
Complex II Biology in Aging, Health, and Disease
Abstract
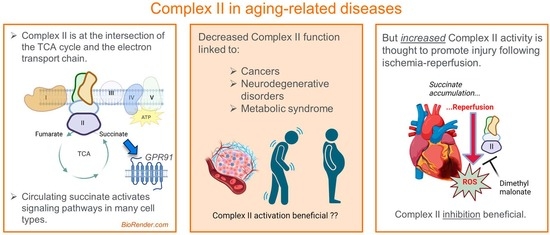
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
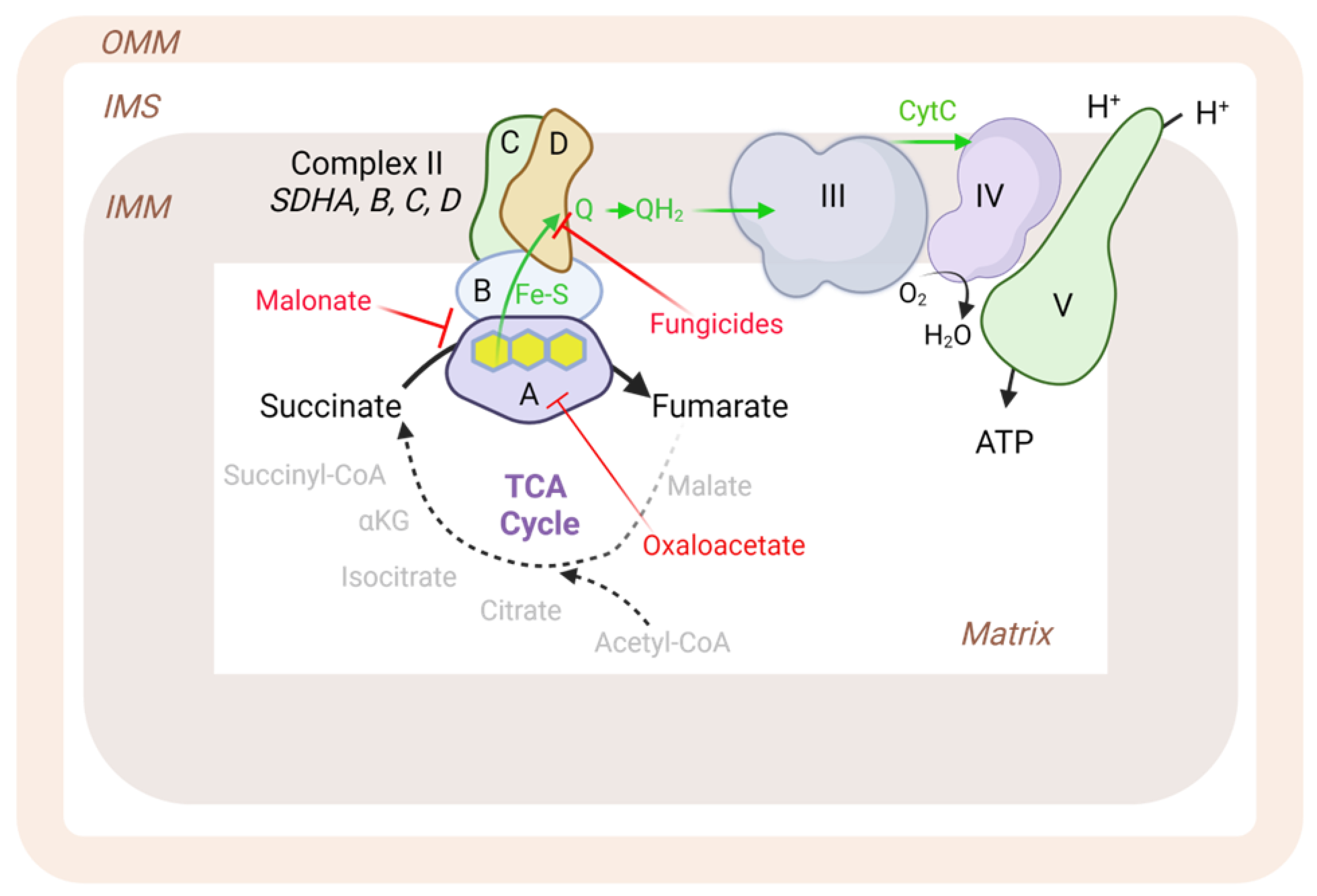
2. Complex II: Structure and Function
2.1. Overall Structure and Assembly
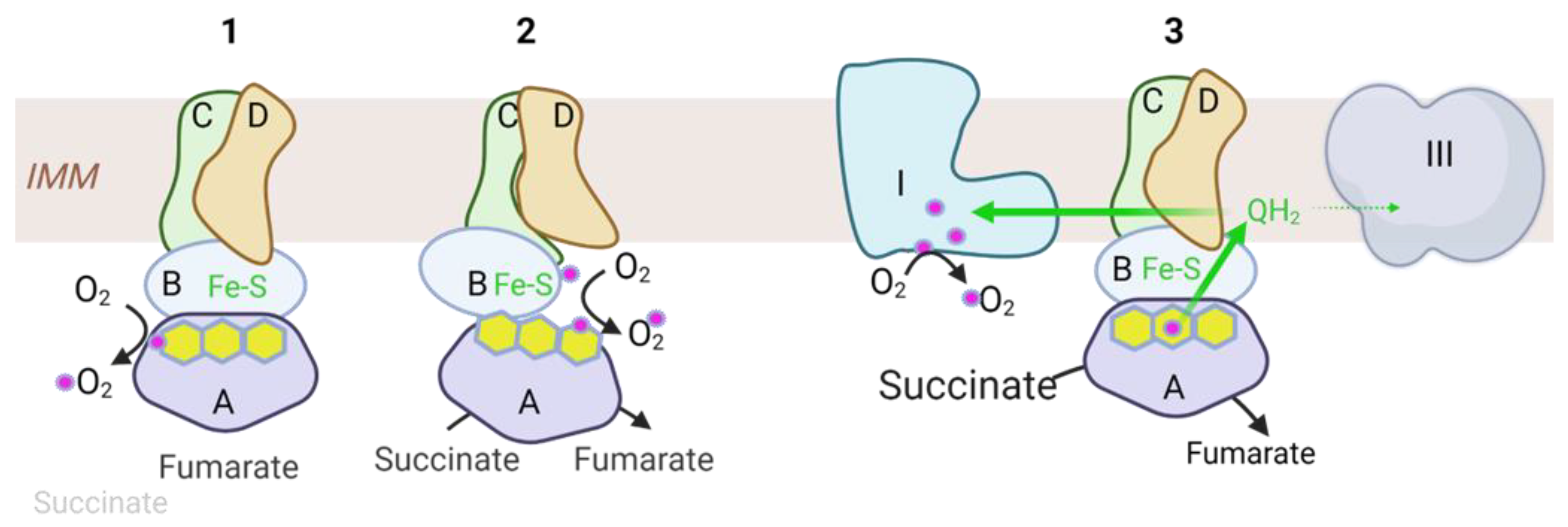
2.2. Inhibitors and Production of Reactive Oxygen Species (ROS)
3. Complex II and Disease
3.1. Inherited Disorders of Complex II Dysfunction
3.2. Complex II in Aging-Associated Neurodegenerative Diseases
3.3. Complex II Dysfunction in Metabolic Syndrome
3.4. Complex II and Succinate in Cardiac Disease
4. Succinate as an Endocrine/Paracrine Factor
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Zhou, X.; Lai, Y.; Xu, J.; Zhang, Y.; Zhou, S.; Feng, Z.; Yu, L.; Tang, Y.; Wang, W.; et al. Structure of the human respiratory complex II. Proc. Natl. Acad. Sci. USA 2023, 120, e2216713120. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2021, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Bénit, P.; Goncalves, J.; El Khoury, R.; Rak, M.; Favier, J.; Gimenez-Roqueplo, A.-P.; Rustin, P. Succinate Dehydrogenase, Succinate, and Superoxides: A Genetic, Epigenetic, Metabolic, Environmental Explosive Crossroad. Biomedicines 2022, 10, 1788. [Google Scholar] [CrossRef]
- Moosavi, B.; Berry, E.A.; Zhu, X.-L.; Yang, W.-C.; Yang, G.-F. The assembly of succinate dehydrogenase: A key enzyme in bioenergetics. Cell. Mol. Life Sci. 2019, 76, 4023–4042. [Google Scholar] [CrossRef]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Bezawork-Geleta, A.; Wen, H.; Dong, L.; Yan, B.; Vider, J.; Boukalova, S.; Krobova, L.; Vanova, K.; Zobalova, R.; Sobol, M.; et al. Alternative assembly of respiratory complex II connects energy stress to metabolic checkpoints. Nat. Commun. 2018, 9, 2221. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Moreno-Sanchez, R.; Neuzil, J.; Rodriguez-Enriquez, S. Inhibitors of succinate: Quinone reductase/Complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm. Res. 2011, 28, 2695–2730. [Google Scholar] [CrossRef]
- Huang, L.-S.; Sun, G.; Cobessi, D.; Wang, A.C.; Shen, J.T.; Tung, E.Y.; Anderson, V.E.; Berry, E.A. 3-Nitropropionic Acid Is a Suicide Inhibitor of Mitochondrial Respiration That, upon Oxidation by Complex II, Forms a Covalent Adduct with a Catalytic Base Arginine in the Active Site of the Enzyme. J. Biol. Chem. 2006, 281, 5965–5972. [Google Scholar] [CrossRef] [Green Version]
- Kruspig, B.; Valter, K.; Skender, B.; Zhivotovsky, B.; Gogvadze, V. Targeting succinate:ubiquinone reductase potentiates the efficacy of anticancer therapy. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2065–2071. [Google Scholar] [CrossRef]
