

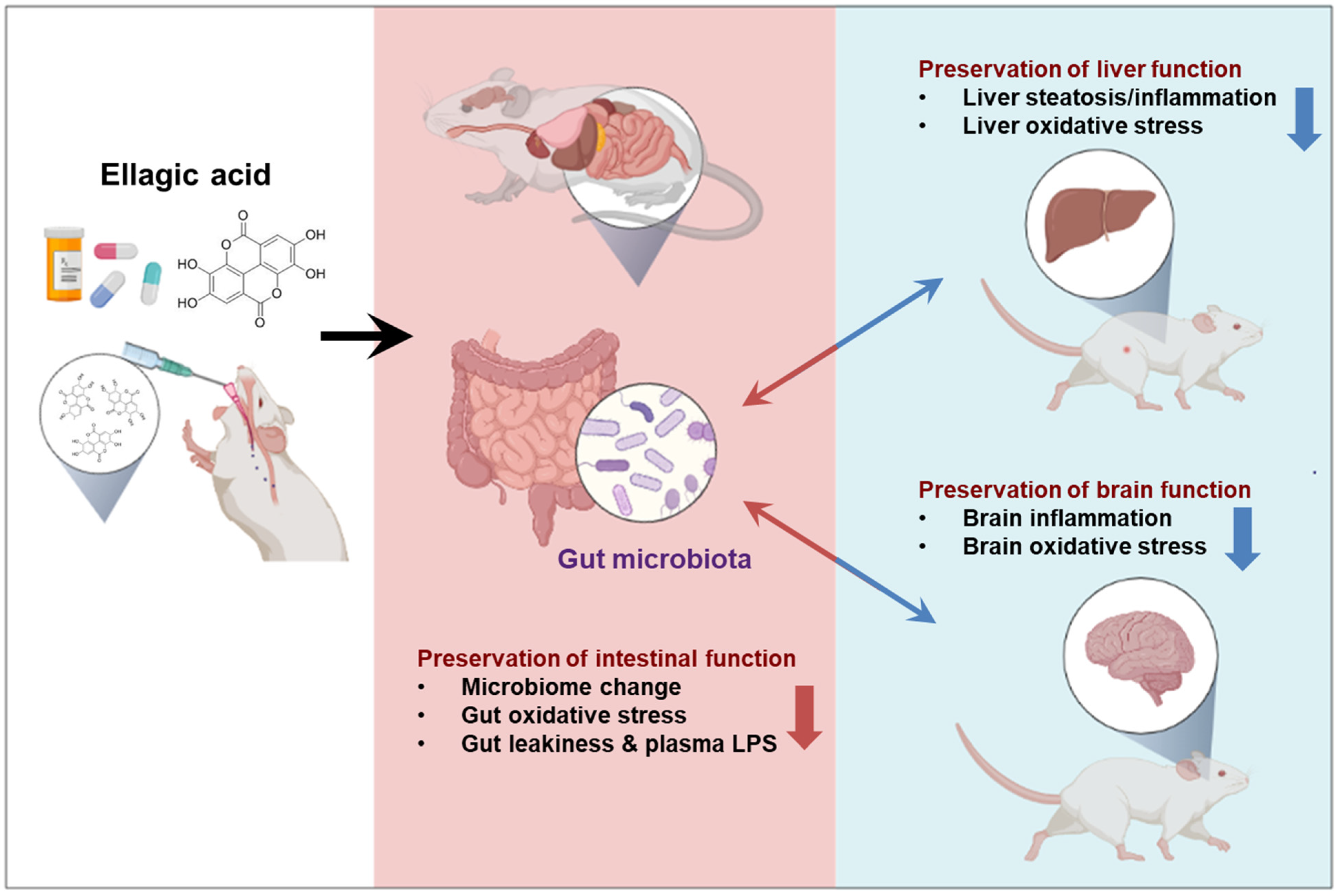
Ellagic Acid Prevented Dextran-Sodium-Sulfate-Induced Colitis, Liver, and Brain Injury through Gut Microbiome Changes
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Assessment of Disease Activity Index
2.4. Histological Analysis and Plasma AST and ALT Measurement
2.5. Endotoxin, Triglyceride, and Reactive Oxygen Species Measurement
2.6. Inflammatory Markers Messurment
2.7. Immunoblot Analysis
2.8. Apoptosis Assay
2.9. Microbial 16S Sequencing and Bioinformatics
2.10. Cell Culture and Confocal Microscopy
2.11. Statistical Analysis
3. Results
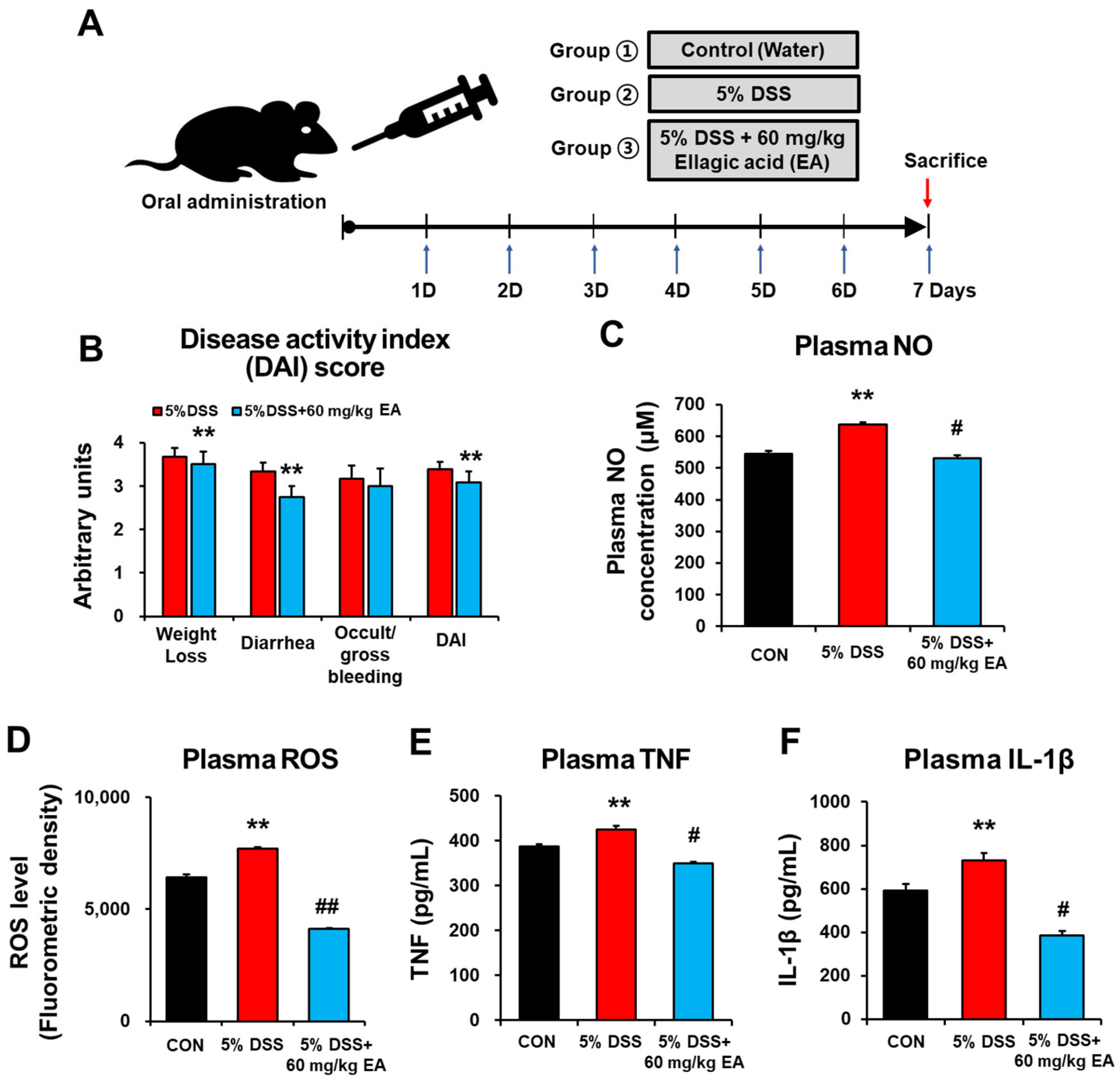
3.1. EA Attenuated the Inflammatory and Oxidative Stress of DSS-Induced Acute Colitis Mice
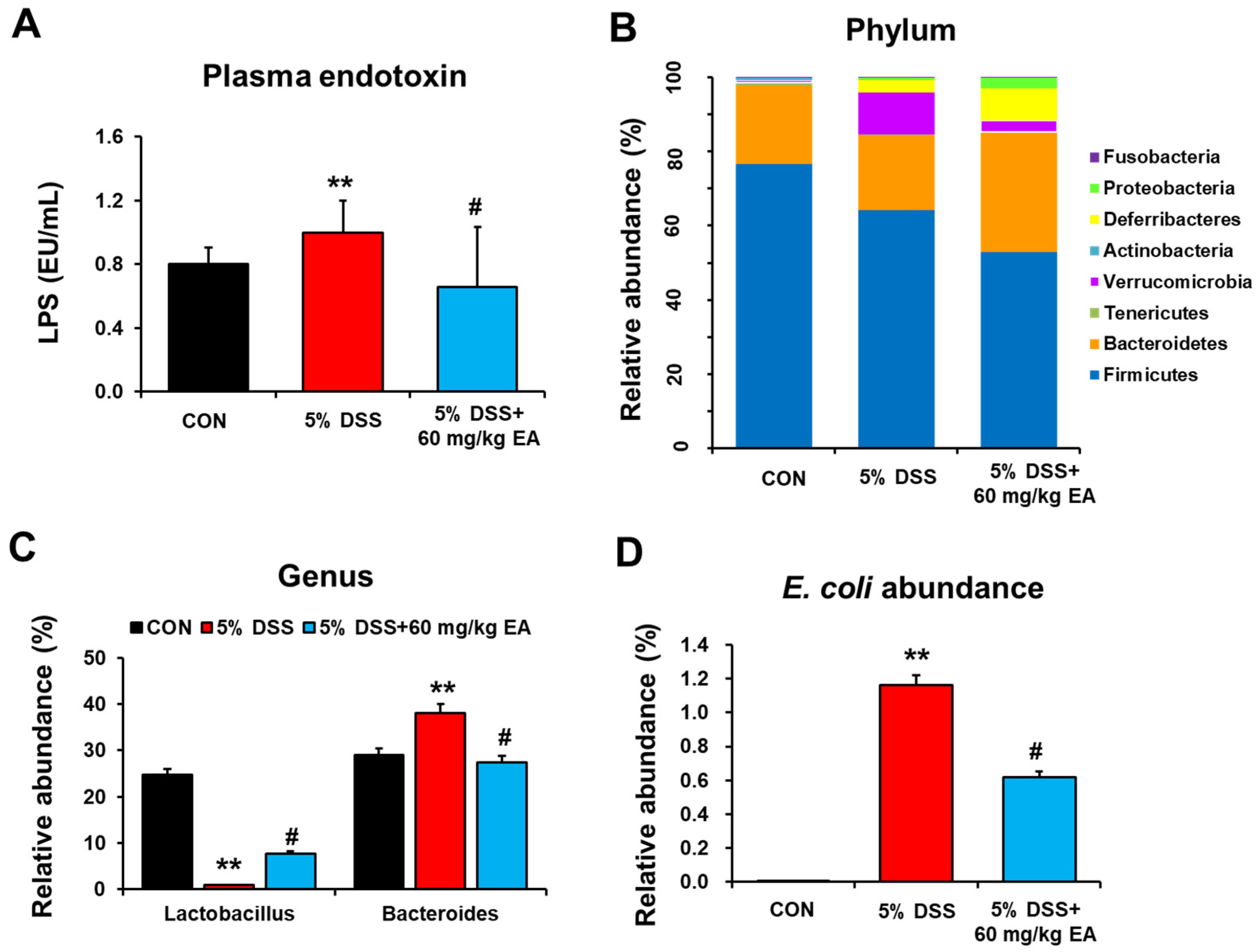
3.2. EA Reduced Endotoxin Levels and Changed the Gut Microbiota of DSS-Induced Acute Colitis in Mice
