
Escalating Bi-Directional Feedback Loops between Proinflammatory Microglia and Mitochondria in Ageing and Post-Diagnosis of Parkinson’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
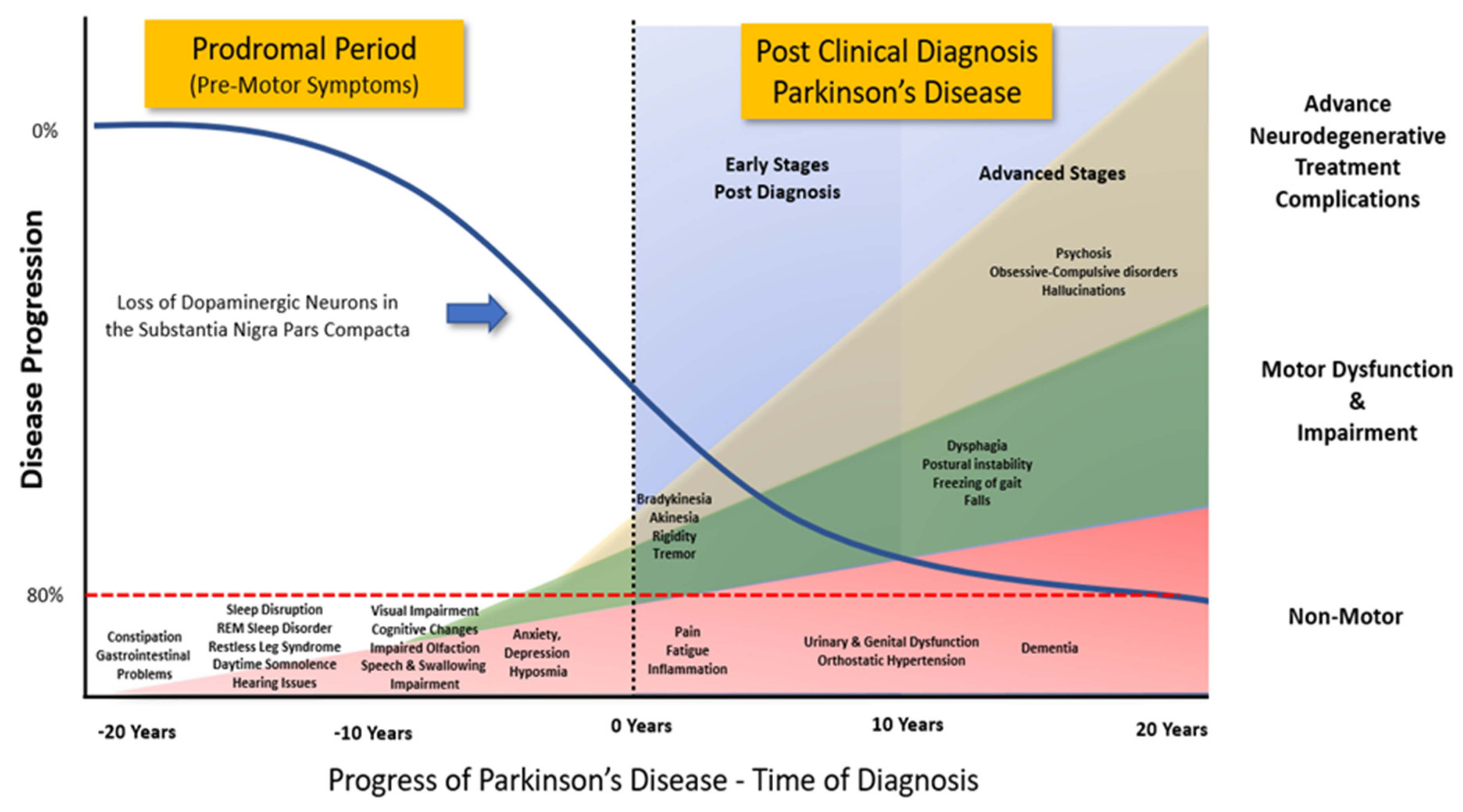
:1. Introduction

2. Epidemiology of PD
3. Loss of Dopaminergic and Adrenergic Neurons in PD
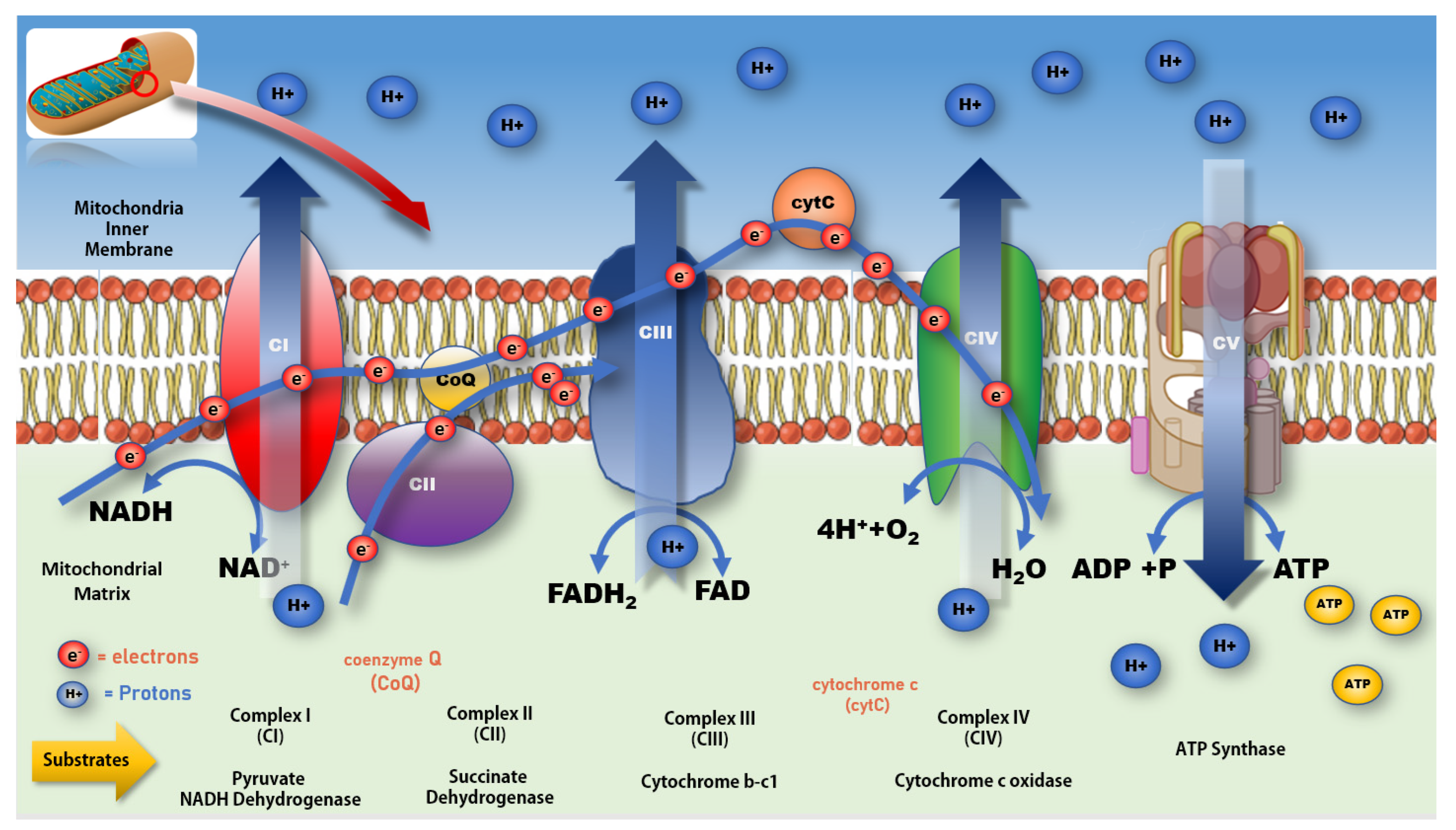
4. Mitochondrial Function in Ageing and PD

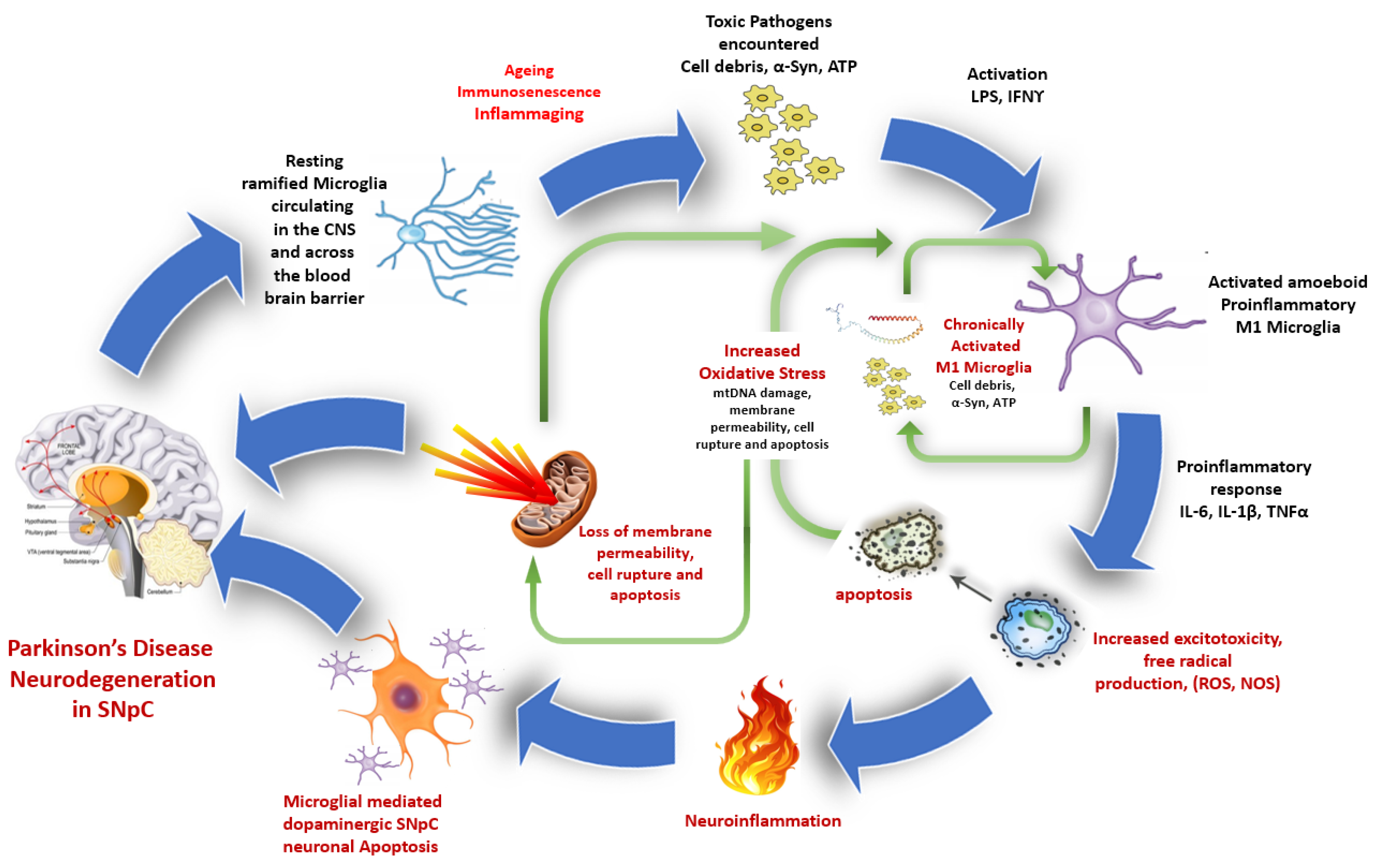
5. Microglia Activation and Neurodegeneration in Ageing and Parkinson’s Disease
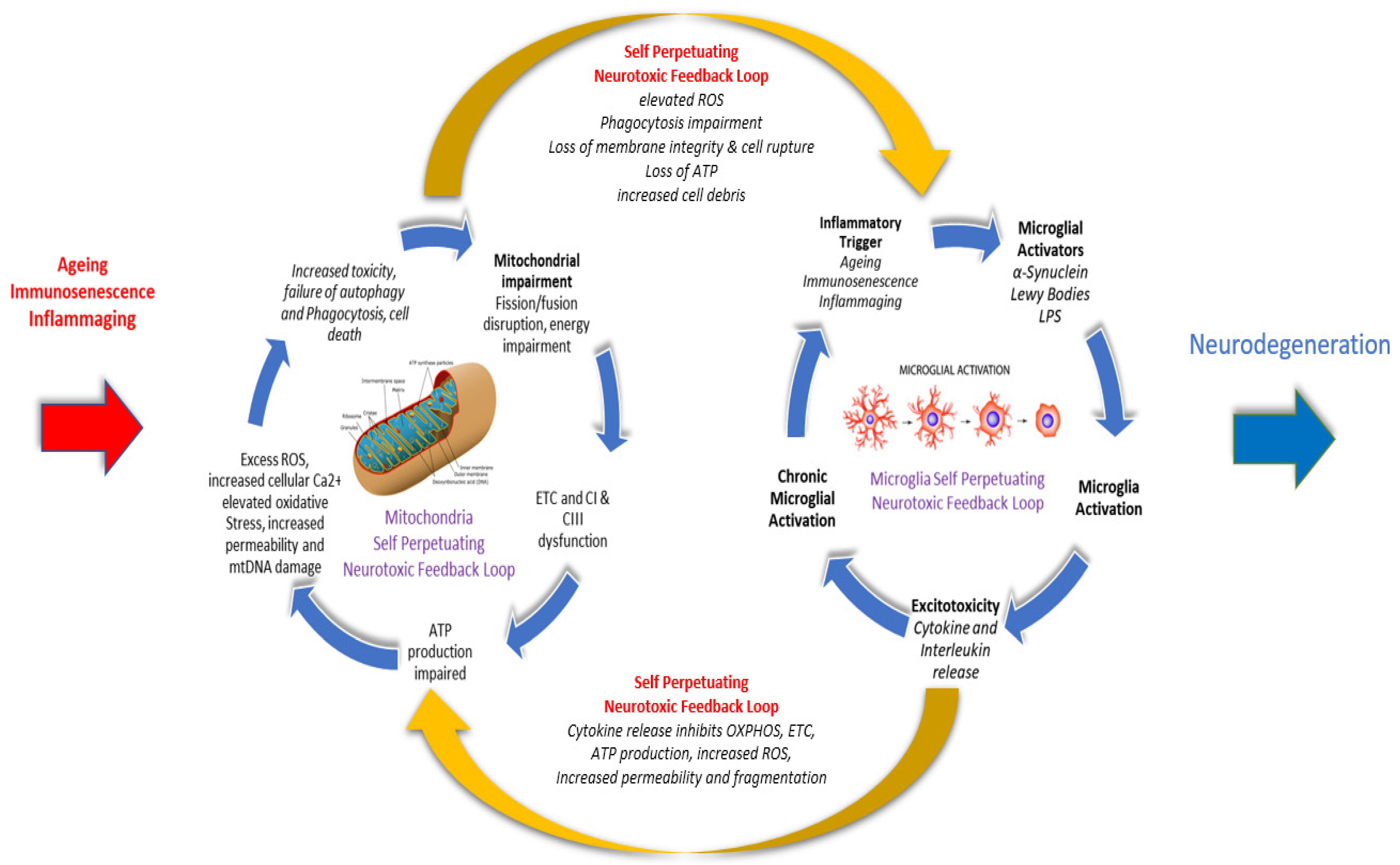
6. Influence of Ageing on Microglia and Mitochondria in PD Neurodegeneration
6.1. Age as the Key Risk Factor of PD
6.2. Inflammation and Inflammaging in Ageing
6.3. Mitochondrial Dysfunction and PD
7. Limitations
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, S.D.; Allen, N.E.; Canning, C.G.; Fung, V.S.C. Parkinson Disease; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018; Volume 159, pp. 173–193. [Google Scholar]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.A.; Troyanova, N.I.; Kovalenko, E.I.; Sapozhnikov, A.M. Similarity and Differences in Inflammation-Related Characteristics of the Peripheral Immune System of Patients with Parkinson’s and Alzheimer’s Diseases. Int. J. Mol. Sci. 2017, 18, 2633. [Google Scholar] [CrossRef] [PubMed]
- Deumens, R.; Blokland, A.; Prickaerts, J. Modeling Parkinson’s disease in rats: An evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol. 2002, 175, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 16. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef]
- Koziorowski, D.; Figura, M.; Milanowski, Ł.M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of Neurodegeneration in Various Forms of Parkinsonism, Similarities and Differences. Cells 2021, 10, 656. [Google Scholar] [CrossRef]
- Cooper, C.A.; Chahine, L.M. Biomarkers in Prodromal Parkinson Disease: A Qualitative Review. J. Int. Neuropsychol. Soc. 2016, 22, 956–967. [Google Scholar] [CrossRef]
- Gaig, C.; Tolosa, E. When does Parkinson’s disease begin? Mov. Disord. 2009, 24 (Suppl. S2), S656–S664. [Google Scholar] [CrossRef]
- Berg, D.; Borghammer, P.; Fereshtehnejad, S.M.; Heinzel, S.; Horsager, J.; Schaeffer, E.; Postuma, R.B. Prodromal Parkinson disease subtypes—Key to understanding heterogeneity. Nat. Rev. Neurol. 2021, 17, 349–361. [Google Scholar] [CrossRef]
- Muslimovic, D.; Post, B.; Speelman, J.D.; De Haan, R.J.; Schmand, B. Cognitive decline in Parkinson’s disease: A prospective longitudinal study. J. Int. Neuropsychol. Soc. 2009, 15, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhou, X.; Zhou, X.; Xiang, Y.; Zhu, L.; Qin, L.; Wang, Y.; Pan, H.; Zhao, Y.; Sun, Q.; et al. Characteristics of Autonomic Dysfunction in Parkinson’s Disease: A Large Chinese Multicenter Cohort Study. Front. Aging Neurosci. 2021, 13, 761044. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Lee, J.Y.; Kim, Y.K.; Yoon, E.J.; Kim, H.; Nam, H.; Jeon, B. Parkinson Disease-Related Brain Metabolic Patterns and Neurodegeneration in Isolated REM Sleep Behavior Disorder. Neurology 2021, 97, e378–e388. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Iranzo, A.; Ostergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Borghammer, P.; Santamaria, J.; et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol. 2017, 16, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Schrag, A.; Sauerbier, A.; Chaudhuri, K.R. New clinical trials for non-motor manifestations of Parkinson’s disease. Mov. Disord. 2015, 30, 1490–1504. [Google Scholar] [CrossRef]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Schrag, A.; Horsfall, L.; Walters, K.; Noyce, A.; Petersen, I. Prediagnostic presentations of Parkinson’s disease in primary care: A case-control study. Lancet Neurol. 2015, 14, 57–64. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Empadinhas, N. The Microbiome-Mitochondria Dance in Prodromal Parkinson’s Disease. Front. Physiol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Dogra, N.; Mani, R.J.; Katare, D.P. The Gut-Brain Axis: Two Ways Signaling in Parkinson’s Disease. Cell. Mol. Neurobiol. 2022, 42, 315–332. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Moca, E.N.; Lecca, D.; Hope, K.T.; Etienne, F.; Schaler, A.W.; Espinoza, K.; Chappell, M.S.; Gray, D.T.; Tweedie, D.; Sidhu, S.; et al. Microglia Drive Pockets of Neuroinflammation in Middle Age. J. Neurosci. 2022, 42, 3896–3918. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflammaging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Castillo, S.; Eugenin, J.; von Bernhardi, R. Impact of Aging in Microglia-Mediated D-Serine Balance in the CNS. Mediat. Inflamm. 2018, 2018, 7219732. [Google Scholar] [CrossRef]
