
NAC Pre-Administration Prevents Cardiac Mitochondrial Bioenergetics, Dynamics, Biogenesis, and Redox Alteration in Folic Acid-AKI-Induced Cardio-Renal Syndrome Type 3
 ,
,  , , ,
, , ,  , and
, and
Abstract
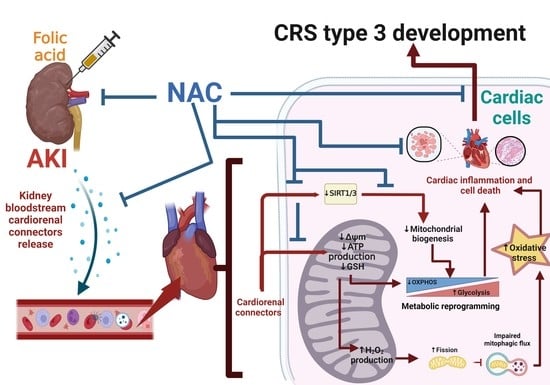
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Experimental Design
2.3. Renal, Liver, and Heart Damage Markers
2.4. Kidney and Heart Histology and Immunohistochemistry
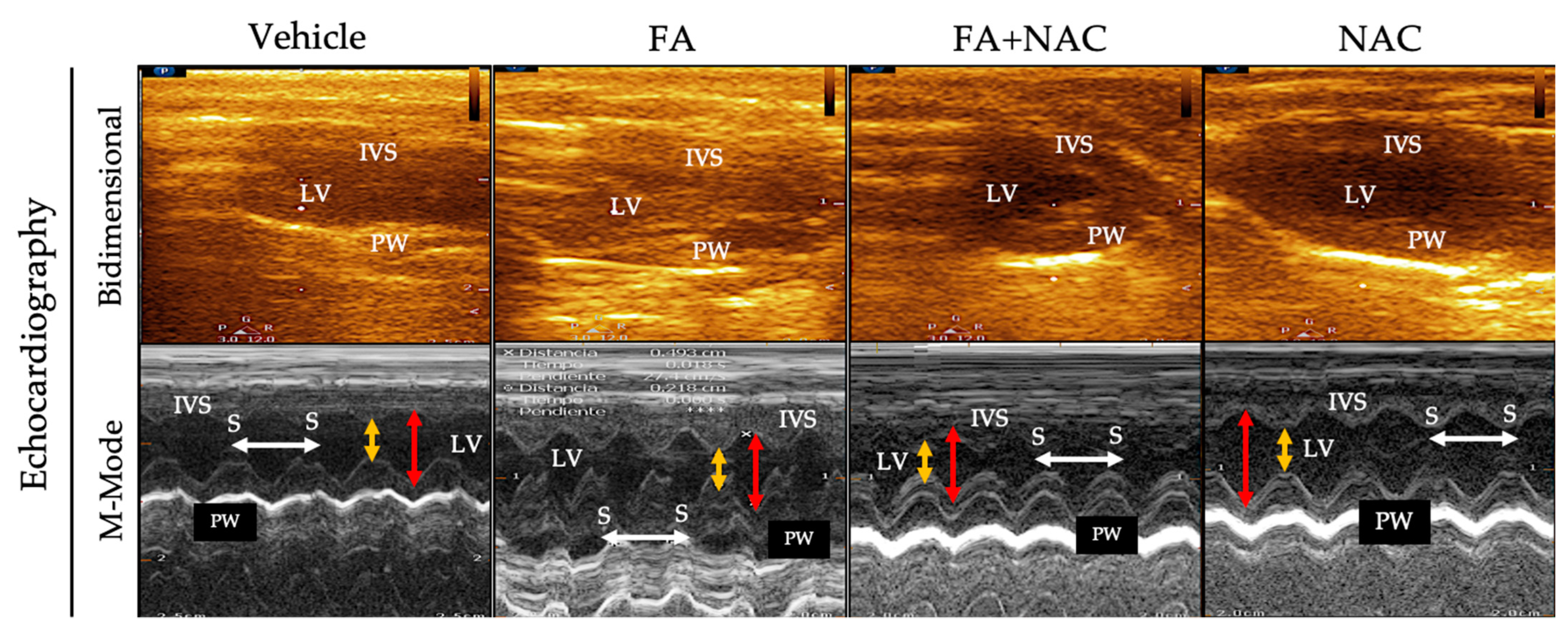
2.5. Evaluation of Cardiac Function by Echocardiography
2.6. Protein Extraction and WB
2.7. Isolation of Heart Mitochondria
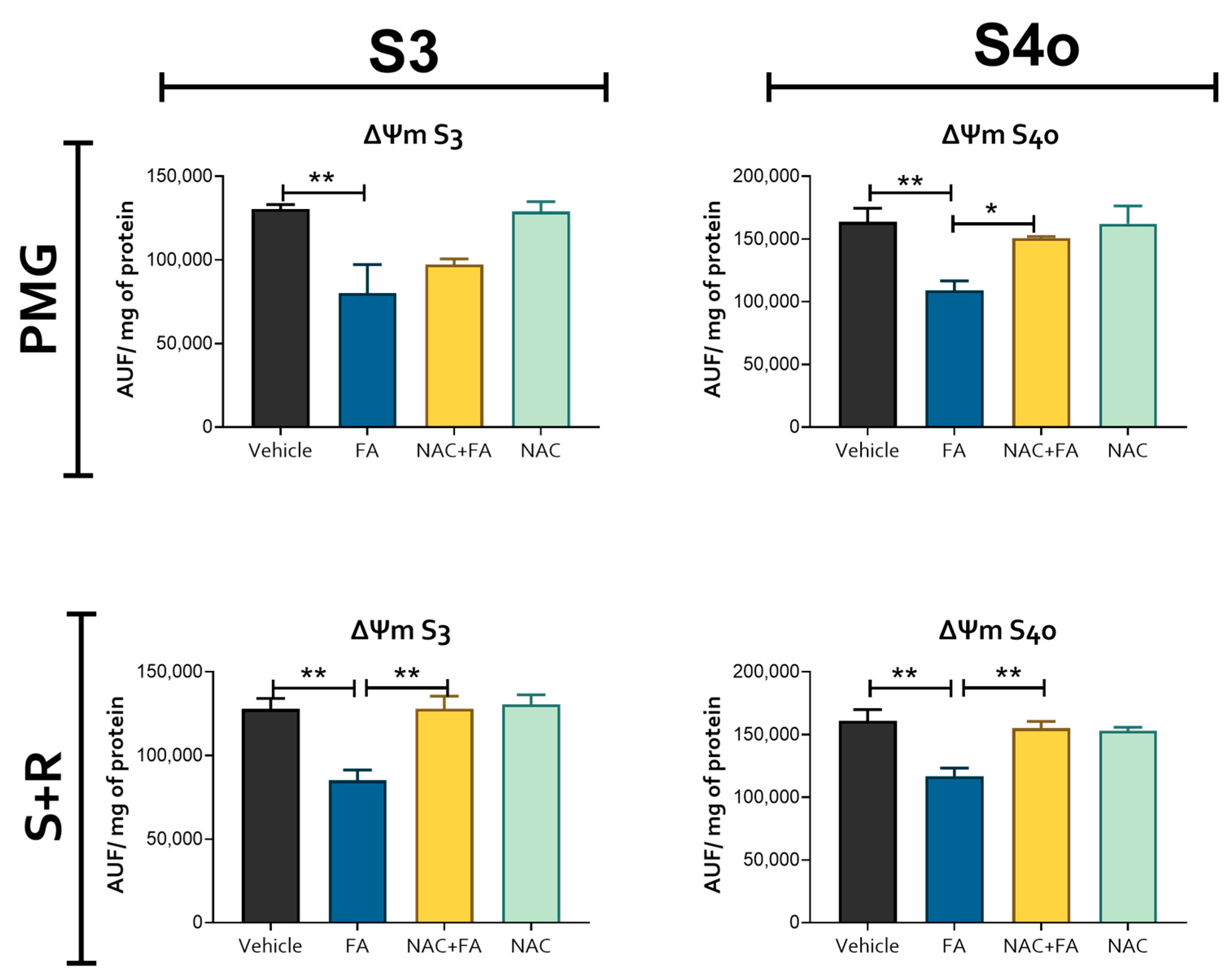
2.8. Mitochondrial Membrane Potential (ΔΨm)
2.9. The Activity of Mitochondrial Respiratory Complexes
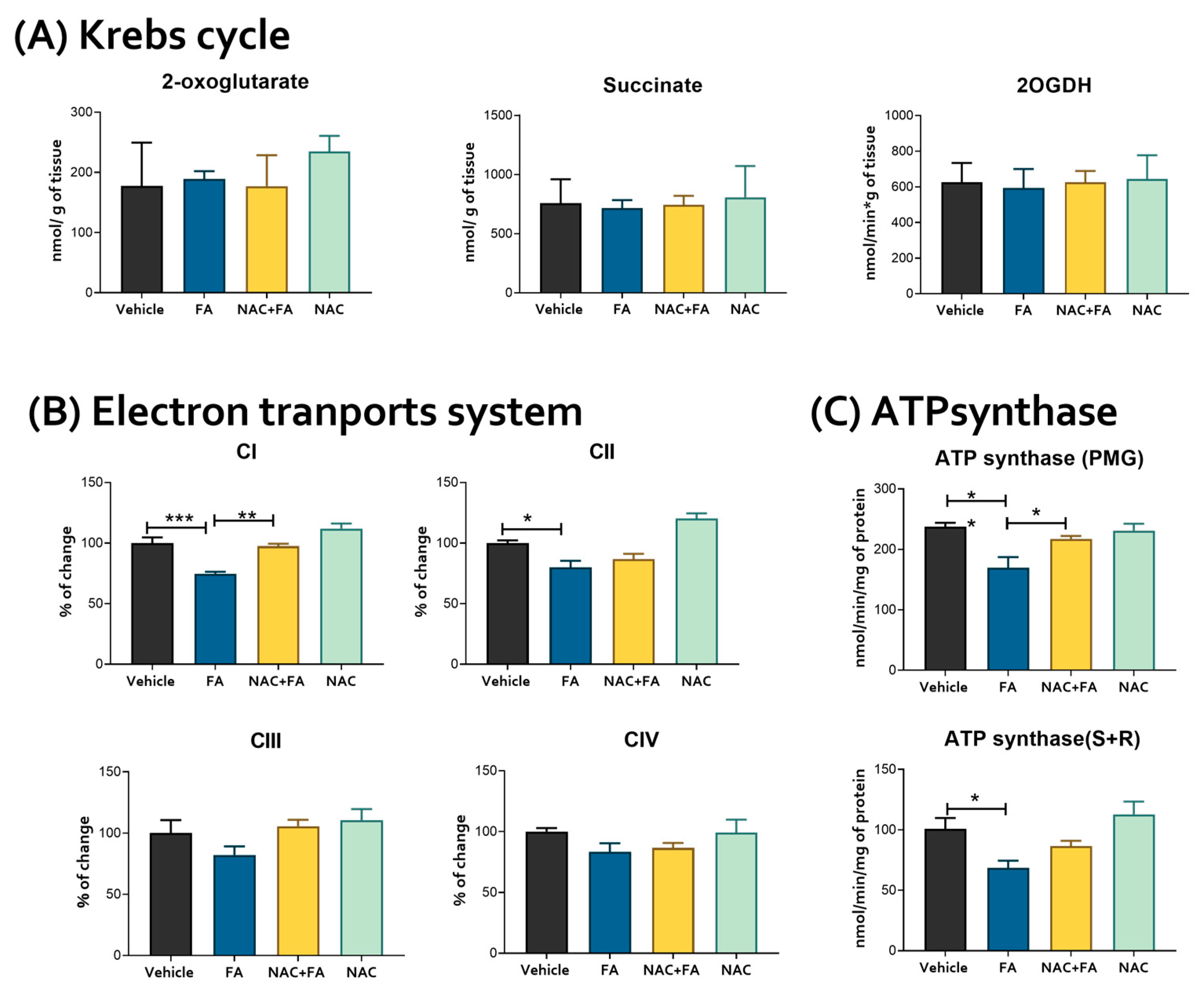
2.10. Krebs Cycle Metabolites and Enzyme Activity
2.11. Mitochondrial H2O2 Production and 4HNE Lipoperoxidation Levels
2.12. Activity of Antioxidant Enzymes
2.13. Mitochondrial Glutathione, GRX Activity, Mitochondrial Protein S-Glutathionylation Levels, and Cardiolipin Levels
2.14. Electron Microscopy
2.15. Statistics
3. Results
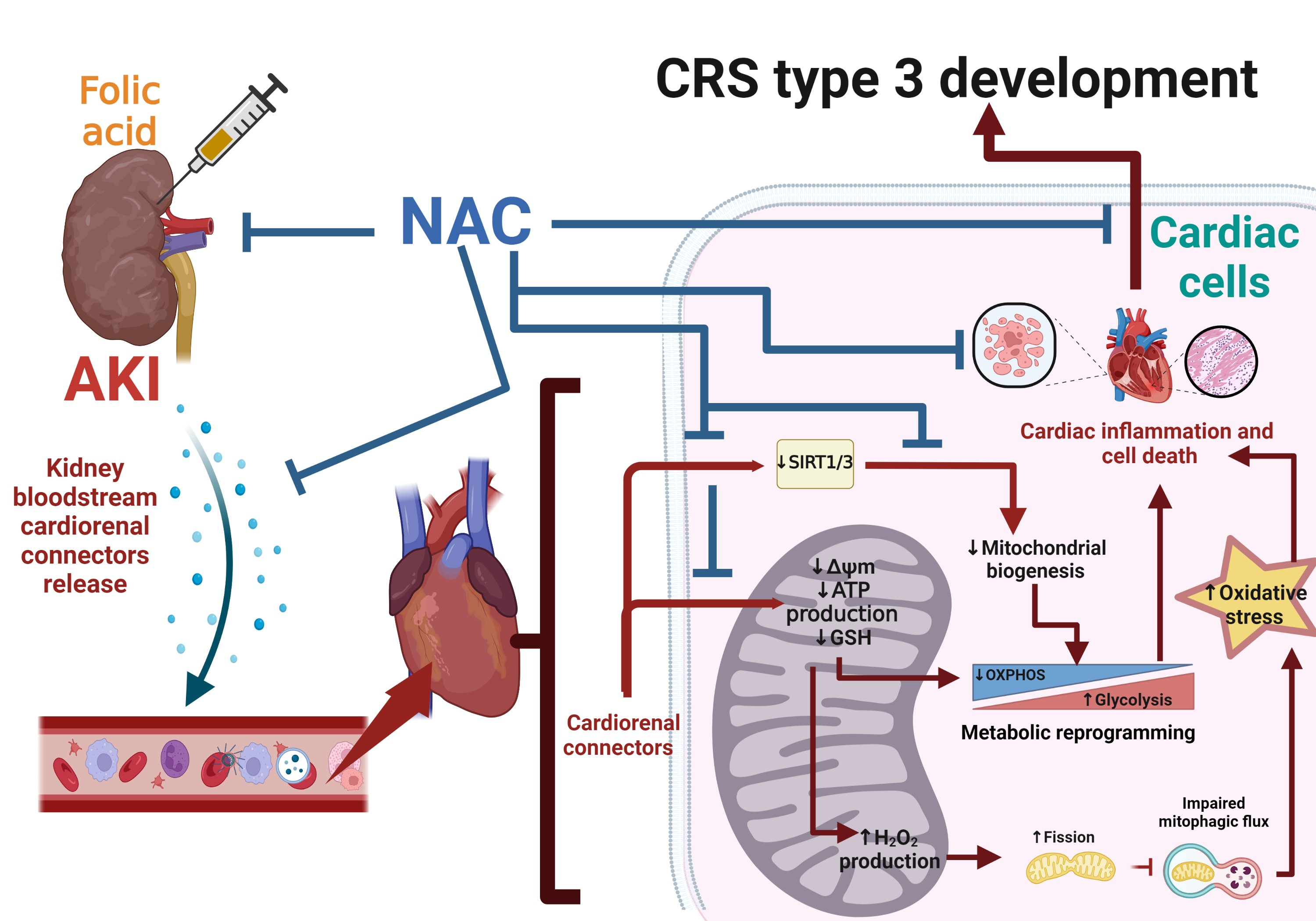
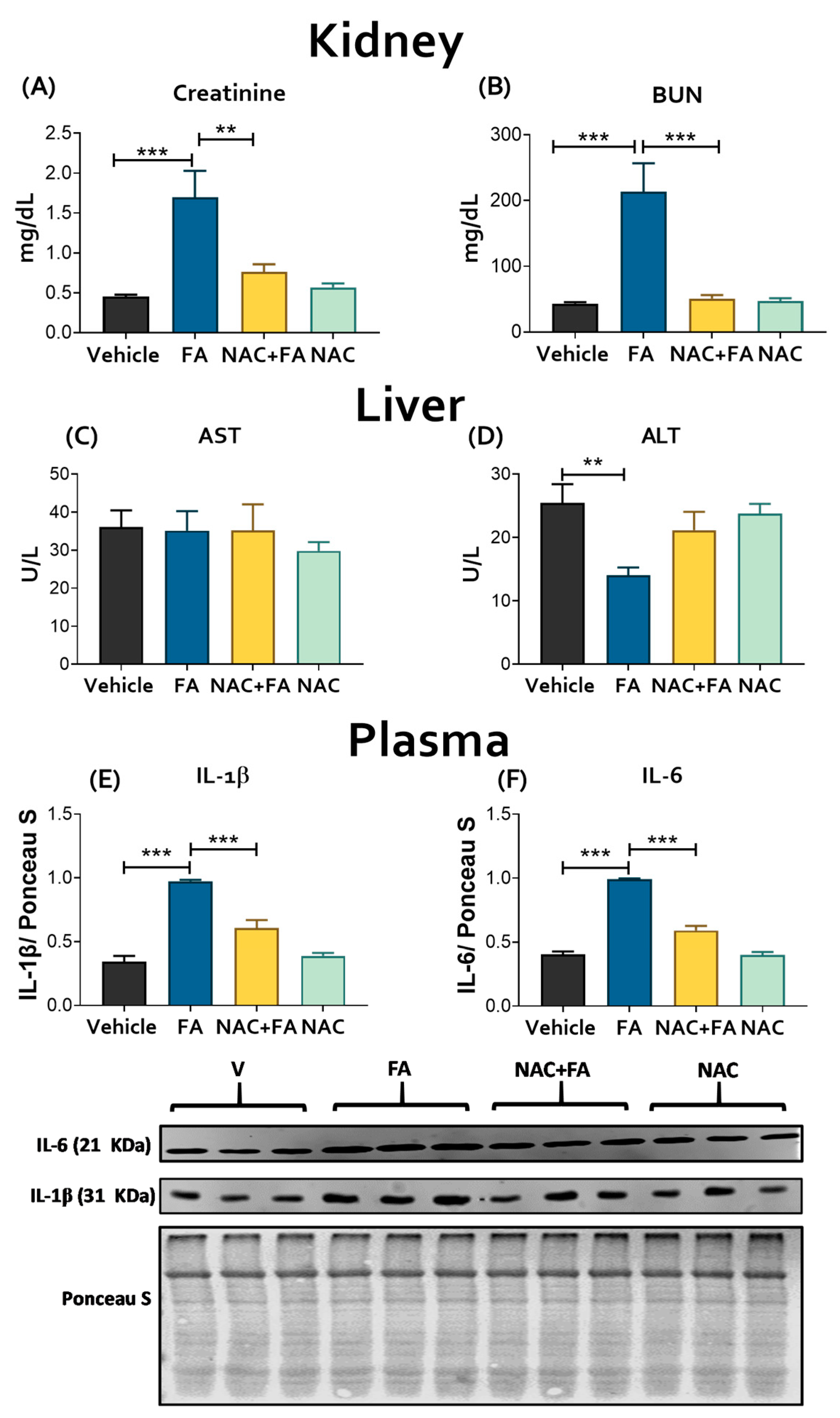
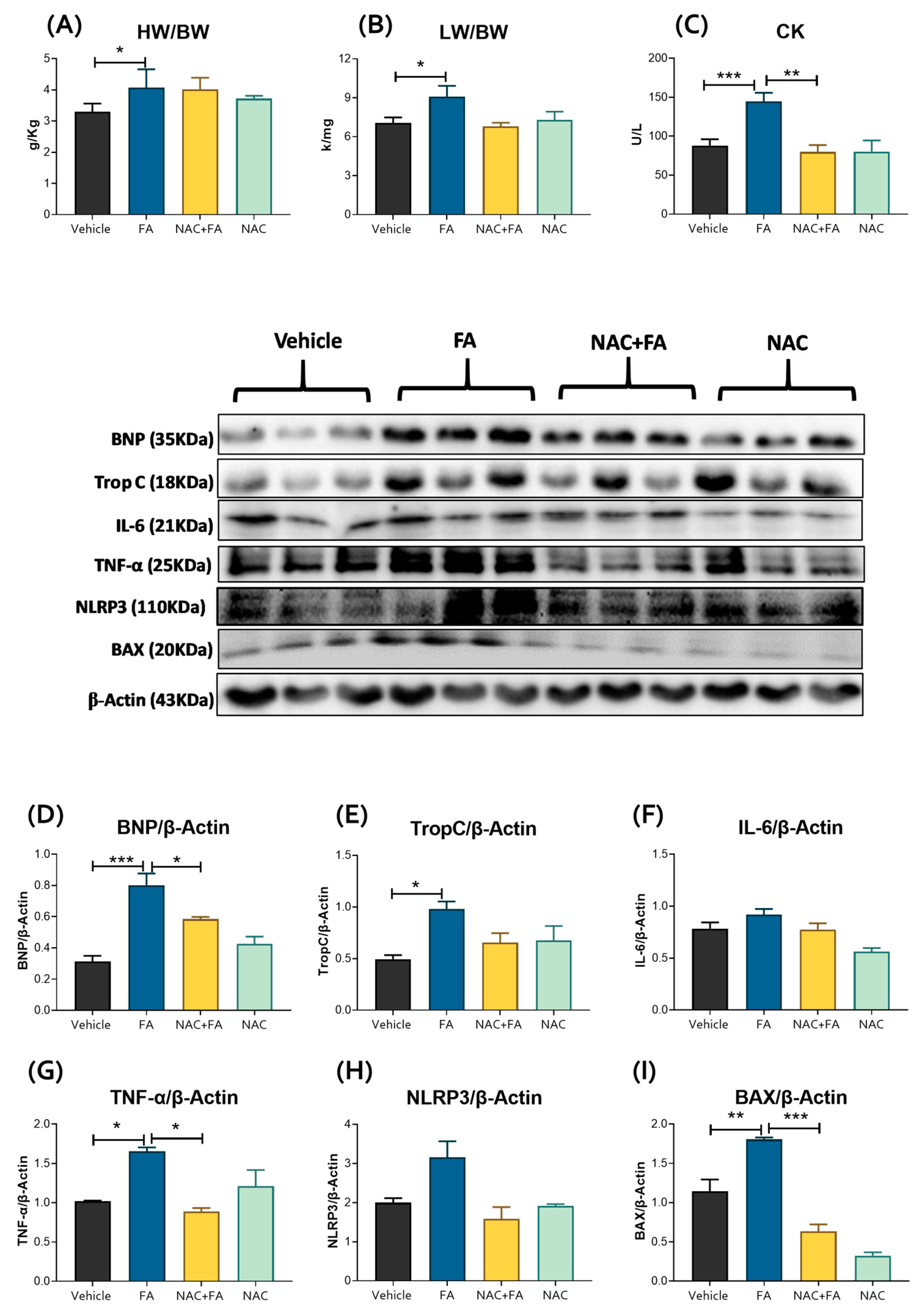
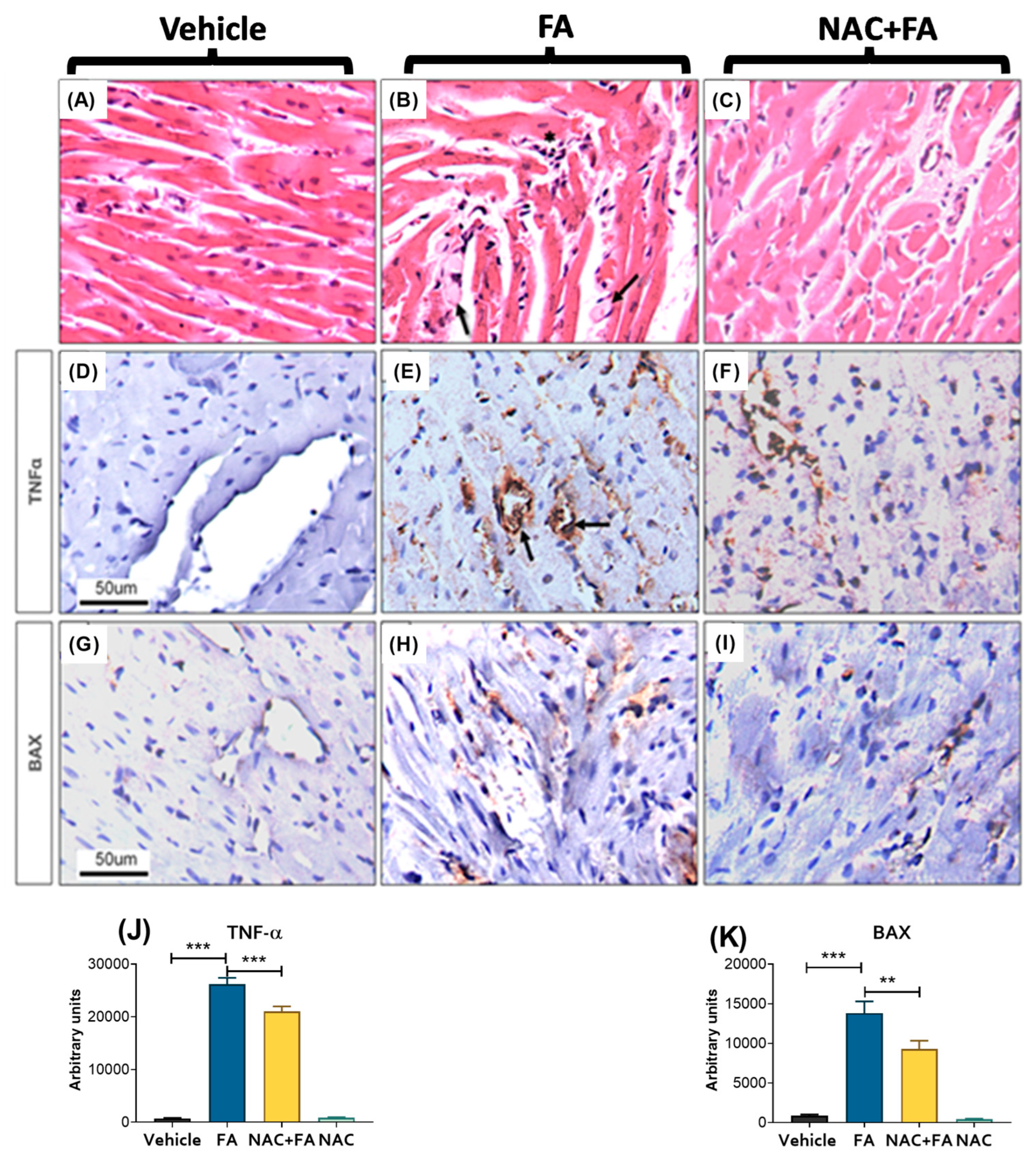
3.1. NAC Prevented CRS Type III Triggers by FA-Induced AKI
3.2. CRS Triggered by FA-Induced AKI Is Related to Mitochondrial Bioenergetics Alterations in the Heart
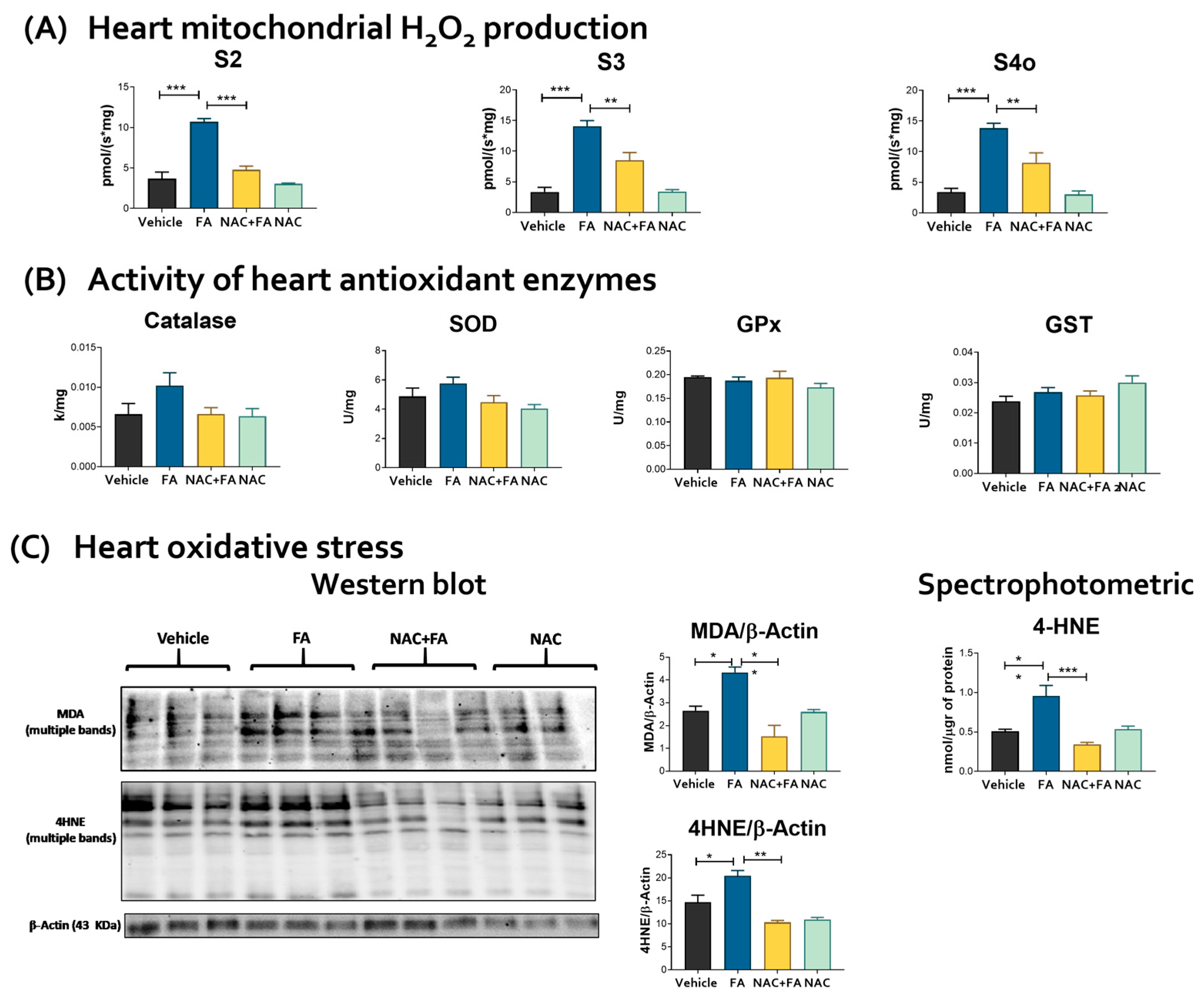
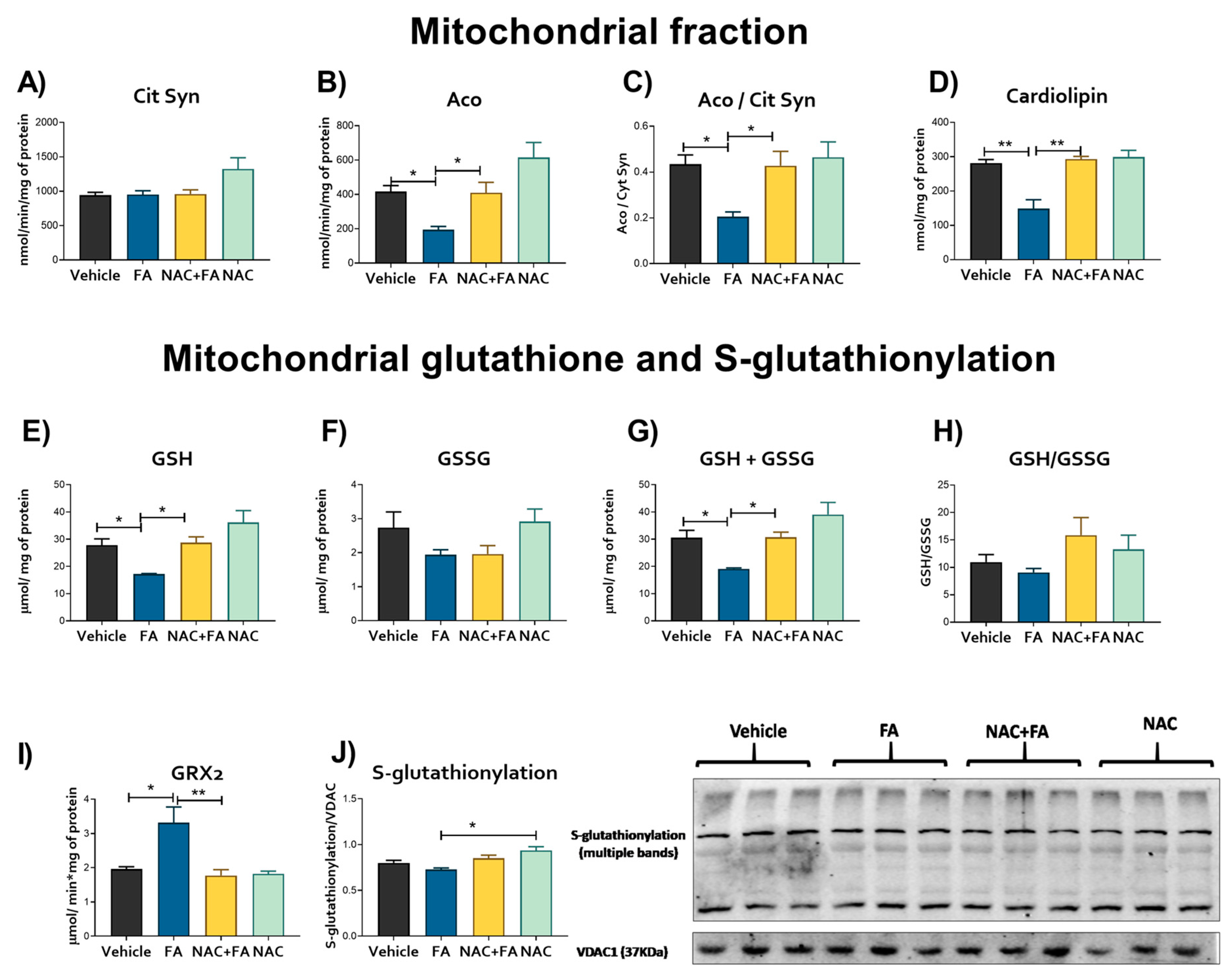
3.3. CRS Triggered by FA-AKI Is Related to Mitochondrial Oxidative Stress and GSH Reduction in the Heart
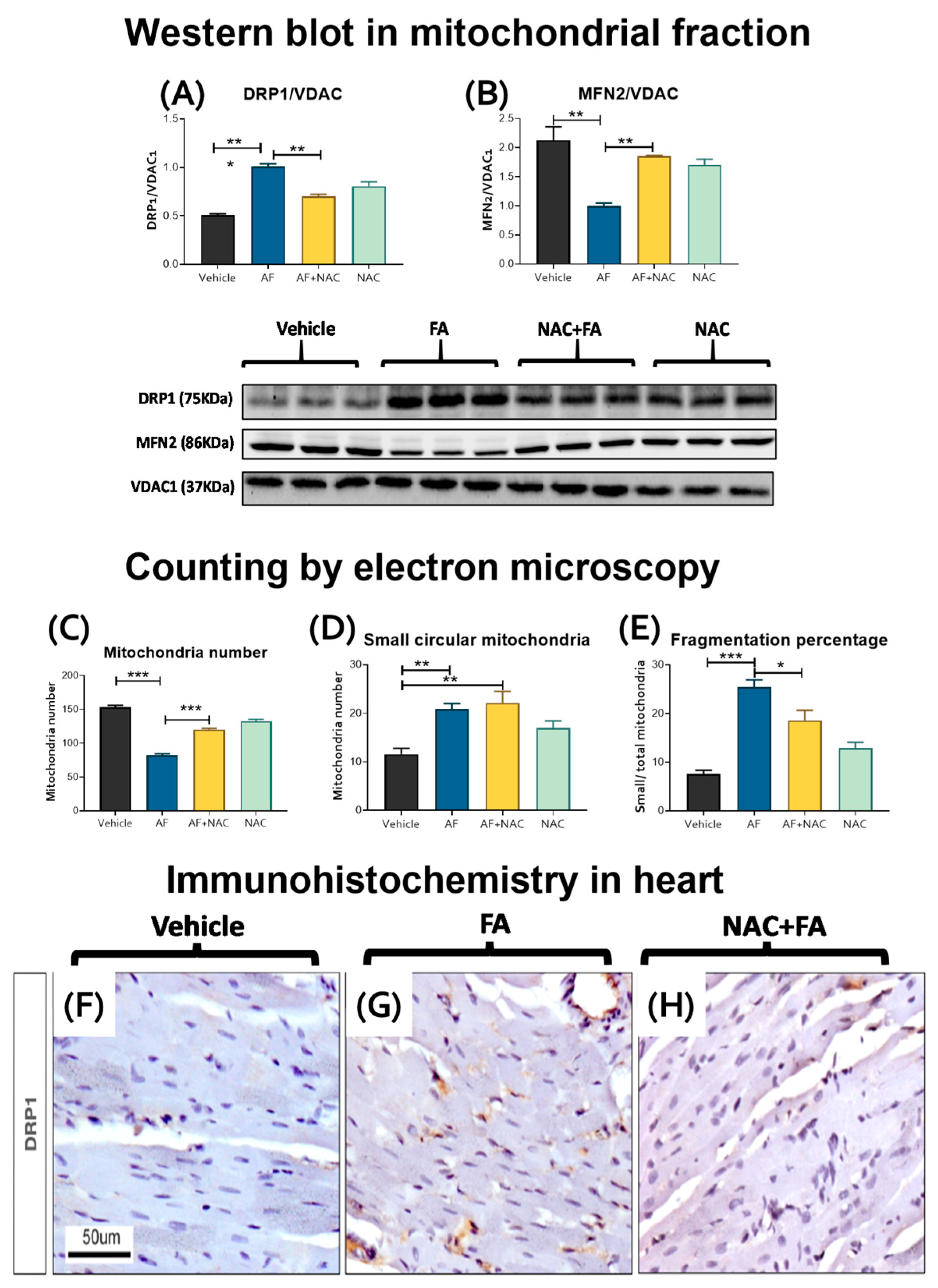
3.4. CRS Triggers by FA-AKI Induce Mitochondrial Fission in the Heart
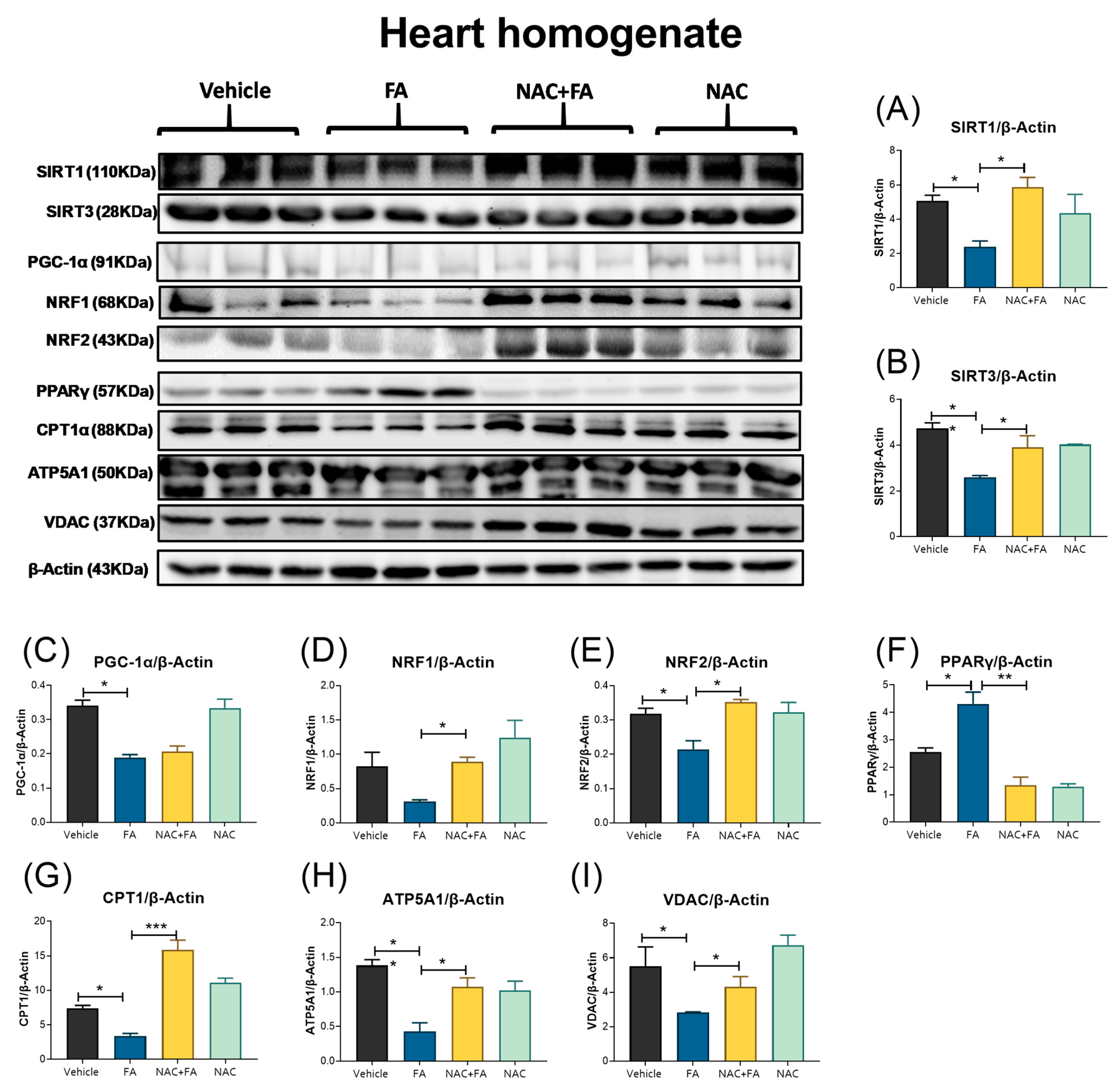
3.5. NAC Prevented Mitochondrial Biogenesis Reduction in the Heart Induced by FA-AKI
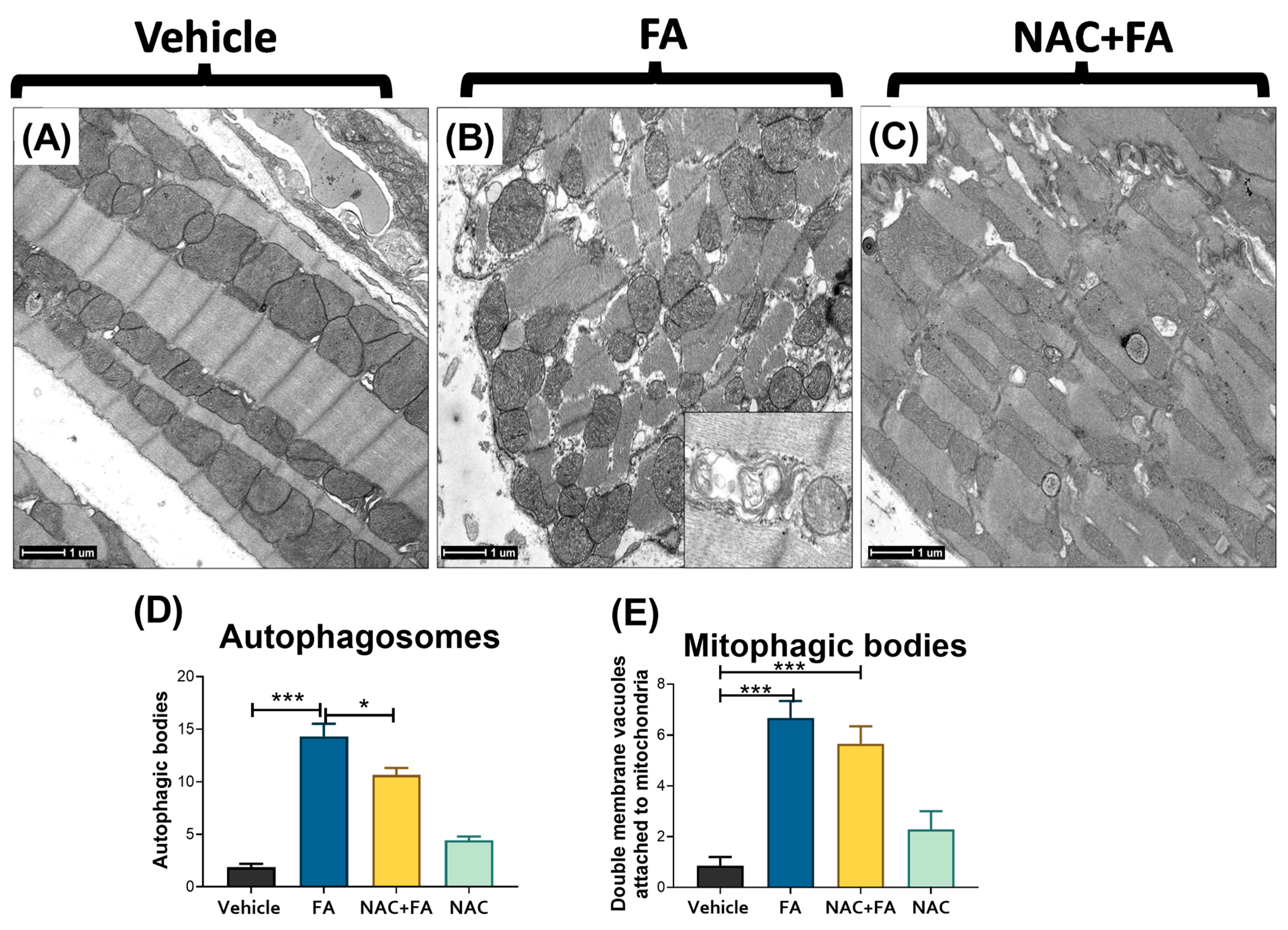
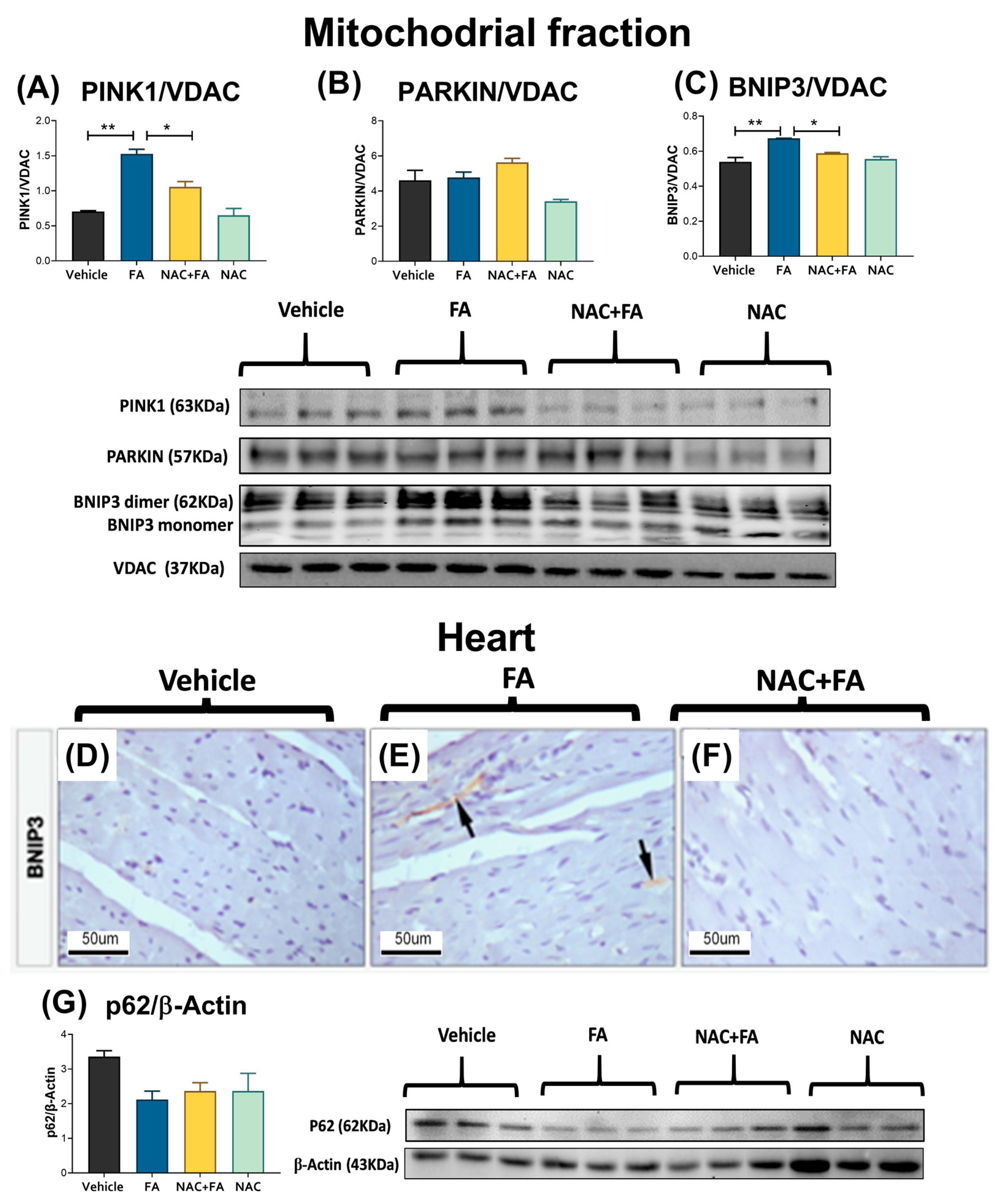
3.6. NAC Prevented Mitophagy Induction in Heart Triggers by FA-AKI
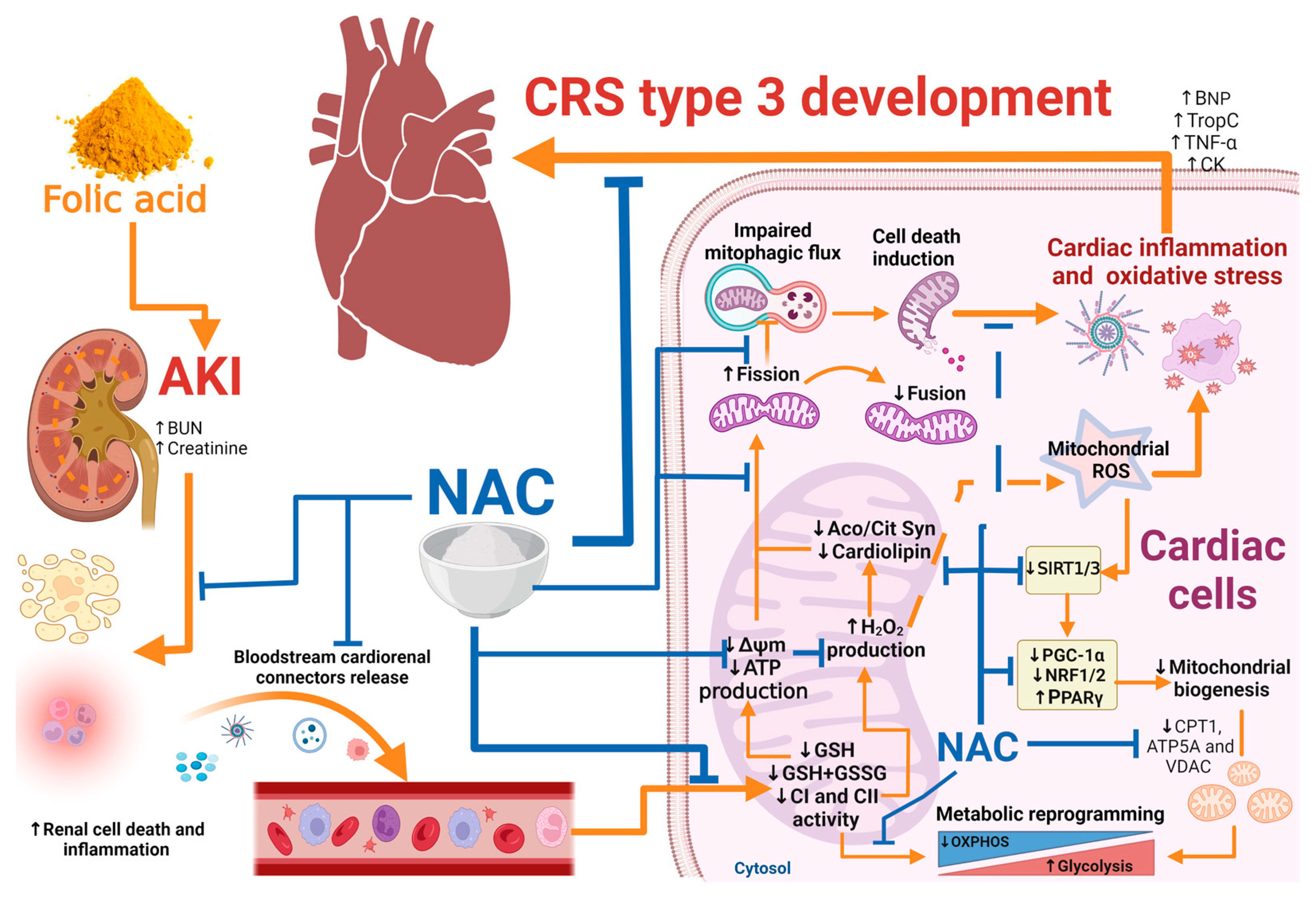
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global Epidemiology and Outcomes of Acute Kidney Injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute Kidney Injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute Kidney Injury. Nat. Rev. Dis. Prim. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Fogo, A.B.; Cohen, A.H.; Colvin, R.B.; Jennette, J.C.; Alpers, C.E. Fundamentals of Renal Pathology, 1st ed.; Springer: Berlin/Heidelberg, German, 2014; ISBN 9783642390807. [Google Scholar]
- Shafi, T.; Coresh, J. Chronic Kidney Disease: Definition, Epidemiology, Cost, and Outcomes. In Chronic Kidney Disease, Dialysis, and Transplantation; Himmelfarb, J., Sayegh, M.H., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 3–21. ISBN 9781437709872. [Google Scholar]