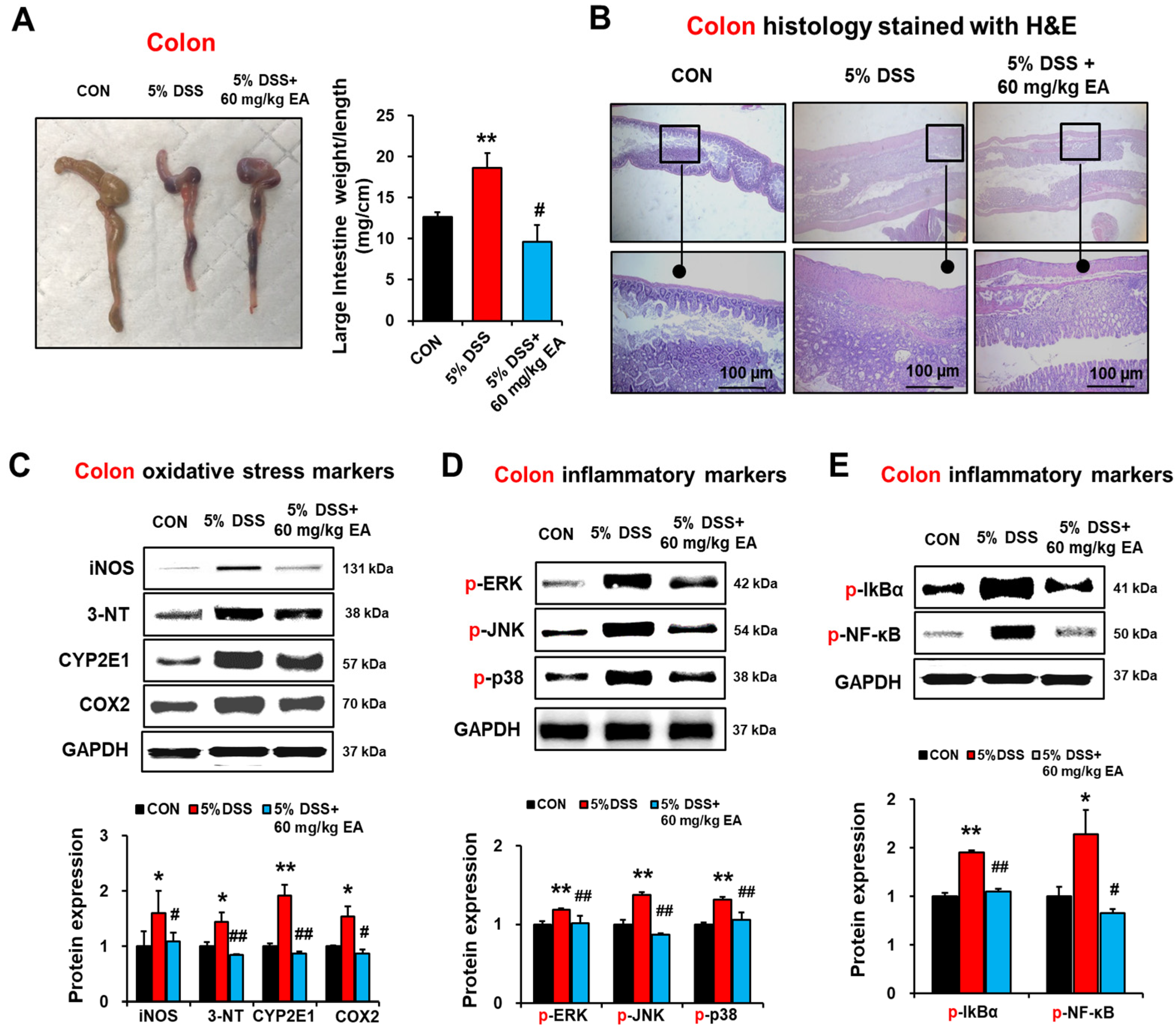
3.3. EA Reduced Oxidative Stress Marker Proteins and p38 MAPK Phosphorylation in the Colon of DSS-Induced Acute Colitis
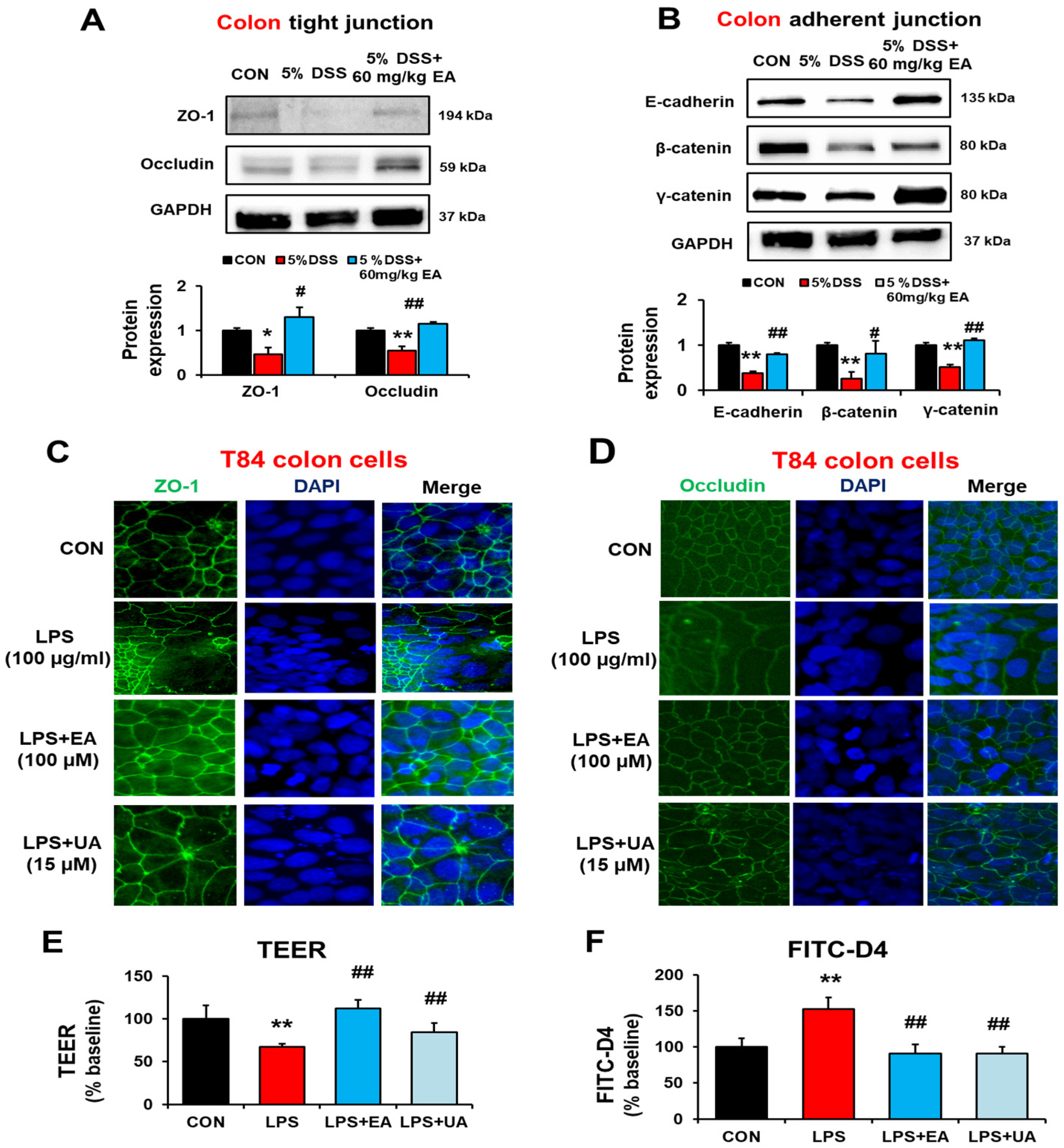
3.4. EA Restored the Gut Tight Junction and Adherent Junction Proteins in DSS-Induced Acute Colitis Mice
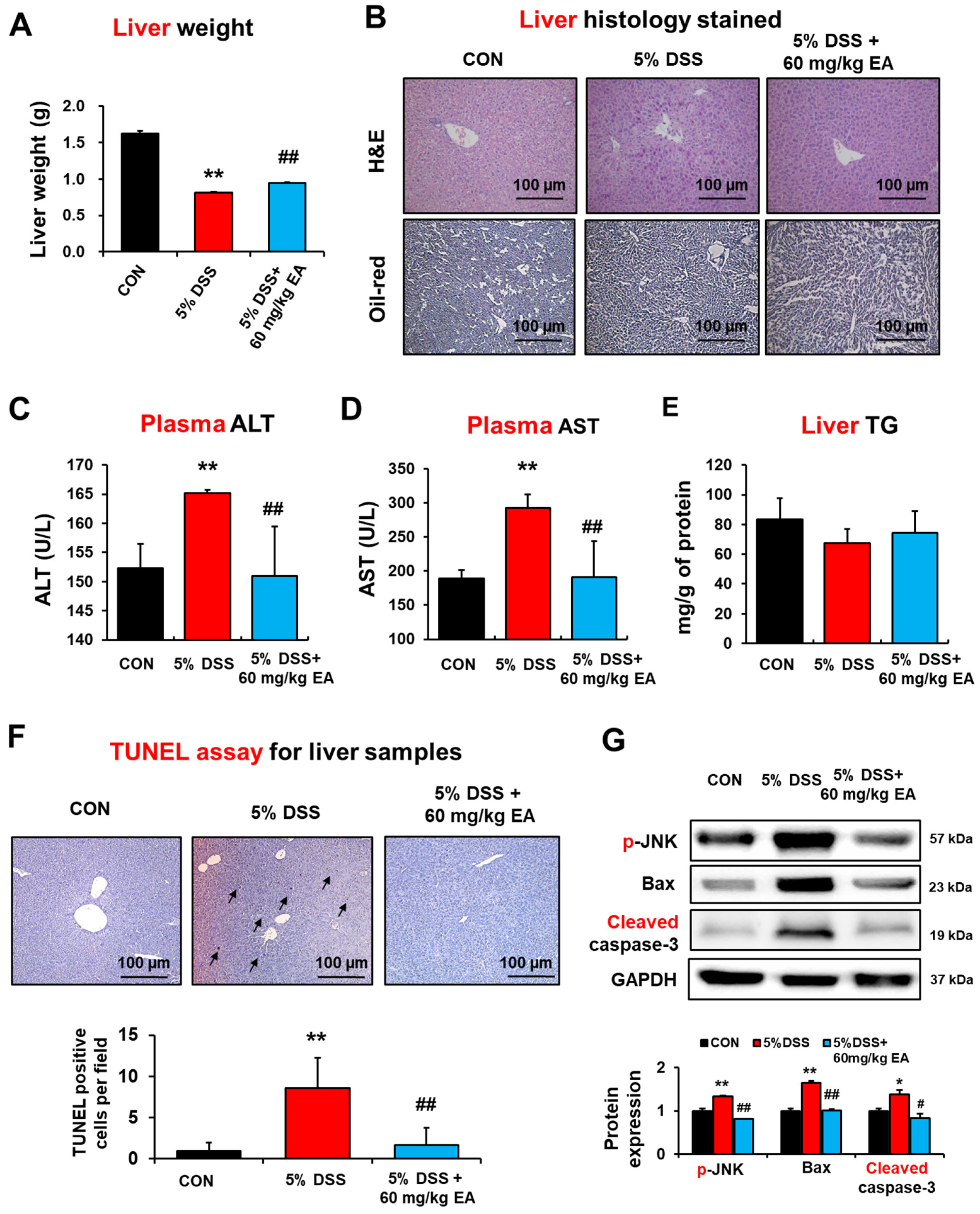
3.5. EA Prevented Hepatic Injury and Apoptosis in DSS-Induced Acute IBD Mice
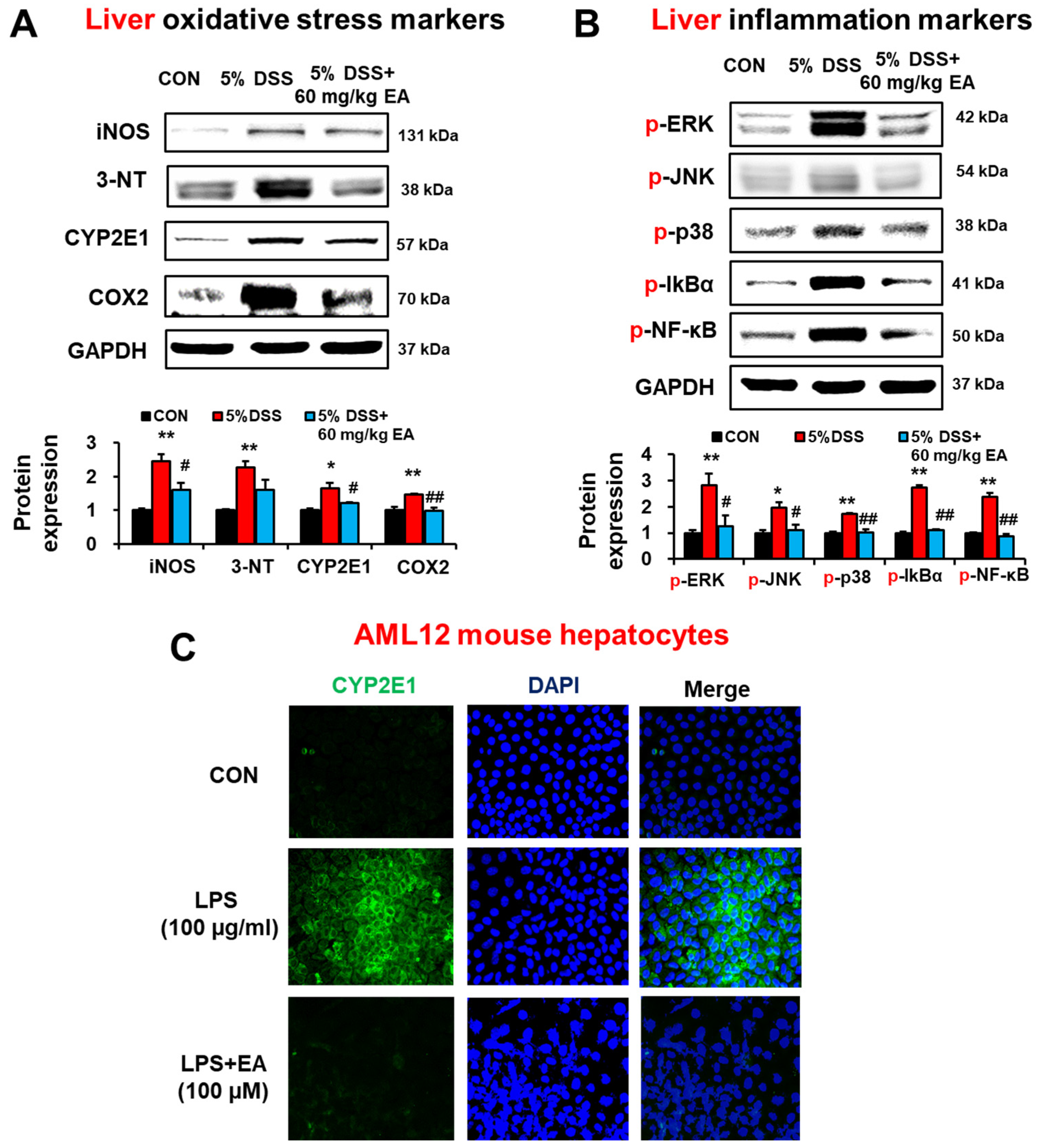
3.6. EA Reduced the Expression of Oxidative/Nitrative Stress Marker Proteins and p38 MAPK Phosphorylation in the Liver of DSS-Induced Acute Colitis Mice
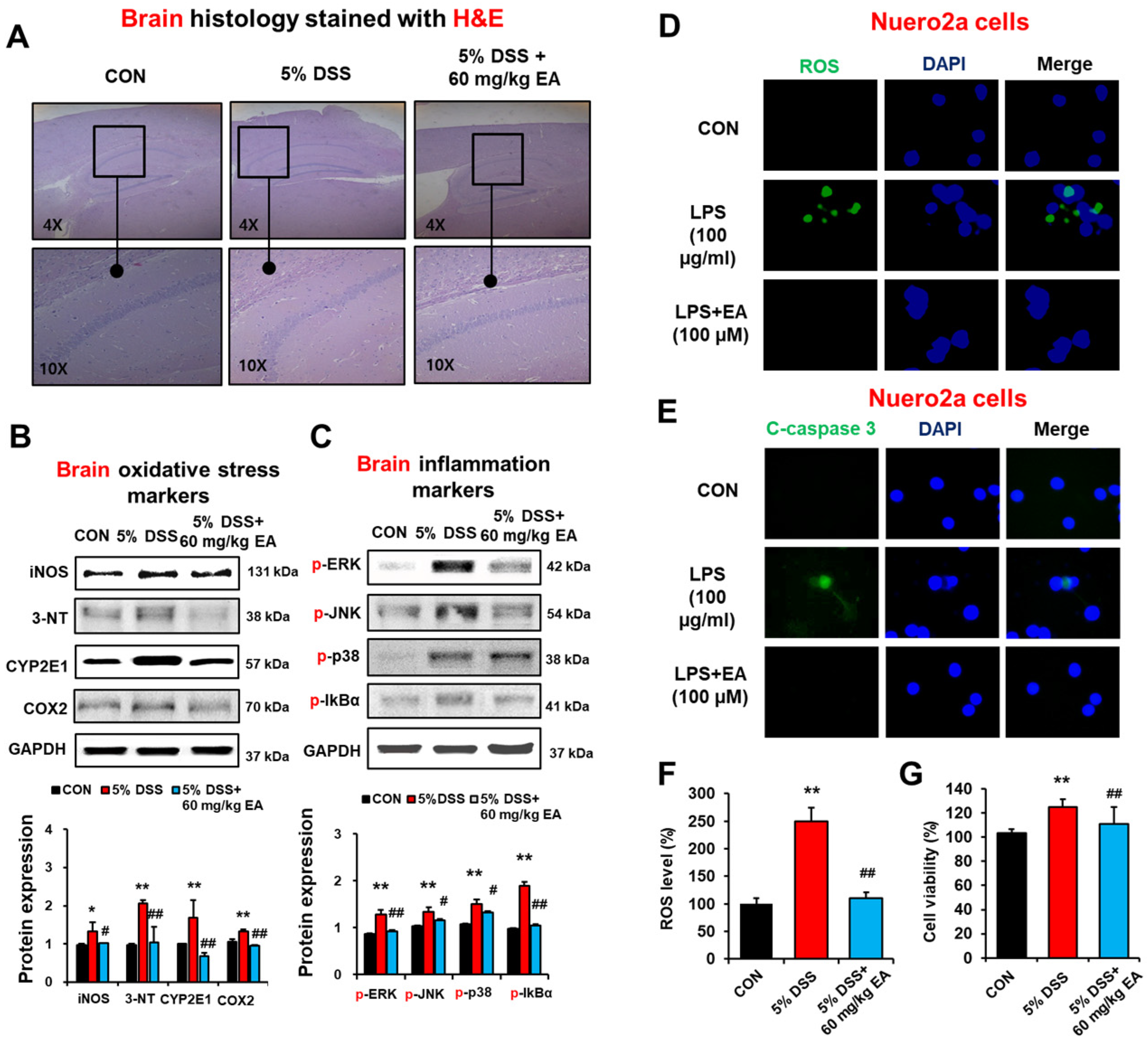
3.7. EA Reduced the Expression of Oxidative Stress Marker Proteins and Phosphorylation of MAPKs in the Brains of DSS-Induced Acute Colitis Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sodagari, H.R.; Aryan, Z.; Abdolghaffari, A.H.; Rezaei, N.; Sahebkar, A. Immunomodulatory