- Tangestani, F.M.; Stough, C. A Review and Hypothesized Model of the Mechanisms That Underpin the Relationship between Inflammation and Cognition in the Elderly. Front. Aging Neurosci. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Batatinha, H.A.P.; Diniz, T.A.; de Souza Teixeira, A.A.; Kruger, K.; Rosa-Neto, J.C. Regulation of autophagy as a therapy for immunosenescence-driven cancer and neurodegenerative diseases: The role of exercise. J. Cell. Physiol. 2019, 234, 14883–14895. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Galtier, I.; Nieto, A.; Lorenzo, J.N.; Barroso, J. Mild cognitive impairment in Parkinson’s disease: Diagnosis and progression to dementia. J. Clin. Exp. Neuropsychol. 2016, 38, 40–50. [Google Scholar] [CrossRef]
- Safarpour, D.; Willis, A.W. Clinical Epidemiology, Evaluation, and Management of Dementia in Parkinson Disease. Am. J. Alzheimers Dis. Other Dement. 2016, 31, 585–594. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Urbani, A.; Babu, M. Mitochondria in Health and in Sickness, 1st ed.; Springer: Singapore, 2019. [Google Scholar]
- Larsson, N.G.; Wedell, A. Mitochondria in human disease. J. Intern. Med. 2020, 287, 589–591. [Google Scholar] [CrossRef]
- Duchen, M.R.; Szabadkai, G. Roles of mitochondria in human disease. Essays Biochem. 2010, 47, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Clark, A.; Svoboda, D.S.; MacLaurin, J.G.; Lagace, D.C.; Park, D.S.; Slack, R.S. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum. Mol. Genet. 2017, 26, 3327–3341. [Google Scholar] [CrossRef] [PubMed]
- Las, G.; Oliveira, M.F.; Shirihai, O.S. Emerging roles of beta-cell mitochondria in type-2-diabetes. Mol. Asp. Med. 2020, 71, 100843. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Forte, M.; Palmerio, S.; Bianchi, F.; Volpe, M.; Rubattu, S. Mitochondrial complex I deficiency and cardiovascular diseases: Current evidence and future directions. J. Mol. Med. 2019, 97, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Giummarra, L.; Crewther, S.G.; Riddell, N.; Murphy, M.J.; Crewther, D.P. Pathway analysis identifies altered mitochondrial metabolism, neurotransmission, structural pathways and complement cascade in retina/RPE/choroid in chick model of form-deprivation myopia. PeerJ 2018, 6, e5048. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef]
- Evans, A.H. Parkinson’s during COVID-19: Then, Now and Next. In Parkinson’s during COVID-19: Managing Treatments and Stress during a Pandemic; 2020. [Google Scholar]
- Marchetti, B.; Giachino, C.; Tirolo, C.; Serapide, M.F. “Reframing” dopamine signaling at the intersection of glial networks in the aged Parkinsonian brain as innate Nrf2/Wnt driver: Therapeutical implications. Aging Cell 2022, 21, e13575. [Google Scholar] [CrossRef]
- Pezzoli, G.; Cereda, E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 2013, 80, 2035–2041. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; Weuve, J.; Nie, H.; Saint-Hilaire, M.H.; Sudarsky, L.; Simon, D.K.; Hersh, B.; Schwartz, J.; Wright, R.O.; Hu, H. Association of cumulative lead exposure with Parkinson’s disease. Environ. Health Perspect. 2010, 118, 1609–1613. [Google Scholar] [CrossRef]
- Cova, I.; Markova, A.; Campini, I.; Grande, G.; Mariani, C.; Pomati, S. Worldwide trends in the prevalence of dementia. J. Neurol. Sci. 2017, 379, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Badanjak, K.; Fixemer, S.; Smajic, S.; Skupin, A.; Grunewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef] [PubMed]
- Suescun, J.; Chandra, S.; Schiess, M.C. The Role of Neuroinflammation in Neurodegenerative Disorders. In Translational Inflammation; Academic Press: Cambridge, MA, USA, 2019; pp. 241–267. [Google Scholar]
- Kasap, M.; Akpinar, G.; Kanli, A. Proteomic studies associated with Parkinson’s disease. Expert Rev. Proteom. 2017, 14, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Bhimani, R. Understanding the Burden on Caregivers of People with Parkinson’s: A Scoping Review of the Literature. Rehabil. Res. Pract. 2014, 2014, 718527. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef]
- Dorsey, E.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.; Ansha, M.; Brayne, C.; Choi, J.-Y.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Schulz, R.; Beach, S. Caregiving as a risk factor for mortality: The Caregiver Health Effects Study. JAMA 1999, 282, 2215–2219. [Google Scholar] [CrossRef]