- Vanova, K.H.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bharathi, S.S.; Beck, M.E.; Goetzman, E.S. The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol. 2019, 26, 101253. [Google Scholar] [CrossRef]
- Rodrigues, J.V.; Gomes, C.M. Mechanism of superoxide and hydrogen peroxide generation by human electron-transfer flavoprotein and pathological variants. Free. Radic. Biol. Med. 2012, 53, 12–19. [Google Scholar] [CrossRef]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, A.S.; Bayley, J.-P. The role of complex II in disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S. Leigh syndrome. Handb. Clin. Neurol. 2023, 194, 43–63. [Google Scholar]
- Jain-Ghai, S.; Cameron, J.M.; Al Maawali, A.; Blaser, S.; MacKay, N.; Robinson, B.; Raiman, J. Complex II deficiency-A case report and review of the literature. Am. J. Med Genet. Part A 2013, 161, 285–294. [Google Scholar] [CrossRef]
- Baysal, B.E.; Maher, E.R. 15 YEARS OF PARAGANGLIOMA: Genetics and mechanism of pheochromocytoma-paraganglioma syndromes characterized by germline SDHB and SDHD mutations. Endocr.-Relat. Cancer 2015, 22, T71–T82. [Google Scholar] [CrossRef] [Green Version]
- Pitsava, G.; Settas, N.; Faucz, F.R.; Stratakis, C.A. Carney Triad, Carney-Stratakis Syndrome, 3PAS and Other Tumors Due to SDH Deficiency. Front. Endocrinol. 2021, 12, 680609. [Google Scholar] [CrossRef]
- Benn, D.E.; Gimenez-Roqueplo, A.-P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.M.; Elston, M.; Gimm, O.; et al. Clinical Presentation and Penetrance of Pheochromocytoma/Paraganglioma Syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically Guided Genetic Screening in a Large Cohort of Italian Patients with Pheochromocytomas and/or Functional or Nonfunctional Paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anttila, T.; Häyry, V.; Nicoli, T.; Hagström, J.; Aittomäki, K.; Vikatmaa, P.; Niemelä, M.; Saarilahti, K.; Mäkitie, A.; Bäck, L.J. A two-decade experience of head and neck paragangliomas in a whole population-based single centre cohort. Eur. Arch. Oto-Rhino-Laryngology 2014, 272, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Oldenburg, R.A.; Nuk, J.; Hoekstra, A.S.; van der Meer, C.A.; Korpershoek, E.; McGillivray, B.; Corssmit, E.P.; Dinjens, W.N.; de Krijger, R.R.; et al. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med. Genet. 2014, 15, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Wojtovich, A.P.; Smith, C.O.; Haynes, C.M.; Nehrke, K.W.; Brookes, P.S. Physiological consequences of complex II inhibition for aging, disease, and the mKATP channel. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Kumaran, S.; Subathra, M.; Balu, M.; Panneerselvam, C. Age-associated decreased activities of mitochondrial electron transport chain complexes in heart and skeletal muscle: Role of l-carnitine. Chem. Interactions 2004, 148, 11–18. [Google Scholar] [CrossRef]