- Li, P.K.T.; Burdmann, E.A.; Mehta, R.L. Acute Kidney Injury: Global Health Alert. Hong Kong J. Nephrol. 2013, 15, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, M.; Doi, K.; Ogasawara, E.; Yamashita, T.; Hamasaki, Y.; Kariya, T.; Takimoto, E.; Yahagi, N.; Nangaku, M.; Noiri, E. Regulation of Mitochondrial Dynamics by Dynamin- Related Protein-1 in Acute Cardiorenal Syndrome. J. Am. Soc. Nephrol. 2015, 26, 2378–2387. [Google Scholar] [CrossRef] [Green Version]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, E840–E878. [Google Scholar] [CrossRef]
- Doi, K.; Noiri, E. Mitochondrial Dysfunction in Cardiorenal Syndrome. Antioxid. Redox Signal. 2016, 25, 200–207. [Google Scholar] [CrossRef]
- Chuasuwan, A.; Kellum, J.A. Cardio-Renal Syndrome Type 3: Epidemiology, Pathophysiology, and Treatment. Semin. Nephrol. 2012, 32, 31–39. [Google Scholar] [CrossRef]
- Kingma, J.G.; Simard, D.; Rouleau, J.R.; Drolet, B.; Simard, C. The Physiopathology of Cardiorenal Syndrome: A Review of the Potential Contributions of Inflammation. J. Cardiovasc. Dev. Dis. 2017, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, X.; Wang, X.; Cui, S.; Liu, R.; Liu, J.; Fu, B.; Gong, M.; Wang, C.; Shi, Y.; et al. Grb2 Induces Cardiorenal Syndrome Type 3: Roles of IL-6, Cardiomyocyte Bioenergetics, and Akt/MTOR Pathway. Front. Cell Dev. Biol. 2021, 9, 630412. [Google Scholar] [CrossRef]
- Bigelman, E.; Cohen, L.; Aharon-Hananel, G.; Levy, R.; Rozenbaum, Z.; Saada, A.; Keren, G.; Entin-Meer, M. Pathological Presentation of Cardiac Mitochondria in a Rat Model for Chronic Kidney Disease. PLoS ONE 2018, 13, e0198196. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1 β and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Fermín, L.M.; Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Tapia, E.; Rivero, I.; Pedraza-Chaverri, J. The Protective Effect of Alpha-Mangostin against Cisplatin-Induced Cell Death in LLC-PK1 Cells Is Associated to Mitochondrial Function Preservation. Antioxidants 2019, 8, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila-Rojas, S.H.; Tapia, E.; Briones-Herrera, A.; Aparicio-Trejo, O.E.; León-Contreras, J.C.; Hernández-Pando, R.; Pedraza-Chaverri, J. Curcumin Prevents Potassium Dichromate (K2Cr2O7)-Induced Renal Hypoxia. Food Chem. Toxicol. 2018, 121, 472–482. [Google Scholar] [CrossRef]
- Molina-Jijón, E.; Aparicio-Trejo, O.E.; Rodríguez-Muñoz, R.; León-Contreras, J.C.; del Carmen Cárdenas-Aguayo, M.; Medina-Campos, O.N.; Tapia, E.; Sánchez-Lozada, L.G.; Hernández-Pando, R.; Reyes, J.L.; et al. The Nephroprotection Exerted by Curcumin in Maleate-Induced Renal Damage Is Associated with Decreased Mitochondrial Fission and Autophagy. BioFactors 2016, 42, 686–702. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial Bioenergetics, Redox State, Dynamics and Turnover Alterations in Renal Mass Reduction Models of Chronic Kidney Diseases and Their Possible Implications in the Progression of This Illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Aranda-Rivera, A.K.; Osorio-Alonso, H.; Martínez-Klimova, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Extracellular Vesicles in Redox Signaling and Metabolic Regulation in Chronic Kidney Disease. Antioxidants 2022, 11, 356. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez- Lozada, L.G.; Pedraza-Chaverri, J. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Gómez-Sierra, T.; Jiménez-Uribe, A.P.; Bellido, B.; Pedraza-Chaverri, J. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in the Promotion of Fibrosis in Obstructive Nephropathy Induced by Unilateral Ureteral Obstruction. BioFactors 2020, 46, 716–733. [Google Scholar] [CrossRef]
- Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Mitochondrial Dysfunction and Inflammaging in Heart Failure: Novel Roles of CYP-Derived Epoxylipids. Cells 2020, 9, 1565. [Google Scholar] [CrossRef] [PubMed]
- Doi, K. Kidney-Heart Interactions in Acute Kidney Injury. Nephron 2016, 134, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Troy, J.L.; Daugharty, T. Dynamics of Glomerular Ultrafiltration in the Rat. J. Clin. Investig. 1971, 50, 1776–1780. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Sowers, J.R. Estrogen and Mitochondria Function in Cardiorenal Metabolic Syndrome, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 127, ISBN 9780123946256. [Google Scholar]
- Correa, F.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; García-Niño, W.R.; Roldán, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Curcumin Maintains Cardiac and Mitochondrial Function in Chronic Kidney Disease. Free Radic. Biol. Med. 2013, 61, 119–129. [Google Scholar] [CrossRef]
- Tamaki, M.; Miyashita, K.; Wakino, S.; Mitsuishi, M.; Hayashi, K.; Itoh, H. Chronic Kidney Disease Reduces Muscle Mitochondria and Exercise Endurance and Its Exacerbation by Dietary Protein through Inactivation of Pyruvate Dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.; Sanchez-Niño, M.D.; Izquierdo, M.C.; Martin-Cleary, C.; Garcia-Bermejo, L.; Moreno, J.A.; Ruiz-Ortega, M.; Draibe, J.; Cruzado, J.M.; Garcia-Gonzalez, M.A.; et al. Translational Value of Animal Models of Kidney Failure. Eur. J. Pharmacol. 2015, 759, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Chunsun, D.; Lawrence, P.K.; Youhua, L. Animal Models of Kidney Diseases. In Sourcebook of Models for Biomedical Research; Humana Press: Totowa, NJ, USA, 2008; pp. 657–664. [Google Scholar]
- Szczypka, M.S.; Westover, A.J.; Clouthier, S.G.; Ferrara, J.L.M.; Humes, H.D. Rare Incorporation of Bone Marrow-Derived Cells into Kidney after Folic Acid-Induced Injury. Stem Cells 2005, 23, 44–54. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, N.; Bayliss, G.; Zhuang, S. EGFR Activity Is Required for Renal Tubular Cell Dedifferentiation and Proliferation in a Murine Model of Folic Acid-Induced Acute Kidney Injury. AJP Ren. Physiol. 2013, 304, F356–F366. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zou, Y.; Fu, Y.; Xing, J.; Wang, K.; Wan, P.; Wang, M. Ibudilast Attenuates Folic Acid—Induced Acute Kidney Injury by Blocking Pyroptosis Through TLR4-Mediated NF- κ B and MAPK Signaling Pathways. Front. Pharmacol. 2021, 12, 650283. [Google Scholar] [CrossRef]