- Beauchamp, L.C.; Finkelstein, D.I.; Bush, A.I.; Evans, A.H.; Barnham, K.J. Parkinsonism as a Third Wave of the COVID-19 Pandemic? J. Park. Dis. 2020, 10, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Helmich, R.C.; Bloem, B.R. The Impact of the COVID-19 Pandemic on Parkinson’s Disease: Hidden Sorrows and Emerging Opportunities. J. Park. Dis. 2020, 10, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, T.; Crewther, S. Post-COVID 19 Neurological Syndrome (PCNS); a novel syndrome with challenges for the global neurology community. J. Neurol. Sci. 2020, 419, 117179. [Google Scholar] [CrossRef] [PubMed]
- Delenclos, M.; Jones, D.R.; McLean, P.J.; Uitti, R.J. Biomarkers in Parkinson’s disease: Advances and strategies. Park. Relat. Disord. 2016, 22 (Suppl. S1), S106–S110. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Pathogenesis-targeted therapeutic strategies in Parkinson’s disease. Mov. Disord. 2019, 34, 41–44. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Stojkovic, T.; Stefanova, E.; Soldatovic, I.; Markovic, V.; Stankovic, I.; Petrovic, I.; Agosta, F.; Galantucci, S.; Filippi, M.; Kostic, V. Exploring the relationship between motor impairment, vascular burden and cognition in Parkinson’s disease. J. Neurol. 2018, 265, 1320–1327. [Google Scholar] [CrossRef]
- Kim, I.; Shin, N.Y.; Yunjin, B.; Hyu Lee, P.; Lee, S.K.; Mee Lim, S. Early-onset mild cognitive impairment in Parkinson’s disease: Altered corticopetal cholinergic network. Science 2017, 7, 2381. [Google Scholar] [CrossRef]
- Galtier, I.; Nieto, A.; Barroso, J. Cognitive Impairment in Parkinson’s Disease: Historical Review, Past, and Present; Dorszewska, J., Ed.; IntechOpen: London, UK, 2016; p. 400. [Google Scholar]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Zarow, C.; Lyness, S.; Mortimer, J.; Chui, H. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Rüb, U.; De Vos, R.; Bohl, J.; Braak, H. Where does Parkinson disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. 2002, 61, 413–426. [Google Scholar] [CrossRef]
- Yao, N.; Wu, Y.; Zhou, Y.; Ju, L.; Liu, Y.; Ju, R.; Duan, D.; Xu, Q. Lesion of the locus coeruleus aggravates dopaminergic neuron degeneration by modulating microglial function in mouse models of Parkinson’s disease. Brain Res. 2015, 1625, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; di Poggio, A.B.; Pellegrini, A.; Ruggieri, S.; Paparelli, A. Noradrenaline in Parkinson’s disease: From disease progression to current therapeutics. Curr. Med. Chem. 2007, 14, 2330–2334. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; LeWitt, P.A.; Kaufmann, H. Norepinephrine deficiency in Parkinson’s disease: The case for noradrenergic enhancement. Mov. Disord. 2014, 29, 1710–1719. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Kalinin, S.; Braun, D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J. Neurochem. 2016, 139 (Suppl. S2), 154–178. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Sevanan, M.; Krishnamoorthy, A.; Sekar, S. A systematic review of molecular approaches that link mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurol. Sci. 2021, 42, 4459–4469. [Google Scholar] [CrossRef]
- Saravanan, J. Role of microgliosis, oxidative stress and associated neuroinflammation in the pathogenesis of Parkinson’s disease: The therapeutic role of Nrf2 activators. Neurochem. Int. 2021, 145, 105014. [Google Scholar]
- Spano, M.; Signorelli, M.; Vitaliani, R.; Giometto, B. The possible involvement of mitochondrial dysfunctions in Lewy body dementia A systematic Review. Funct. Neurol. 2015, 30, 151–158. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B.; Zhang, Q. NADPH and NADPH Oxidase. In Encyclopedia of Respiratory Medicine; Laurent, G.J., Shapiro, S.D., Eds.; Academic Press: Oxford, UK, 2006; pp. 77–84. [Google Scholar]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122 Pt 8, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, R.F.; Wang, G. Impact of dopamine oxidation on dopaminergic neurodegeneration. ACS Chem. 2018, 10, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–S120. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef]
