) and have been implicated as main targets/substrates for lipid peroxidation under oxidative stress in studies of other intestinal disease models [
25]. Lipid radicals that form as a result of this peroxidation can be downstream signaling molecules for cell demise (i.e., oxidized arachidonic and adrenic phosphatidylethanolamines (PEs) in ferroptosis, an iron-dependent form of regulatory cell death that is triggered by accumulation of lipid radicals), as well as have the ability to form protein adducts and modify protein function inside the cell, with eventual cytotoxic consequences (i.e., 4-HNE and 4-HHE) [
26,
27,
28]. Downstream formation of toxic lipid aldehydes, 4-HHE (4-hydroxy-2-hexenal) and 4-HNE (4-hydroxy-2-nonenal) stems from peroxidation products of n-3 and n-6 PUFAs, respectively [
29]. Interestingly, we have noted an uptrend in 4-HHE in our immunoblotting more so than 4-HNE. Furthermore, blotting for 4-HNE in active NEC intestinal specimens actually resulted in multiple bands on the membrane (
Appendix A). Identification of these bands can help us determine if these multiple bands are a result of 4-HNE adducts and what proteins are affected by these modifications. Theoretically, given 4-HNE is a result of peroxidation of n6 PUFAs, while 4-HHE is a result of n3 PUFA peroxidation, we can likely augment the levels of these lipid aldehydes by changing the baseline supply of PUFAs. Dietary modification/supplementation of PUFAs (i.e., omega-3 docosahexaenoic acid (DHA), omega-6 arachidonic acid) may have a role in the tendency for production of one toxic lipid aldehyde over the other in the face of oxidative stress, depending on which fatty acids have been incorporated into the lipid profiles of the cells [
30]. Additionally, when comparing oxidation products of PUFAs in infant formula versus human breastmilk, formula was found to have higher levels of 4-HHE/n-3 PUFA ratios than breastmilk [
31]. Levels of malondialdehyde (MDA, another lipid peroxidation marker) were also elevated. Further investigation into lipid profiling of formulas and fortification mixes will help further elucidate if the potential source of oxidant substrates stems from nutritional sources. Supplementation of readily oxidizable PUFAs may exacerbate intestinal inflammation that is seen in NEC. As a comparison to other intestinal disorders, inflammatory bowel disease such as Crohn’s has also been recently tied with westernization of diets. Western diets are rich in inflammatory PUFAs such as omega-6 arachidonic acid. Mayr et al. have shown that intestinal epithelial cells with Crohn’s have Gpx4 dysfunction and signs of lipid peroxidation [
32]. Furthermore, the group has been able to trigger Crohn’s-like inflammation similar to the human phenotype in mice that lack one allele of Gpx4 (murine double knockouts are lethal).
Nutritional supplementation of key antioxidants (i.e., anti-inflammatory omega 3 fatty acids, Vitamin E, n-acetylcysteine) can, on the contrary, promote adequate antioxidant defense responses. Breastfeeding with human milk is an example of a nutritional strategy that has been shown to promote a healthy redox environment [
10]. HMOs (human milk oligosaccharides) have been extensively studied for their benefits to gut health, promoting a healthy microbiome balance and controlling pathogens, increasing cellular proliferation, modulating inflammatory responses, and with indirect antioxidant function [
33,
34,
35,
36]. Targeted supplementation with GSH, for example, as the principal antioxidant for premature infants with antioxidant deficiencies, is being investigated in literature for its therapeutic and preventative potential. Mode of supplementation is also imperative as GSH addition to nutritional modalities such as parenteral nutrition or infant formula has the potential for oxidation of the antioxidant leading to decreased availability [
37].