- Durlacher-betzer, K.; Hassan, A.; Levi, R.; Axelrod, J.; Silver, J.; Naveh-many, T. Interleukin-6 Contributes to the Increase in Fibroblast Growth Factor 23 Expression in Acute And Chronic Kidney Disease. Kidney Int. 2018, 94, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid–Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, T.; Petrovic, D.; Matic, S.; Turnic, T.N.; Jeremic, J.; Radonjic, K.; Srejovic, I.; Zivkovic, V.; Bolevich, S.; Bolevich, S.; et al. The Influence of Folic Acid-Induced Acute Kidney Injury on Cardiac Function and Redox Status in Rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sud, A.; Kaur, T.; Tandon, C.; Singla, S.K. N-Acetylcysteine with Apocynin Prevents Hyperoxaluria-Induced Mitochondrial Protein Perturbations in Nephrolithiasis. Free Radic. Res. 2016, 50, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Amini, A.; Masoumi-Moghaddam, S.; Morris, D.L. Utility of Bromelain and N-Acetylcysteine in Treatment of Peritoneal Dissemination of Gastrointestinal Mucin-Producing Malignancies, 1st ed.; Springer International Publishing: Cham, Switzerland, 2016; ISBN 9783319285702. [Google Scholar]
- Tamma, G.; Valenti, G. Evaluating the Oxidative Stress in Renal Diseases: What Is the Role for S-Glutathionylation? Antioxid. Redox Signal. 2016, 25, 117–118. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Treberg, J.R. Protein S-Glutathionlyation Links Energy Metabolism to Redox Signaling in Mitochondria. Redox Biol. 2016, 8, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.L.; Zhang, Y.; Xu, W.; Liang, R.; Zheng, J.; Luo, Y.; Wang, Y.; Huang, K. An ITRAQ-Based Mitoproteomics Approach for Profiling the Nephrotoxicity Mechanisms of Ochratoxin A in HEK 293 Cells. J. Proteomics 2013, 78, 398–415. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Rojas-Morales, P.; Avila-Rojas, S.H.; León-Contreras, J.C.; Hernández-Pando, R.; Jiménez-Uribe, A.P.; Prieto-Carrasco, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. Int. J. Mol. Sci. 2020, 21, 6512. [Google Scholar] [CrossRef]
- Prieto-Carrasco, R.; Silva-Palacios, A.; Rojas-Morales, P.; Aparicio-Trejo, O.E.; Medina-Reyes, E.I.; Hernández-Cruz, E.Y.; Sánchez-Garibay, C.; Salinas-Lara, C.; Pavón, N.; Roldán, F.J.; et al. Unilateral Ureteral Obstruction for 28 Days in Rats Is Not Associated with Changes in Cardiac Function or Alterations in Mitochondrial Function. Biology 2021, 10, 671. [Google Scholar] [CrossRef]