- Barja, G. The Mitochondrial Free Radical Theory of Aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 1–27. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Bennett, J.P., Jr. An evaluation of the role of mitochondria in neurodegenerative diseases: Mitochondrial mutations and oxidative pathology, protective nuclear responses, and cell death in neurodegeneration. Brain Res. Rev. 1999, 29, 1–25. [Google Scholar] [CrossRef]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial involvement in neurodegenerative diseases. IUBMB Life 2013, 65, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Aging Neurosci. 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Rango, M.; Bresolin, N. Brain Mitochondria, Aging, and Parkinson’s Disease. Genes 2018, 9, 250. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Aldana, B.I. Microglia-Specific Metabolic Changes in Neurodegeneration. J. Mol. Biol. 2019, 431, 1830–1842. [Google Scholar] [CrossRef]
- Shin, Y.; Ryall, J.; Britto, J.; Lau, C.; Devenish, R.; Nagley, P.; Beart, P. Inhibition of bioenergetics provides novel insights into recruitment of PINK 1-dependent neuronal mitophagy. J. Neurochem. 2019, 149, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Austin, G.L.; Young, L.E.A.; Johnson, L.A.; Sun, R. Mitochondrial Metabolism in Major Neurological Diseases. Cells 2018, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Park. Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Salmina, A.B.; Kharitonova, E.V.; Gorina, Y.V.; Teplyashina, E.A.; Malinovskaya, N.A.; Khilazheva, E.D.; Mosyagina, A.I.; Morgun, A.V.; Shuvaev, A.N.; Salmin, V.V.; et al. Blood-Brain Barrier and Neurovascular Unit In Vitro Models for Studying Mitochondria-Driven Molecular Mechanisms of Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 4661. [Google Scholar] [CrossRef]
- Agnihotri, A.; Aruoma, O.I. Alzheimer’s Disease and Parkinson’s Disease: A Nutritional Toxicology Perspective of the Impact of Oxidative Stress, Mitochondrial Dysfunction, Nutrigenomics and Environmental Chemicals. J. Am. Coll. Nutr. 2019, 39, 16–27. [Google Scholar] [CrossRef]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharm. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Meng, R.; Zhao, X.; Hua, C.; Kang, K.; Han, Y.; Ma, X. Clinical significance of baseline neutrophil to lymphocyte ratio in patients with ischemic stroke or haemarragic stroke—An updated Meta-Analysis. Front. Neurol. 2019, 10, 1032. [Google Scholar] [CrossRef] [PubMed]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial dysfunction in Parkinson’s disease. Park. Dis. 2011, 2011, 716871. [Google Scholar] [CrossRef] [PubMed]
- McGuire, P.J. Mitochondrial Dysfunction and the Aging Immune System. Biology 2019, 8, 26. [Google Scholar] [CrossRef]
- Kapnick, S.M.; Pacheco, S.E.; McGuire, P.J. The emerging role of immune dysfunction in mitochondrial diseases as a paradigm for understanding immunometabolism. Metabolism 2018, 81, 97–112. [Google Scholar] [CrossRef]
- Culmsee, C.; Michels, S.; Scheu, S.; Arolt, V.; Dannlowski, U.; Alferink, J. Mitochondria, Microglia, and the Immune System-How Are They Linked in Affective Disorders? Front. Psychiatry 2018, 9, 739. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Ahmad, M.H.; Fatima, M.; Mondal, A.C. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J. Clin. Neurosci. 2019, 59, 6–11. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Minakami, S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem. J. 1979, 180, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Wang, Y.D.; Yu, X.M.; Li, D.W.; Li, G.R. Mitochondria-mediated damage to dopaminergic neurons in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 615–623. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Lu, M.H.; Yuan, D.J.; Xu, D.E.; Yao, P.P.; Ji, W.L.; Chen, H.; Liu, W.L.; Yan, C.X.; Xia, Y.Y.; et al. Mitochondrial Dysfunction in Neural Injury. Front. Neurosci. 2019, 13, 30. [Google Scholar] [CrossRef]
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J.I.; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef]
- Harry, G.; McPherson, C. Microglia: Neuroprotective and Neurodestructive Properties. In Handbook of Neurotoxicity; CRC Press: Boca Raton, FL, USA, 2014; pp. 109–132. [Google Scholar]
- Aires, I.D.; Ribeiro-Rodrigues, T.; Boia, R.; Ferreira-Rodrigues, M.; Girão, H.; Ambrósio, A.F.; Santiago, A.R. Microglial extracellular vesicles as vehicles for neurodegeneration spreading. Biomolecules 2021, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.L. Parkinson’s disease: Microglial/macrophage-induced immunoexcitotoxicity as a central mechanism of neurodegeneration. Surg. Neurol. Int. 2017, 8, 65. [Google Scholar] [CrossRef]