- Cocco, T.; Sgobbo, P.; Clemente, M.; Lopriore, B.; Grattagliano, I.; Di Paola, M.; Villani, G. Tissue-specific changes of mitochondrial functions in aged rats: Effect of a long-term dietary treatment with N-acetylcysteine. Free. Radic. Biol. Med. 2005, 38, 796–805. [Google Scholar] [CrossRef]
- Editors, P.O. Expression of Concern: Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats. PLoS ONE 2022, 17, e0263555. [Google Scholar]
- Bowman, A.; Birch-Machin, M.A. Age-Dependent Decrease of Mitochondrial Complex II Activity in Human Skin Fibroblasts. J. Investig. Dermatol. 2016, 136, 912–919. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Long, J.; He, P.; Shen, Y.; Li, R. New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: Deficiency of estrogen receptor-beta. J. Alzheimers Dis. 2012, 30, 545–558. [Google Scholar] [CrossRef]
- Chi, J.; Xie, Q.; Jia, J.; Liu, X.; Sun, J.; Deng, Y.; Yi, L. Integrated Analysis and Identification of Novel Biomarkers in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 178. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.-C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.-M.; Hirsch, E.; Hantraye, P.; et al. Involvement of Mitochondrial Complex II Defects in Neuronal Death Produced by N-Terminus Fragment of Mutated Huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Ngo, D.T.M.; Sverdlov, A.L.; Karki, S.; Macartney-Coxson, D.; Stubbs, R.S.; Farb, M.G.; Carmine, B.; Hess, D.T.; Colucci, W.S.; Gokce, N. Oxidative modifications of mitochondrial complex II are associated with insulin resistance of visceral fat in obesity. Am. J. Physiol. Metab. 2019, 316, E168–E177. [Google Scholar] [CrossRef]
- He, J.; Watkins, S.; Kelley, D.E. Skeletal Muscle Lipid Content and Oxidative Enzyme Activity in Relation to Muscle Fiber Type in Type 2 Diabetes and Obesity. Diabetes 2001, 50, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, B.; Long, L.; Luo, P.; Xiang, W.; Li, X.; Wang, H.; Jiang, Q.; Tan, X.; Luo, S.; et al. Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat. Commun. 2021, 12, 102. [Google Scholar] [CrossRef]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [Green Version]
- Heidorn-Czarna, M.; Heidorn, H.-M.; Fernando, S.; Sanislav, O.; Jarmuszkiewicz, W.; Mutzel, R.; Fisher, P.R. Chronic Activation of AMPK Induces Mitochondrial Biogenesis through Differential Phosphorylation and Abundance of Mitochondrial Proteins in Dictyostelium discoideum. Int. J. Mol. Sci. 2021, 22, 11675. [Google Scholar] [CrossRef]