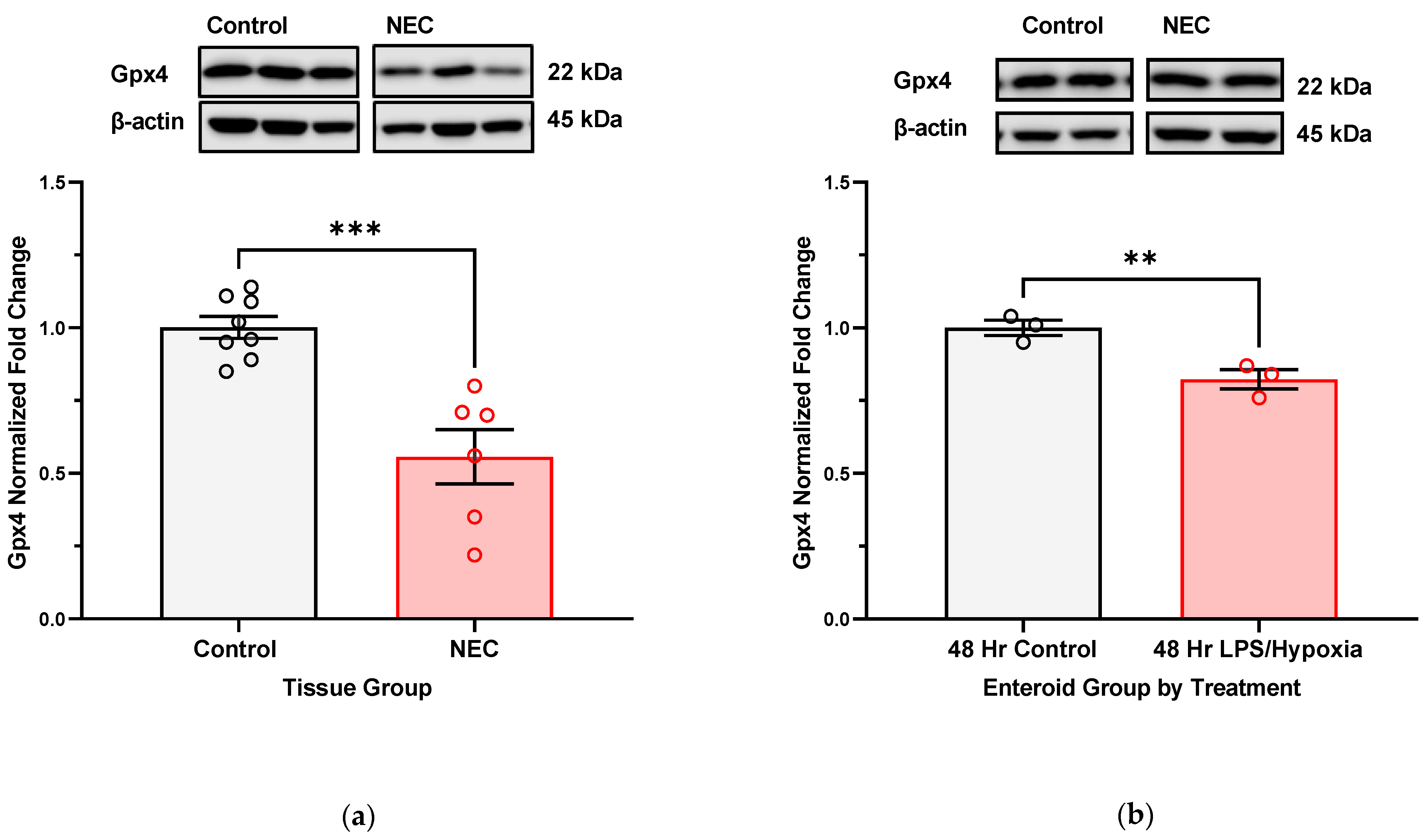
With analysis of Gpx4 expression in NEC-affected human intestinal tissue and in NEC-induced enteroids, we show that with longer exposure to NEC-stimuli, there is likely a more significant deregulation of the innate antioxidant defense mechanisms. In the specific case of Gpx4, we see a downregulation in protein expression of this enzyme. Uniquely, it is one of the only GSH-dependent glutathione peroxidases capable of neutralizing lipid radicals into benign alcohols [
38]. Consequently, we can infer that in the face of oxidative stress and lipid radical accumulation, one of the main detoxifying enzymes becomes deregulated or dysfunctional. This may be due to decreased production of Gpx4 but can also be a result of degradation or modification (i.e., protein glutathionylation or adduct formation with a toxic lipid aldehyde product such as 4-HHE or 4-HNE). Further specialized characterization of targets of protein glutathionylation and or adduct formation will help identify if Gpx4 is affected by either of these modifications.