- Gonzalez, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases:an update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. The effects of statins on microglial cells to protect against neurodegenerative disorders: A mechanistic review. Biofactors 2019, 46, 309–325. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef]
- Ren, M.; Guo, Y.; Wei, X.; Yan, S.; Qin, Y.; Zhang, X.; Jiang, F.; Lou, H. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson’s disease. Exp. Neurol. 2018, 302, 205–213. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.G.; Zhou, X.G.; Qiao, G.; Yu, L.; Tang, Y.; Yan, L.; Qiu, W.Q.; Pan, R.; Yu, C.L.; Law, B.Y.; et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 2021, 65, 101202. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.S.; Azam, S.; Choi, D.K. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 2019, 35, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Pisanu, A.; Lecca, D.; Mulas, G.; Wardas, J.; Simbula, G.; Spiga, S.; Carta, A.R. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 2014, 71, 280–291. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Süβ, P.; Lana, A.J.; Schlachetzki, J.C.M. Chronic peripheral inflammation: A possible contributor to neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1711–1714. [Google Scholar] [CrossRef]
- Ponce, J.; Ulu, A.; Hanson, C.; Cameron-Smith, E.; Bertoni, J.; Wuebker, J.; Fisher, A.; Siu, K.C.; Marmelat, V.; Adamec, J.; et al. Role of Specialized Pro-resolving Mediators in Reducing Neuroinflammation in Neurodegenerative Disorders. Front. Aging Neurosci. 2022, 14, 780811. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Xu, J.; Gao, X.; Yang, C.; Chen, L.; Chen, Z. Resolvin D1 Attenuates Mpp+-Induced Parkinson Disease via Inhibiting Inflammation in PC12 Cells. Med. Sci. Monit. 2017, 23, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Cleveland, D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 2012, 209, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Rehman, M.U.; Sehar, N.; Dar, N.J.; Khan, A.; Arafah, A.; Rashid, S.; Rashid, S.M.; Ganaie, M.A. Mitochondrial dysfunctions, oxidative stress and neuroinflammation as therapeutic targets for neurodegenerative diseases: An update on current advances and impediments. Neurosci. Biobehav. Rev. 2023, 144, 104961. [Google Scholar] [CrossRef]
- Carta, S.; Penco, F.; Lavieri, R.; Martini, A.; Dinarello, C.A.; Gattorno, M.; Rubartelli, A. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc. Natl. Acad. Sci. USA 2015, 112, 2835–2840. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, Q.; Jiang, G.X.; Wang, G.; Cheng, Q. Identification of distinct blood-based biomarkers in early stage of Parkinson’s disease. Neurol. Sci. 2020, 41, 893–901. [Google Scholar] [CrossRef]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef]
- Nonaka, T.; Watanabe, S.T.; Iwatsubo, T.; Hasegawa, M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: Cellular models of neurodegenerative diseases. J. Biol. Chem. 2010, 285, 34885–34898. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K. Functional diversity of microglia–how heterogeneous are they to begin with? Front. Cell. Neurosci. 2013, 7, 65. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxid. Redox Signal. 2020, 9, 743. [Google Scholar] [CrossRef]
- Chausse, B.; Lewen, A.; Poschet, G.; Kann, O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain Behav. Immun. 2020, 88, 802–814. [Google Scholar] [CrossRef]
- Gertig, U.; Hanisch, U.K. Microglial diversity by responses and responders. Front. Cell. Neurosci. 2014, 8, 101. [Google Scholar] [CrossRef]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iturbe, B.; Pons, H.; Johnson, R.J. Role of the Immune System in Hypertension. Physiol. Rev. 2017, 97, 1127–1164. [Google Scholar] [CrossRef] [PubMed]
- Di Stadio, A.; Angelini, C. Microglia polarization by mitochondrial metabolism modulation: A therapeutic opportunity in neurodegenerative diseases. Mitochondrion 2019, 46, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Rojo, A.I.; Innamorato, N.G.; Martin-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Ageing, neuroinflammation and neurodegeneration. Front. Biosci. 2015, 7, 189–204. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and Peripheral Atypical Inflammatory Mediators Potentiate Neuroinflammation and Neurodegeneration. Front. Cell Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Litteljohn, D.; Mangano, E.; Clarke, M.; Bobyn, J.; Moloney, K.; Hayley, S. Inflammatory mechanisms of neurodegeneration in toxin-based models of Parkinson’s disease. Park. Dis. 2011, 2011, 713517. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Botran, R.; Ahmed, Z.; Crespo, F.A.; Gatenbee, C.; Gonzalez, J.; Dickson, D.W.; Litvan, I. Cytokine expression and microglial activation in progressive supranuclear palsy. Park. Relat. Disord. 2011, 17, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Tiwari, P.C.; Nath, R.; Pant, K.K. Role of neuroinflammation and latent transcription factors in pathogenesis of Parkinson’s disease. Neurol. Res. 2016, 38, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Gill, J.S.; Bansal, P.K.; Deshmukh, R. Neuroinflammation—A major cause for striatal dopaminergic degeneration in Parkinson’s disease. J. Neurol. Sci. 2017, 381, 308–314. [Google Scholar] [CrossRef]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef]
- Davis, A.A.; Inman, C.E.; Wargel, Z.M.; Dube, U.; Freeberg, B.M.; Galluppi, A.; Haines, J.N.; Dhavale, D.D.; Miller, R.; Choudhury, F.A.; et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 2020, 12, eaay3069. [Google Scholar] [CrossRef]
- Smith, J.D. Apolipoprotein E4: An allele associated with many diseases. Ann. Med. 2000, 32, 118–127. [Google Scholar] [CrossRef]
- De Lau, L.M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Serum cholesterol levels and the risk of Parkinson’s disease. Am. J. Epidemiol. 2006, 164, 998–1002. [Google Scholar] [CrossRef]
- Williams-Gray, C.; Goris, A.; Saiki, M.; Foltynie, T.; Compston, D.A.; Sawcer, S.J.; Barker, R.A. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J. Neurol. 2009, 256, 493–498. [Google Scholar] [CrossRef]
- Gao, J.; Huang, X.; Park, Y.; Liu, R.; Hollenbeck, A.; Schatzkin, A.; Mailman, R.B.; Chen, H. Apolipoprotein E genotypes and the risk of Parkinson disease. Neurobiol. Aging 2011, 32, 2106.e1–2106.e6. [Google Scholar] [CrossRef] [PubMed]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Foss, D.; Mahley, R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1987, 917, 148–161. [Google Scholar] [CrossRef]
- Wagner, J.; Neher, J.J. Killing ageing neurons, one cell at a time. Nat. Neurosci. 2021, 24, 759–760. [Google Scholar] [CrossRef]
- Davis, R.L.; Wong, S.L.; Carling, P.J.; Payne, T.; Sue, C.M.; Bandmann, O. Serum FGF-21, GDF-15, and blood mtDNA copy number are not biomarkers of Parkinson disease. Neurol. Clin. Pract. 2020, 10, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Zalocusky, K.A.; Najm, R.; Taubes, A.L.; Hao, Y.; Yoon, S.Y.; Koutsodendris, N.; Nelson, M.R.; Rao, A.; Bennett, D.A.; Bant, J.; et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The Effects of APOE4 on Mitochondrial Dynamics and Proteins in vivo. J. Alzheimers Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef]
- Cebrian, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Neumann, H.; Cavalie, A.; Jenne, D.E.; Wekerle, H. Induction of MHC class I genes in neurons. Science 1995, 269, 549–552. [Google Scholar] [CrossRef]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Disord. 2020, 11, 578. [Google Scholar] [CrossRef]
- Dose, J.; Huebbe, P.; Nebel, A.; Rimbach, G. APOE genotype and stress response-a mini review. Lipids Health Dis. 2016, 15, 121. [Google Scholar] [CrossRef]
- Nakamura, T.; Watanabe, A.; Fujino, T.; Hosono, T.; Michikawa, M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol. Neurodegener. 2009, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.D.; Mahley, R.W. Apolipoprotein E4 Domain Interaction Mediates Detrimental Effects on Mitochondria and Is a Potential Therapeutic Target for Alzheimer Disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Reiman, E.M.; Beach, T.G.; Serrano, G.E.; Sabbagh, M.N.; Nielsen, M.; Caselli, R.J.; Shi, J. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology 2020, 94, e2404–e2411. [Google Scholar] [CrossRef]
- Azam, S.; Haque, M.E.; Kim, I.S.; Choi, D.K. Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression. Cells 2021, 10, 150. [Google Scholar] [CrossRef]
- Oberg, M.; Fabrik, I.; Fabrikova, D.; Zehetner, N.; Hartlova, A. The role of innate immunity and inflammation in Parkinson s disease. Scand. J. Immunol. 2021, 93, e13022. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 2017, 31, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Currais, A. Ageing and inflammation—A central role for mitochondria in brain health and disease. Ageing Res. Rev. 2015, 21, 30–42. [Google Scholar] [CrossRef]
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders-a Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef]