- Brandauer, J.; Andersen, M.A.; Kellezi, H.; Risis, S.; Frã¸sig, C.; Vienberg, S.G.; Treebak, J.T. AMP-activated protein kinase controls exercise training- and AICAR-induced increases in SIRT3 and MnSOD. Front. Physiol. 2015, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stancu, A.L. AMPK activation can delay aging. Discoveries 2015, 3, e53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, M.H.; Gozashti, M.; Aghadavood, M.; Mehdizadeh, M.R.; Hayatbakhsh, M.M. Clinical Significance of Serum IL-6 and TNF-α Levels in Patients with Metabolic Syndrome. Rep. Biochem. Mol. Biol. 2017, 6, 74–79. [Google Scholar] [PubMed]
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat. Metab. 2021, 3, 1302–1312. [Google Scholar] [CrossRef]
- Zell, R.; Geck, P.; Werdan, K.; Boekstegers, P. Tnf-α and IL-1α inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: Evidence for primary impairment of mitochondrial function. Mol. Cell. Biochem. 1997, 177, 61–67. [Google Scholar] [CrossRef]
- Mariappan, N.; Elks, C.M.; Haque, M.; Francis, J. Interaction of TNF with Angiotensin II Contributes to Mitochondrial Oxidative Stress and Cardiac Damage in Rats. PLoS ONE 2012, 7, e46568. [Google Scholar] [CrossRef]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef]
- Chen, X.; Sunkel, B.; Wang, M.; Kang, S.; Wang, T.; Gnanaprakasam, J.N.R.; Liu, L.; Cassel, T.A.; Scott, D.A.; Muñoz-Cabello, A.M.; et al. Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation. Sci. Immunol. 2022, 7, eabm8161. [Google Scholar] [CrossRef]
- Hassanpour, S.H.; Dehghani, M.A.; Karami, S.Z. Study of respiratory chain dysfunction in heart disease. J. Cardiovasc. Thorac. Res. 2018, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol. Neurobiol. 2017, 55, 2547–2564. [Google Scholar] [CrossRef] [Green Version]
- Niatsetskaya, Z.V.; Sosunov, S.A.; Matsiukevich, D.; Utkina-Sosunova, I.V.; Ratner, V.I.; Starkov, A.A.; Ten, V.S. The Oxygen Free Radicals Originating from Mitochondrial Complex I Contribute to Oxidative Brain Injury Following Hypoxia–Ischemia in Neonatal Mice. J. Neurosci. 2012, 32, 3235–3244. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord EN, J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Hochachka, P.W.; Dressendorfer, R.H. Succinate accumulation in man during exercise. Eur. J. Appl. Physiol. 1976, 35, 235–242. [Google Scholar] [CrossRef]
- Hochachka, P.; Owen, T.; Allen, J.; Whittow, G. Multiple end products of anaerobiosis in diving vertebrates. Comp. Biochem. Physiol. Part B Comp. Biochem. 1975, 50, 17–22. [Google Scholar] [CrossRef]
- Benzi, G.; Arrigoni, E.; Marzatico, F.; Villa, R. Influence of some biological pyrimidines on the succinate cycle during and after cerebral ischemia. Biochem. Pharmacol. 1979, 28, 2545–2550. [Google Scholar] [CrossRef]