- Hernández-Reséndiz, S.; Correa, F.; García-Niño, W.R.; Buelna-Chontal, M.; Roldán, F.J.; Ramírez-Camacho, I.; Delgado-Toral, C.; Carbó, R.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Cardioprotection by Curcumin Post-Treatment in Rats with Established Chronic Kidney Disease. Cardiovasc. Drugs Ther. 2015, 29, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin Prevents Mitochondrial Dynamics Disturbances in Early 5/6 Nephrectomy: Relation to Oxidative Stress and Mitochondrial Bioenergetics. BioFactors 2017, 43, 293–310. [Google Scholar] [CrossRef] [PubMed]
- García-Arroyo, F.E.; Gonzaga-Sánchez, G.; Tapia, E.; Muñoz-Jiménez, I.; Manterola-Romero, L.; Osorio-Alonso, H.; Arellano-Buendía, A.S.; Pedraza-Chaverri, J.; Roncal-Jiménez, C.A.; Lanaspa, M.A.; et al. Osthol Ameliorates Kidney Damage and Metabolic Syndrome Induced by a High-Fat/High-Sugar Diet. Int. J. Mol. Sci. 2021, 22, 2431. [Google Scholar] [CrossRef]
- Negrette-Guzmán, M.; García-Niño, W.R.; Tapia, E.; Zazueta, C.; Huerta-Yepez, S.; León-Contreras, J.C.; Hernández-Pando, R.; Aparicio-Trejo, O.E.; Madero, M.; Pedraza-Chaverri, J. Curcumin Attenuates Gentamicin-Induced Kidney Mitochondrial Alterations: Possible Role of a Mitochondrial Biogenesis Mechanism. Evid. Based Complement. Altern. Med. 2015, 2015, 917435. [Google Scholar] [CrossRef] [Green Version]
- Gaudeuille, A.; N’Demanga Kamoune, J.; Ngakoula-Mbangui, D.; Mamadou, N.N. Profile of Surgical Emergencies in Rural Areas: The Regional Hospital of Bambari in Central Africa as an Example. Dakar Médical 2002, 47, 219–223. [Google Scholar] [PubMed]
- Fox, J.T.; Stover, P.J. Folate-Mediated One-Carbon Metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [CrossRef]
- Welzel, T.M.; Katki, H.A.; Sakoda, L.C.; Evans, A.A.; London, W.T.; Chen, G.; O’Broin, S.; Shen, F.M.; Lin, W.Y.; McGlynn, K.A. Blood Folate Levels and Risk of Liver Damage and Hepatocellular Carcinoma in a Prospective High-Risk Cohort. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 1279–1282. [Google Scholar] [CrossRef] [Green Version]
- Medici, V.; Virata, M.C.; Peerson, J.M.; Stabler, S.P.; French, S.W.; Gregory, J.F.; Albanese, A.; Bowlus, C.L.; Devaraj, S.; Panacek, E.A.; et al. S-Adenosyl-L-Methionine Treatment for Alcoholic Liver Disease: A Double-Blinded, Randomized, Placebo-Controlled Trial. Alcohol. Clin. Exp. Res. 2011, 35, 1960–1965. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Day, C.P. S-Adenosylmethionine (SAMe) Therapy in Liver Disease: A Review of Current Evidence and Clinical Utility. J. Hepatol. 2012, 57, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Medici, V.; Halsted, C.H. Folate, Alcohol, and Liver Disease. Mol. Nutr. Food Res. 2013, 57, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-Y.; Siow, Y.L.; Au-Yeung, K.K.W.; House, J.; Karmin, O. Folic Acid Supplementation Inhibits NADPH Oxidase-Mediated Superoxide Anion Production in the Kidney. Am. J. Physiol. Ren. Physiol. 2011, 300, F189–F198. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, C.M.; Spence, J.D. Folic Acid Supplementation and Chronic Kidney Disease Progression. Kidney Int. 2016, 90, 1144–1145. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.H.; Prathapasinghe, G.A.; Siow, Y.L.; Karmin, O. Hyperhomocysteinemia Induces Liver Injury in Rat: Protective Effect of Folic Acid Supplementation. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Zhang, B.; Li, Y.; Xu, X.; Lv, J.; Jia, Q.; Chai, R.; Xue, W.; Li, Y.; Wang, Y.; et al. Mitochondrial Dysfunction: An Emerging Link in the Pathophysiology of Cardiorenal Syndrome. Front. Cardiovasc. Med. 2022, 9, 837270. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The Mechanism of Superoxide Production by the Antimycin-Inhibited Mitochondrial Q-Cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef] [Green Version]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial Dynamics: Regulatory Mechanisms and Emerging Role in Renal Pathophysiology; Nature Publishing Group: London, UK, 2013; Volume 83, pp. 568–581. [Google Scholar]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial Energetics in the Kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed Mitochondrial Biogenesis in Folic Acid-Induced Acute Kidney Injury and Early Fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sánchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The Inflammatory Cytokine TWEAK Decreases PGC-1α Expression and Mitochondrial Function in Acute Kidney Injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Chancy, C.D.; Kekuda, R.; Huang, W.; Prasad, P.D.; Kuhnel, J.M.; Sirotnak, F.M.; Roon, P.; Ganapathy, V.; Smith, S.B. Expression and Differential Polarization of the Reduced-Folate Transporter-1 and the Folate Receptor α in Mammalian Retinal Pigment Epithelium. J. Biol. Chem. 2000, 275, 20676–20684. [Google Scholar] [CrossRef] [Green Version]
- Nazki, F.H.; Sameer, A.S.; Ganaie, B.A. Folate: Metabolism, Genes, Polymorphisms and the Associated Diseases. Gene 2014, 533, 11–20. [Google Scholar] [CrossRef]
- Huguenin, M.E.; Birbaumer, A.; Brunner, F.P.; Thorhorst, J.; Schmidt, U.; Dubach, U.C.; Thiel, G. An Evaluation of the Role of Tubular Obstruction in Folic Acid-Induced Acute Renal Failure in the Rat. Nephron 1978, 22, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Puri, V.; Sharma, R.; Puri, S. Folic Acid Induces Acute Renal Failure (ARF) by Enhancing Renal Prooxidant State. Exp. Toxicol. Pathol. 2012, 64, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.; Lorz, C.; Catalan, M.P.; Danoff, T.M.; Yamasaki, Y.; Egido, J.; Neilson, E.G. Expression of Apoptosis Regulatory Proteins in Tubular Epithelium Stressed in Culture or Following Acute Renal Failure. Kidney Int. 2000, 57, 969–981. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Yang, J.; Liu, Y. Single Injection of Naked Plasmid Encoding Hepatocyte Growth Factor Prevents Cell Death and Ameliorates Acute Renal Failure in Mice. J. Am. Soc. Nephrol. 2002, 13, 411–422. [Google Scholar] [CrossRef]