- Lenaz, G.; D’Aurelio, M.; Merlo Pich, M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial bioenergetics in aging. Biochim. Biophys. Acta (BBA) Bioenerg. 2000, 1459, 397–404. [Google Scholar] [CrossRef]
- Chandra, G.; Shenoi, R.; Anand, R.; Rajamma, U.; Mohanakumar, K. Reinforcing mitochondrial functions in aging brain: An insight into Parkinson’s disease therapeutics. J. Chem. Neuroanat. 2019, 95, 29–42. [Google Scholar] [CrossRef]
- Pence, B.D.; Yarbro, J.R. Aging impairs mitochondrial respiratory capacity in classical monocytes. Exp. Gerontol. 2018, 108, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Geurts, J.J.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion 2010, 10, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nash, Y.; Frenkel, D. New insights on Parkinson’s disease genes: The link between mitochondria impairment and neuroinflammation. J. Neural. Transm. 2015, 122, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Riddell, N.; Crewther, S.G. Novel evidence for complement system activation in chick myopia and hyperopia models: A meta-analysis of transcriptome datasets. Science 2017, 7, 9719. [Google Scholar] [CrossRef]
- Riddell, N.; Faou, P.; Murphy, M.; Giummarra, L.; Downs, R.A.; Rajapaksha, H.; Crewther, S.G. The retina/RPE proteome in chick myopia and hyperopia models: Commonalities with inherited and age-related ocular pathologies. Mol. Vis. 2017, 23, 872–888. [Google Scholar]
- Sun, F.; Deng, Y.; Han, X.; Liu, Q.; Zhang, P.; Manzoor, R.; Ma, H. A secret that underlies Parkinson’s disease: The damaging cycle. Neurochem. Int. 2019, 129, 104484. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- Gyengesi, E.; Rangel, A.; Ullah, F.; Liang, H.; Niedermayer, G.; Asgarov, R.; Venigalla, M.; Gunawardena, D.; Karl, T.; Munch, G. Chronic Microglial Activation in the GFAP-IL6 Mouse Contributes to Age-Dependent Cerebellar Volume Loss and Impairment in Motor Function. Front. Neurosci. 2019, 13, 303. [Google Scholar] [CrossRef]
- Meszaros, A.; Molnar, K.; Nogradi, B.; Hernadi, Z.; Nyul-Toth, A.; Wilhelm, I.; Krizbai, I.A. Neurovascular Inflammaging in Health and Disease. Cells 2020, 9, 1614. [Google Scholar] [CrossRef]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Tronel, C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Dupont, A.C.; Arlicot, N. Molecular Targets for PET Imaging of Activated Microglia: The Current Situation and Future Expectations. Int. J. Mol. Sci. 2017, 18, 802. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef]
- Santoro, A.; Spinelli, C.C.; Martucciello, S.; Nori, S.L.; Capunzo, M.; Puca, A.A.; Ciaglia, E. Innate immunity and cellular senescence: The good and the bad in the developmental and aged brain. J. Leukoc. Biol. 2018, 103, 509–524. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and inflammaging as two sides of the same coin: Friends or foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef]
- Krief, S.; Lonnqvist, F.; Raimbault, S.; Baude, B.; Van Spronsen, A.; Arner, P.; Strosberg, A.D.; Ricquier, D.; Emorine, L.J. Tissue distribution of beta 3-adrenergic receptor mRNA in man. J. Clin. Investig. 1993, 91, 344–349. [Google Scholar] [CrossRef]
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Biological and Clinical Implications of Comorbidities in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 394. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.A.; Potashkin, J.A. System-based approaches to decode the molecular links in Parkinson’s disease and diabetes. Neurobiol. Dis. 2014, 72 Pt A, 84–91. [Google Scholar] [CrossRef]
- Fuger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermuller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Presta, I.; Vismara, M.; Novellino, F.; Donato, A.; Zaffino, P.; Scali, E.; Pirrone, K.C.; Spadea, M.F.; Malara, N.; Donato, G. Innate Immunity Cells and the Neurovascular Unit. Int. J. Mol. Sci. 2018, 19, 3856. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.; Davis, R.; Sue, C.M. New diagnostic pathways for mitochondrial disease. J. Transl. Genet. Genom. 2020, 4, 188–202. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ. 2007, 14, 1261–1266. [Google Scholar] [CrossRef]
- Schapira, A.H.; Gegg, M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Park. Dis. 2011, 2011, 159160. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, J.L. The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection. (Archives of Otolaryngology—Head & Neck Surgery). JAMA J. Am. Med. Assoc. 2002, 288, 296. [Google Scholar]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Deeb, W.; Nozile-Firth, K.; Okun, M.S. Parkinson’s disease: Diagnosis and appreciation of comorbidities. Handb. Clin. Neurol. 2019, 167, 257–277. [Google Scholar] [CrossRef]