and Anti-Inflammatory Phytochemicals for the Treatment of Inflammatory Bowel Disease (IBD)-Turning Strong Rationale into Strong Evidence? J. Pharmacopunct. 2018, 21, 294–295. [Google Scholar] [CrossRef]
- Choi, E.Y.; Cho, K.K.; Choi, I.S. Inflammatory Bowel Disease and Cytokine. J. Life Sci. 2013, 23, 448–461. [Google Scholar] [CrossRef]
- Abiodun, O.O.; Rodriguez-Nogales, A.; Algieri, F.; Gomez-Caravaca, A.M.; Segura-Carretero, A.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Galvez, J. Antiinflammatory and immunomodulatory activity of an ethanolic extract from the stem bark of Terminalia catappa L. (Combretaceae): In vitro and in vivo evidences. J. Ethnopharmacol. 2016, 192, 309–319. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Nho, J.H.; Kang, B.M.; Jung, W.S. Anti-inflammatory effect of the Robinia pseudoacacia L. high temperature extract. Korean J. Plant Resour. 2018, 31, 294–302. [Google Scholar]
- Park, S.B.; Song, H.M.; Kim, H.N.; Park, G.H.; Son, H.-J.; Um, Y.; Park, J.A.; Jeong, J.B. Anti-inflammatory effect of Biji (Soybean curd residue) on LPS-stimulated RAW264. 7 cells. Korean J. Plant Resour. 2018, 31, 117–123. [Google Scholar]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Suzuki, K.; Honma, T. Recent advances in basic and clinical aspects of inflammatory bowel disease: Which steps in the mucosal inflammation should we block for the treatment of inflammatory bowel disease? World J. Gastroenterol. WJG 2007, 13, 2145. [Google Scholar] [CrossRef] [PubMed]
- Vakili, S.T.T.; Taher, M.; Daryani, N.E. Update on the management of ulcerative colitis. Acta Medica Iran. 2012, 363–372. [Google Scholar]