- Hamel, D.; Sanchez, M.; Duhamel, F.; Roy, O.; Honoré, J.-C.; Noueihed, B.; Zhou, T.; Nadeau-Vallée, M.; Hou, X.; Lavoie, J.-C.; et al. G-Protein–Coupled Receptor 91 and Succinate Are Key Contributors in Neonatal Postcerebral Hypoxia-Ischemia Recovery. Arter. Thromb. Vasc. Biol. 2014, 34, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Sahni, P.; Zhang, J.; Sosunov, S.; Galkin, A.; Niatsetskaya, Z.; Starkov, A.; Brookes, P.; Ten, V.S. Krebs cycle metabolites and preferential succinate oxidation following neonatal hypoxic-ischemic brain injury in mice. Pediatr. Res. 2017, 83, 491–497. [Google Scholar] [CrossRef]
- Prag, H.A.; Gruszczyk, A.V.; Huang, M.M.; Beach, T.E.; Young, T.; Tronci, L.; Nikitopoulou, E.; Mulvey, J.F.; Ascione, R.; Hadjihambi, A.; et al. Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc. Res. 2021, 117, 1188–1201. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.T.; Miller, J.H.; Day, M.M.; Munger, J.C.; Brookes, P.S. Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 2018, 23, 2617–2628. [Google Scholar] [CrossRef] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Leite, L.N.; Gonzaga, N.A.; Simplicio, J.A.; Vale, G.T.D.; Carballido, J.M.; Alves-Filho, J.C.; Tirapelli, C.R. Pharmacological characterization of the mechanisms underlying the vascular effects of succinate. Eur. J. Pharmacol. 2016, 789, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, J.; Jouret, F.; Pirotte, B.; Hanson, J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 2016, 159, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2015, 109, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottahedin, A.; Prag, H.A.; Dannhorn, A.; Mair, R.; Schmidt, C.; Yang, M.; Sorby-Adams, A.; Lee, J.J.; Burger, N.; Kulaveerasingam, D.; et al. Targeting succinate metabolism to decrease brain injury upon mechanical thrombectomy treatment of ischemic stroke. Redox Biol. 2023, 59, 102600. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.E.; Prag, H.A.; Pala, L.; Logan, A.; Huang, M.M.; Gruszczyk, A.V.; Martin, J.L.; Mahbubani, K.; Hamed, M.O.; Hosgood, S.A.; et al. Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol. 2020, 36, 101640. [Google Scholar] [CrossRef]
- Xu, J.; Pan, H.; Xie, X.; Zhang, J.; Wang, Y.; Yang, G. Inhibiting Succinate Dehydrogenase by Dimethyl Malonate Alleviates Brain Damage in a Rat Model of Cardiac Arrest. Neuroscience 2018, 393, 24–32. [Google Scholar] [CrossRef]
- Kula-Alwar, D.; Prag, H.A.; Krieg, T. Targeting Succinate Metabolism in Ischemia/Reperfusion Injury. Circulation 2019, 140, 1968–1970. [Google Scholar] [CrossRef]
- Prag, H.A.; Pala, L.; Kula-Alwar, D.; Mulvey, J.F.; Luping, D.; Beach, T.E.; Booty, L.M.; Hall, A.R.; Logan, A.; Sauchanka, V.; et al. Ester Prodrugs of Malonate with Enhanced Intracellular Delivery Protect Against Cardiac Ischemia-Reperfusion Injury In Vivo. Cardiovasc. Drugs Ther. 2022, 36, 1–13. [Google Scholar] [CrossRef]