- Doi, K.; Okamoto, K.; Negishi, K.; Suzuki, Y.; Nakao, A.; Fujita, T.; Toda, A.; Yokomizo, T.; Kita, Y.; Kihara, Y.; et al. Attenuation of Folic Acid-Induced Renal Inflammatory Injury in Platelet-Activating Factor Receptor-Deficient Mice. Am. J. Pathol. 2006, 168, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Ortega, A.; Rámila, D.; Izquierdo, A.; González, L.; Barat, A.; Gazapo, R.; Bosch, R.J.; Esbrit, P. Role of the Renin-Angiotensin System on the Parathyroid Hormone-Related Protein Overexpression Induced by Nephrotoxic Acute Renal Failure in the Rat. J. Am. Soc. Nephrol. 2005, 16, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.-C. Proliferation of Bone Marrow-Derived Cells Contributes to Regeneration after Folic Acid-Induced Acute Tubular Injury. J. Am. Soc. Nephrol. 2005, 16, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Lucock, M. Folic Acid: Nutritional Biochemistry, Molecular Biology, and Role in Disease Processes. Mol. Genet. Metab. 2000, 71, 121–138. [Google Scholar] [CrossRef]
- Edelstein, C.L.; Akcay, A.; Nguyen, Q. Mediators of Inflammation in Acute Kidney Injury. Mediators Inflamm. 2009, 2009, 137072. [Google Scholar] [CrossRef] [Green Version]
- De Deyn, P.P.; Vanholder, R.; D’Hooge, R. Nitric Oxide in Uremia: Effects of Several Potentially Toxic Guanidino Compounds. Kidney Int. Suppl. 2003, 63 (Suppl. 84), S25–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeremy BS, M.P. Renal Failure Is Associated with Driving of Gene Expression towards Cardiac Hypertrophy and Reduced Mitochondrial Activity. J. Clin. Exp. Cardiolog. 2012, 3, 1–9. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, Y.; Inagi, R. Mitochondria: A Therapeutic Target in Acute Kidney Injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial Dysfunction in the Pathophysiology of Renal Diseases. Am. J. Physiol. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Kazachenko, A.V.; Vyssokikh, M.Y.; Vasileva, A.K.; Tcvirkun, D.V.; Isaev, N.K.; Kirpatovsky, V.I.; Zorov, D.B. The Role of Mitochondria in Oxidative and Nitrosative Stress during Ischemia/Reperfusion in the Rat Kidney. Kidney Int. 2007, 72, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J. Protein S-Glutathionylation Reactions as a Global Inhibitor of Cell Metabolism for the Desensitization of Hydrogen Peroxide Signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef]
- Ramírez-Camacho, I.; Correa, F.; El Hafidi, M.; Silva-Palacios, A.; Ostolga-Chavarría, M.; Esparza-Perusquía, M.; Olvera-Sánchez, S.; Flores-Herrera, O.; Zazueta, C. Cardioprotective Strategies Preserve the Stability of Respiratory Chain Supercomplexes and Reduce Oxidative Stress in Reperfused Ischemic Hearts. Free Radic. Biol. Med. 2018, 129, 407–417. [Google Scholar] [CrossRef]