- Rufo, P.A.; Bousvaros, A. Current therapy of inflammatory bowel disease in children. Paediatr. Drugs 2006, 8, 279–302. [Google Scholar] [CrossRef]
- Triantafillidis, J.K.; Merikas, E.; Georgopoulos, F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des. Dev. Ther. 2011, 5, 185–210. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Villegas, I.; La Casa, C.; de la Lastra, C.A. Resveratrol, a polyphenol found in grapes, suppresses oxidative damage and stimulates apoptosis during early colonic inflammation in rats. Biochem. Pharmacol. 2004, 67, 1399–1410. [Google Scholar] [PubMed]
- Martín, A.R.; Villegas, I.; Sánchez-Hidalgo, M.; De La Lastra, C.A. The effects of resveratrol, a phytoalexin derived from red wines, on chronic inflammation induced in an experimentally induced colitis model. Br. J. Pharmacol. 2006, 147, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P.; Sahebkar, A. Ellagic Acid and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 473–479. [Google Scholar] [CrossRef]
- Devipriya, N.; Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Effect of ellagic acid, a natural polyphenol, on alcohol-induced prooxidant and antioxidant imbalance: A drug dose dependent study. Singap. Med. J. 2007, 48, 311–318. [Google Scholar]
- Shukla, M.; Gupta, K.; Rasheed, Z.; Khan, K.A.; Haqqi, T.M. Bioavailable constituents/metabolites of pomegranate (Punica granatum L) preferentially inhibit COX2 activity ex vivo and IL-1beta-induced PGE2 production in human chondrocytes in vitro. J. Inflamm. 2008, 5, 9. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 2006, 71, 1397–1421. [Google Scholar] [CrossRef]
- Tresca, A.J. IBD and Liver Disease. Available online: https://www.verywellhealth.com/cirrhosis-of-the-liver-1941713 (accessed on 22 August 2023).