While our study focuses on the consequences of intestinal oxidative injury of NEC, the disease is a multisystemic disorder, with the redox aberrations that start in the intestine but that have the potential to spread and lead to ultimate systemic septic shock and multiorgan failure. The previously mentioned clinical study that measured total GSH in blood samples of infants with and without NEC did not show a significant drop in the antioxidant between healthy infants and those affected by NEC, but it did show a trend towards lower GSH levels with more extensive disease [
12]. This prompts further needed research into the temporal relationship of accumulating oxidative distress and cellular signaling and defense response in NEC-affected infants. Deregulation of the antioxidant response is likely a later pathologic dysfunction that occurs as the redox tone shifts towards a more oxidized environment. Furthermore, Bindi et al. have showed that oxidative injury of NEC leads to mitochondrial dysfunction in the liver in a murine model [
39]. After experimental induction of NEC with hypoxia, hyperosmolar formula, and LPS administration, mouse pups in the experimental NEC group were compared to breastfed control pups. Interestingly, NEC pups demonstrated depletion of viable and functional liver mitochondria and increased levels of NRF2 in the liver (nuclear factor erythroid 2-related factor 2), a marker of corollary oxidative stress in this study. NRF2 is upregulated in the face of oxidative stress to induce expression of an array of antioxidant genes for downstream function of the antioxidant defense system [
40]. The Bindi study group suggests that mitochondrial damage is a key pathologic response to NEC injury on a systemic level. Investigation of mitochondrial response is imperative in the intestine as well, as function of mitochondria is closely tied to cell death pathways and disruption in energy production within affected cells. The increased metabolic requirements of fighting an active infection and hyperinflammation are likely compromised by the mitochondrial damage that occurs with NEC, ultimately leading to systemic shock pathophysiology where the energy demands of the tissue are unable to be met by compromised energy production. This is especially true for preterm infants who already have dysfunctional or immature mitochondria, predisposing them to prematurity-related disorders, such as bronchopulmonary dysplasia [
41].
Lipid radical accumulation within a cell and dysfunctional Gpx4 are two of the three inciting criteria that can prompt a cell towards undergoing a relatively newly described form of cell death, ferroptosis [
42]. Ferroptosis is an iron-dependent form of programmed cell death that is triggered by three hallmark events, with lipid peroxidation and overwhelming of intracellular antioxidant defense machinery (downregulation of Gpx4) being two of them [
43]. The third factor is accumulation of iron within the affected cell, which triggers auto-oxidative injury through catalysis of Fenton chemistry [
44]. Free ferrous iron has the potential to form extremely reactive hydroxyl radicals in the presence of hydrogen peroxide. Hydroxyl radicals are nonspecific and have the potential to react with biomolecules such as PUFAs, amplifying the redox aberrations that follow lipid peroxidation, as formed lipid radicals go on to participate in autooxidation [
9]. Our future investigation will continue characterizing the role of the glutathione-dependent defense system in NEC, the implications of redox imbalance on cellular signaling with increasing oxidative distress in the disease, the effects of lipid peroxidation on the intracellular redox environment (as well as the potential cytotoxic protein modifications that are possible through lipid aldehyde adduct formation and glutathionylation), modulation of lipid peroxidation byproducts through changes in dietary PUFA supplementation, and the role for ferroptosis as another cell death pathway to describe the terminal pathophysiology associated with necrotizing enterocolitis.
The results of our study are unique in that they are based on analysis of human infant tissue and human-derived enteroid cultures. This provides representative samples that are closest to the patient groups that are most susceptible to NEC. The intestinal samples we worked with were obtained from infants undergoing clinically indicated bowel resections for either active NEC or other gastrointestinal conditions requiring surgical correction. Although these samples provide an opportunity for us to study NEC in the most appropriate experimental setting, there are limitations to this as well. Surgical correction is relatively rare and seldom indicated in the newborn. The amount of tissue available is limited. Basic clinical characteristics (i.e., age, prematurity, clinical conditions) depend on the unique case of a patient requiring surgery, and, thus, while NEC resections may come from patients with relatively similar histories, comparative control samples come from cases that also have surgical pathology, but which do not have to be tied to certain characteristics such as prematurity or birth weight. This results in subject populations that can be relatively similar but not fully matched in all their clinical histories. Additionally, working with fragile NEC-affected tissue poses its own challenges given that the necrotic bowel has altered genetic/protein profiles affected by pathologic degradation and may be difficult to propagate when deriving enteroid cultures. To address these concerns, we worked to include all available control tissue (i.e., increase number of controls to case samples) as a comparison when the experiment required an adequate comparison.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