- Smith, J.A.; Stallons, L.J.; Collier, J.B.; Chavin, K.D.; Schnellmann, R.G. Suppression of Mitochondrial Biogenesis through Toll-like Receptor 4-Dependent Mitogen-Activated Protein Kinase Kinase/Extracellular Signal-Regulated Kinase Signaling in Endotoxin-Induced Acute Kidney Injury. J. Pharmacol. Exp. Ther. 2014, 352, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Aroor, A.R.; Mandavia, C.; Ren, J.; Sowers, J.R.; Pulakat, L. Mitochondria and Oxidative Stress in the Cardiorenal Metabolic Syndrome. Cardiorenal Med. 2012, 2, 87–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, S.J.; Choudhary, A.; Kalsi, A.K.; Zhao, S.; Alex, R.; Abraham, N.G. Ox-Hdl: A Starring Role in Cardiorenal Syndrome and the Effects of Heme Oxygenase-1 Intervention. Diagnostics 2020, 10, 976. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Cohen, K.; Yadin, D.; Nudelman, V.; Gorfil, D.; Laniado-Schwartzman, M.; Kornwoski, R.; Aravot, D.; Abraham, N.G.; Arad, M.; et al. Regulation of Diabetic Cardiomyopathy by Caloric Restriction Is Mediated by Intracellular Signaling Pathways Involving “SIRT1 and PGC-1α”. Cardiovasc. Diabetol. 2018, 17, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Vehicle | FA | NAC+FA | NAC |
|---|---|---|---|---|
| IVS (mm) | 2 ± 0.19 | 2.02 ± 0.25 | 2.0 ± 0.15 | 1.9 ± 0.19 |
| LVDd (mm) | 5.3 ± 0.33 | 5.1 ± 0.38 | 4.6 ± 0.31 * | 5.1 ± 0.45 |
| LVDs (mm) | 2.6± 0.23 | 2.5± 0.5 | 2.04 ± 0.11 * | 2.2± 0.14 |
| LVPW (mm) | 1.9 ± 0.14 | 1.8 ± 0.17 | 1.8 ± 0.13 | 1.8 ± 0.08 |
| EF(%) | 83.8 ± 6.16 | 90.4 ± 1.19 | 83.3 ± 1.5 | 90.9 ± 1.52 |
| FS (%) | 49.7 ± 3.2 | 51.3 ± 9.2 | 55.7 ± 1.9 | 56.7 ± 2.5 |
| HR (bpm) | 369.7 ± 67.9 | 424.3 ± 58.9 | 351.6 ± 45.8 | 356.2 ± 22.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas-López, B.; Romero-Ramirez, E.I.; García-Arroyo, F.E.; Tapia, E.; León-Contreras, J.C.; Silva-Palacios, A.; Roldán, F.-J.; Campos, O.N.M.; Hernandez-Esquivel, L.; Marín-Hernández, A.; et al. NAC Pre-Administration Prevents Cardiac Mitochondrial Bioenergetics, Dynamics, Biogenesis, and Redox Alteration in Folic Acid-AKI-Induced Cardio-Renal Syndrome Type 3. Antioxidants 2023, 12, 1592. https://doi.org/10.3390/antiox12081592
Cuevas-López B, Romero-Ramirez EI, García-Arroyo FE, Tapia E, León-Contreras JC, Silva-Palacios A, Roldán F-J, Campos ONM, Hernandez-Esquivel L, Marín-Hernández A, et al. NAC Pre-Administration Prevents Cardiac Mitochondrial Bioenergetics, Dynamics, Biogenesis, and Redox Alteration in Folic Acid-AKI-Induced Cardio-Renal Syndrome Type 3. Antioxidants. 2023; 12(8):1592. https://doi.org/10.3390/antiox12081592
Chicago/Turabian StyleCuevas-López, Belén, Edgar Ignacio Romero-Ramirez, Fernando E. García-Arroyo, Edilia Tapia, Juan Carlos León-Contreras, Alejandro Silva-Palacios, Francisco-Javier Roldán, Omar Noel Medina Campos, Luz Hernandez-Esquivel, Alvaro Marín-Hernández, and et al. 2023. "NAC Pre-Administration Prevents Cardiac Mitochondrial Bioenergetics, Dynamics, Biogenesis, and Redox Alteration in Folic Acid-AKI-Induced Cardio-Renal Syndrome Type 3" Antioxidants 12, no. 8: 1592. https://doi.org/10.3390/antiox12081592