- Cho, Y.-E.; Lee, M.-H.; Song, B.-J. Neuronal cell death and degeneration through increased nitroxidative stress and tau phosphorylation in HIV-1 transgenic rats. PLoS ONE 2017, 12, e0169945. [Google Scholar] [CrossRef]
- Casella, G.; Tontini, G.E.; Bassotti, G.; Pastorelli, L.; Villanacci, V.; Spina, L.; Baldini, V.; Vecchi, M. Neurological disorders and inflammatory bowel diseases. World J. Gastroenterol. WJG 2014, 20, 8764. [Google Scholar]
- Cho, Y.-E.; Yu, L.-R.; Abdelmegeed, M.A.; Yoo, S.-H.; Song, B.-J. Apoptosis of enterocytes and nitration of junctional complex proteins promote alcohol-induced gut leakiness and liver injury. J. Hepatol. 2018, 69, 142–153. [Google Scholar] [CrossRef]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.H.; Song, B.J. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1–Mediated Oxidative and Nitrative Stress. Hepatology 2021, 73, 2180–2195. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sim, Y.; Hwang, J.H.; Kwun, I.S.; Lim, J.H.; Kim, J.; Kim, J.I.; Baek, M.C.; Akbar, M.; Seo, W.; et al. Ellagic Acid Prevents Binge Alcohol-Induced Leaky Gut and Liver Injury through Inhibiting Gut Dysbiosis and Oxidative Stress. Antioxidants 2021, 10, 1386. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Song, B.J. Pomegranate prevents binge alcohol-induced gut leakiness and hepatic inflammation by suppressing oxidative and nitrative stress. Redox Biol. 2018, 18, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Mohebali, N.; Norhaizan, M.E.; Looi, C.Y. Gallic acid attenuates dextran sulfate sodium-induced experimental colitis in BALB/c mice. Drug Des. Dev. Ther. 2015, 9, 3923. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.S.; Murthy, S.; Shah, R.; Sedergran, D. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. A J. Tech. Methods Pathol. 1993, 69, 238–249. [Google Scholar]
- Cho, Y.E.; Mezey, E.; Hardwick, J.P.; Salem, N., Jr.; Clemens, D.L.; Song, B.J. Increased ethanol-inducible cytochrome P450-2E1 and cytochrome P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism through oxidative and endoplasmic reticulum stress. Hepatol. Commun. 2017, 1, 675–690. [Google Scholar] [CrossRef]
- Panpetch, W.; Hiengrach, P.; Nilgate, S.; Tumwasorn, S.; Somboonna, N.; Wilantho, A.; Chatthanathon, P.; Prueksapanich, P.; Leelahavanichkul, A. Additional Candida albicans administration enhances the severity of dextran sulfate solution induced colitis mouse model through leaky gut-enhanced systemic inflammation and gut-dysbiosis but attenuated by Lactobacillus rhamnosus L34. Gut Microbes 2020, 11, 465–480. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Gao, X.; Nie, L.; Xie, J.; Dai, T.; Shi, C.; Tao, L.; Wang, Y.; Tian, Y.; Sheng, J. Astragalin Attenuates Dextran Sulfate Sodium (DSS)-Induced Acute Experimental Colitis by Alleviating Gut Microbiota Dysbiosis and Inhibiting NF-kappaB Activation in Mice. Front. Immunol. 2020, 11, 2058. [Google Scholar] [CrossRef] [PubMed]
- Poritz, L.S.; Garver, K.I.; Green, C.; Fitzpatrick, L.; Ruggiero, F.; Koltun, W.A. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J. Surg. Res. 2007, 140, 12–19. [Google Scholar] [CrossRef]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Boulete, I.M.; Thadi, A.; Beaufrand, C.; Patwa, V.; Joshi, A.; Foss, J.A.; Eddy, E.P.; Eutamene, H.; Palejwala, V.A.; Theodorou, V.; et al. Oral treatment with plecanatide or dolcanatide attenuates visceral hypersensitivity via activation of guanylate cyclase-C in rat models. World J. Gastroenterol. 2018, 24, 1888–1900. [Google Scholar] [CrossRef] [PubMed]
- Gabele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Scholmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef]
- Farzaei, M.H.; Rahimi, R.; Abdollahi, M. The role of dietary polyphenols in the management of inflammatory bowel disease. Curr. Pharm. Biotechnol. 2015, 16, 196–210. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Nutritional Management of Inflammatory Bowel Diseases: A Comprehensive Guide; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Li, X.; Xu, L.; Peng, X.; Zhang, H.; Kang, M.; Jiang, Y.; Shi, H.; Chen, H.; Zhao, C.; Yu, Y. The alleviating effect of ellagic acid on DSS-induced colitis via regulating gut microbiomes and gene expression of colonic epithelial cells. Food Funct. 2023, 14, 7550–7561. [Google Scholar] [CrossRef]
- Gonzalez-Sarrias, A.; Espin, J.C.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T. Gene expression, cell cycle arrest and MAPK signalling regulation in Caco-2 cells exposed to ellagic acid and its metabolites, urolithins. Mol. Nutr. Food Res. 2009, 53, 686–698. [Google Scholar] [CrossRef]
- Veiga, P.; Gallini, C.A.; Beal, C.; Michaud, M.; Delaney, M.L.; DuBois, A.; Khlebnikov, A.; van Hylckama Vlieg, J.E.; Punit, S.; Glickman, J.N.; et al. Bifidobacterium animalis subsp. lactis fermented milk product reduces inflammation by altering a niche for colitogenic microbes. Proc. Natl. Acad. Sci. USA 2010, 107, 18132–18137. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Interactions of gut microbiota with dietary polyphenols and consequences to human health. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 471–476. [Google Scholar] [CrossRef]
- Ballway, J.W.; Song, B.-J. Translational approaches with antioxidant phytochemicals against alcohol-mediated oxidative stress, gut dysbiosis, intestinal barrier dysfunction, and fatty liver disease. Antioxidants 2021, 10, 384. [Google Scholar] [CrossRef]
- Roberts, B.J.; Shoaf, S.E.; Jeong, K.-S.; Song, B.J. Induction of CYP2E1 in liver, kidney, brain and intestine during chronic ethanol administration and withdrawal: Evidence that CYP2E1 possesses a rapid phase half-life of 6 hours or less. Biochem. Biophys. Res. Commun. 1994, 205, 1064–1071. [Google Scholar] [CrossRef]