- Bae, J.; Salamon, R.J.; Brandt, E.B.; Paltzer, W.G.; Zhang, Z.; Britt, E.C.; Hacker, T.A.; Fan, J.; Mahmoud, A.I. Malonate Promotes Adult Cardiomyocyte Proliferation and Heart Regeneration. Circulation 2021, 143, 1973–1986. [Google Scholar] [CrossRef]
- Jespersen, N.R.; Hjortbak, M.V.; Lassen, T.R.; Støttrup, N.B.; Johnsen, J.; Tonnesen, P.T.; Larsen, S.; Kimose, H.-H.; Bøtker, H.E. Cardioprotective effect of succinate dehydrogenase inhibition in rat hearts and human myocardium with and without diabetes mellitus. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.-P.; Lin, D.C.-H.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.-L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Cho, S.W.; Saxena, D.; Li, X. Multifaceted Actions of Succinate as a Signaling Transmitter Vary with Its Cellular Locations. Endocrinol. Metab. 2020, 35, 36–43. [Google Scholar] [CrossRef]
- Robben, J.H.; Fenton, R.A.; Vargas, S.L.; Schweer, H.; Peti-Peterdi, J.; Deen, P.M.; Milligan, G. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009, 76, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef]
- Ariza, A.C.; Deen, P.M.T.; Robben, J.H. The Succinate Receptor as a Novel Therapeutic Target for Oxidative and Metabolic Stress-Related Conditions. Front. Endocrinol. 2012, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Correa, P.R.A.; Kruglov, E.A.; Thompson, M.; Leite, M.F.; Dranoff, J.A.; Nathanson, M.H. Succinate is a paracrine signal for liver damage. J. Hepatol. 2007, 47, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.; Bozi, L.H.M.; Yaghi, O.K.; Mills, E.L.; Xiao, H.; Nicholson, H.E.; Paschini, M.; Paulo, J.A.; Garrity, R.; Laznik-Bogoslavski, D.; et al. pH-Gated Succinate Secretion Regulates Muscle Remodeling in Response to Exercise. Cell 2020, 183, 62–75.e17. [Google Scholar] [CrossRef]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Favret, S.; Binet, F.; Lapalme, E.; Leboeuf, D.; Carbadillo, J.; Rubic, T.; Picard, E.; Mawambo, G.; Tetreault, N.; Joyal, J.-S.; et al. Deficiency in the metabolite receptor SUCNR1 (GPR91) leads to outer retinal lesions. Aging 2013, 5, 427–444. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goetzman, E.; Gong, Z.; Zhang, B.; Muzumdar, R. Complex II Biology in Aging, Health, and Disease. Antioxidants 2023, 12, 1477. https://doi.org/10.3390/antiox12071477
Goetzman E, Gong Z, Zhang B, Muzumdar R. Complex II Biology in Aging, Health, and Disease. Antioxidants. 2023; 12(7):1477. https://doi.org/10.3390/antiox12071477
Chicago/Turabian StyleGoetzman, Eric, Zhenwei Gong, Bob Zhang, and Radhika Muzumdar. 2023. "Complex II Biology in Aging, Health, and Disease" Antioxidants 12, no. 7: 1477. https://doi.org/10.3390/antiox12071477