- Roberts, B.J.; Song, B.-J.; Soh, Y.; Park, S.S.; Shoaf, S.E. Ethanol Induces CYP2E1 by Protein Stabilization: Role of ubiquitin conjugation in the rapid degradation of cyp2e1 (∗). J. Biol. Chem. 1995, 270, 29632–29635. [Google Scholar] [CrossRef]
- Rosillo, M.A.; Sanchez-Hidalgo, M.; Cardeno, A.; de la Lastra, C.A. Protective effect of ellagic acid, a natural polyphenolic compound, in a murine model of Crohn’s disease. Biochem. Pharmacol. 2011, 82, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Vinayak, M. Ellagic acid inhibits PKC signaling by improving antioxidant defense system in murine T cell lymphoma. Mol. Biol. Rep. 2014, 41, 4187–4197. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.Y.; Mong, M.C.; Chan, K.C.; Yin, M.C. Anti-glycative and anti-inflammatory effects of caffeic acid and ellagic acid in kidney of diabetic mice. Mol. Nutr. Food Res. 2010, 54, 388–395. [Google Scholar] [CrossRef]
- Mo, J.; Panichayupakaranant, P.; Kaewnopparat, N.; Songkro, S.; Reanmongkol, W. Topical anti-inflammatory potential of standardized pomegranate rind extract and ellagic acid in contact dermatitis. Phytother. Res. 2014, 28, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.E.; Bozdag, Z.; Ibiloglu, I.; Arikanoglu, Z.; Yazgan, U.C.; Kaplan, I.; Gumus, M.; Atamanalp, S.S. Therapeutic effects of ellagic acid on L-arginin induced acute pancreatitis. Acta Cir. Bras. 2016, 31, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.; Maria Giner, R.; Rios, J.L.; Recio, M.C. Intestinal anti-inflammatory activity of ellagic acid in the acute and chronic dextrane sulfate sodium models of mice colitis. J. Ethnopharmacol. 2013, 150, 925–934. [Google Scholar] [CrossRef]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Bosca, L.; Zeini, M.; Traves, P.G.; Hortelano, S. Nitric oxide and cell viability in inflammatory cells: A role for NO in macrophage function and fate. Toxicology 2005, 208, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Ahad, A.; Ganai, A.A.; Mujeeb, M.; Siddiqui, W.A. Ellagic acid, an NF-kappaB inhibitor, ameliorates renal function in experimental diabetic nephropathy. Chem. Biol. Interact. 2014, 219, 64–75. [Google Scholar] [CrossRef]
- Li, Z.-R.; Jia, R.-B.; Cai, X.; Luo, D.; Chen, C.; Zhao, M. Characterizations of food-derived ellagic acid-Undaria pinnatifida polysaccharides solid dispersion and its benefits on solubility, dispersity and biotransformation of ellagic acid. Food Chem. 2023, 413, 135530. [Google Scholar] [CrossRef]
- Romier, B.; Van De Walle, J.; During, A.; Larondelle, Y.; Schneider, Y.J. Modulation of signalling nuclear factor-kappaB activation pathway by polyphenols in human intestinal Caco-2 cells. Br. J. Nutr. 2008, 100, 542–551. [Google Scholar] [CrossRef]
- Rosillo, M.A.; Sanchez-Hidalgo, M.; Cardeno, A.; Aparicio-Soto, M.; Sanchez-Fidalgo, S.; Villegas, I.; de la Lastra, C.A. Dietary supplementation of an ellagic acid-enriched pomegranate extract attenuates chronic colonic inflammation in rats. Pharmacol. Res. 2012, 66, 235–242. [Google Scholar] [CrossRef]
- Allahverdi, T.D.; Allahverdi, E.; Yayla, S.; Deprem, T.; Merhan, O.; Vural, S. The comparison of the effects of ellagic acid and diclofenac sodium on intra-abdominal adhesion: An in vivo study in the rat model. Int. Surg. 2014, 99, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.; Fu, Y.; Wei, Z.; Yang, Z. Inhibition of allergic airway inflammation through the blockage of NF-κB activation by ellagic acid in an ovalbumin-induced mouse asthma model. Food Funct. 2014, 5, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi-Niri, S.F.; Maqbool, F.; Baeeri, M.; Gholami, M.; Abdollahi, M. Phosalone-induced inflammation and oxidative stress in the colon: Evaluation and treatment. World J. Gastroenterol. 2016, 22, 4999–5011. [Google Scholar] [CrossRef]
- Yao, H.; Hu, C.; Yin, L.; Tao, X.; Xu, L.; Qi, Y.; Han, X.; Xu, Y.; Zhao, Y.; Wang, C.; et al. Dioscin reduces lipopolysaccharide-induced inflammatory liver injury via regulating TLR4/MyD88 signal pathway. Int. Immunopharmacol. 2016, 36, 132–141. [Google Scholar] [CrossRef]
- Doğanyiğit, Z.; Okan, A.; Kaymak, E.; Pandır, D.; Silici, S. Investigation of protective effects of apilarnil against lipopolysaccharide induced liver injury in rats via TLR 4/HMGB-1/NF-κB pathway. Biomed. Pharmacother. 2020, 125, 109967. [Google Scholar] [CrossRef]
- Lykhmus, O.; Mishra, N.; Koval, L.; Kalashnyk, O.; Gergalova, G.; Uspenska, K.; Komisarenko, S.; Soreq, H.; Skok, M. Molecular mechanisms regulating LPS-induced inflammation in the brain. Front. Mol. Neurosci. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective effect of quercetin against the detrimental effects of LPS in the adult mouse brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-h.; Kim, J.-S.; Kwon, J.-H.; Kwun, I.-S.; Baek, M.-C.; Kwon, G.-S.; Rungratanawanich, W.; Song, B.-J.; Kim, D.-K.; Kwon, H.-J.; et al. Ellagic Acid Prevented Dextran-Sodium-Sulfate-Induced Colitis, Liver, and Brain Injury through Gut Microbiome Changes. Antioxidants 2023, 12, 1886. https://doi.org/10.3390/antiox12101886
Kim D-h, Kim J-S, Kwon J-H, Kwun I-S, Baek M-C, Kwon G-S, Rungratanawanich W, Song B-J, Kim D-K, Kwon H-J, et al. Ellagic Acid Prevented Dextran-Sodium-Sulfate-Induced Colitis, Liver, and Brain Injury through Gut Microbiome Changes. Antioxidants. 2023; 12(10):1886. https://doi.org/10.3390/antiox12101886
Chicago/Turabian StyleKim, Dong-ha, Ji-Su Kim, Jae-Hee Kwon, In-Sook Kwun, Moon-Chang Baek, Gi-Seok Kwon, Wiramon Rungratanawanich, Byoung-Joon Song, Do-Kyun Kim, Hyo-Jung Kwon, and et al. 2023. "Ellagic Acid Prevented Dextran-Sodium-Sulfate-Induced Colitis, Liver, and Brain Injury through Gut Microbiome Changes" Antioxidants 12, no. 10: 1886. https://doi.org/10.3390/antiox12101886