- Yu, J.W.; Walter, B.L. Addressing critical care gaps in inpatient Parkinson’s care–Minimizing the impact of comorbidities and developing new care delivery models. Park. Relat. Disord. 2022, 104, 121–122. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Rabaneda-Lombarte, N.; Xicoy-Espaulella, E.; Serratosa, J.; Saura, J.; Sola, C. Parkinsonian Neurotoxins Impair the Pro-inflammatory Response of Glial Cells. Front. Mol. Neurosci. 2018, 11, 479. [Google Scholar] [CrossRef]
- Moya, G.E.; Rivera, P.D.; Dittenhafer-Reed, K.E. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 7030. [Google Scholar] [CrossRef]
- Harper, M.E.; Monemdjou, S.; Ramsey, J.J.; Weindruch, R. Age-related increase in mitochondrial proton leak and decrease in ATP turnover reactions in mouse hepatocytes. Am. J. Physiol.-Endocrinol. Metab. 1998, 275, E197–E206. [Google Scholar] [CrossRef]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia Using Dendrimer-Based Therapeutics. Theranostics 2018, 8, 5529–5547. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sánchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523. [Google Scholar] [CrossRef]
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45. [Google Scholar] [CrossRef]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef]
- Reichenbach, J.; Schubert, R.; Horvath, R.; Petersen, J.; Futterer, N.; Malle, E.; Stumpf, A.; Gebhardt, B.R.; Koehl, U.; Schraven, B.; et al. Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr. Res. 2006, 60, 321–326. [Google Scholar] [CrossRef]
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.P.; Ngoei, K.R.; et al. Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech. 2016, 9, 1295–1305. [Google Scholar] [CrossRef]
- Requejo-Aguilar, R.; Bolanos, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. 2016, 100, 123–137. [Google Scholar] [CrossRef]
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 100. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.; Tanaka, A.; Suen, D.; Gautier, C.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.T.; Sheng, Z.H. Developmental regulation of microtubule-based trafficking and anchoring of axonal mitochondria in health and diseases. Dev. Neurobiol. 2021, 81, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Bhat, A.; Mahalakshmi, A.M.; Tuladhar, S.; Bishir, M.; Mohan, S.K.; Veeraraghavan, V.P.; Chandra, R.; Essa, M.M.; Chidambaram, S.B.; et al. Mitochondrial and Organellar Crosstalk in Parkinson’s Disease. Am. Soc. Neurochem. 2021, 13, 17590914211028364. [Google Scholar] [CrossRef]
- Trushina, E.; McMurray, C.T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef]
- Mishra, A.K.; Dixit, A. Dopaminergic Axons: Key Recitalists in Parkinson’s Disease. Neurochem Res. 2022, 47, 234–248. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Reiman, E.M.; Foster, N.L.; Aisen, P.S.; Petersen, R.C.; Shaw, L.M.; Trojanowski, J.Q.; Jack, C.R., Jr.; et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010, 75, 230–238. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Munoz-Manchado, A.B.; Villadiego, J.; Romo-Madero, S.; Suarez-Luna, N.; Bermejo-Navas, A.; Rodriguez-Gomez, J.A.; Garrido-Gil, P.; Labandeira-Garcia, J.L.; Echevarria, M.; Lopez-Barneo, J.; et al. Chronic and progressive Parkinson’s disease MPTP model in adult and aged mice. J. Neurochem. 2016, 136, 373–387. [Google Scholar] [CrossRef]
- van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A.C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, K.; Nakagawa, R.; Ishido, M.; Yoshinaga, Y.; Watanabe, J.; Hayashi, Y.; Mishima, T.; Fujioka, S.; Tsuboi, Y. Impact of motor and nonmotor symptoms in Parkinson disease for the quality of life: The Japanese Quality-of-Life Survey of Parkinson Disease (JAQPAD) study. J. Neurol. Sci. 2020, 419, 117172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, T.; Chen, Y.; Zheng, D.; Sun, D.; Tu, Q.; Huang, J.; Zhang, J.; Li, Z. Cognitive Deficit and Aberrant Intrinsic Brain Functional Network in Early-Stage Drug-Naive Parkinson’s Disease. Front. Neurosci. 2022, 16, 150. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef]
- Ye, J.; Jiang, Z.; Chen, X.; Liu, M.; Li, J.; Liu, N. Electron transport chain inhibitors induce microglia activation through enhancing mitochondrial reactive oxygen species production. Exp. Cell Res. 2016, 340, 315–326. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravenhill, S.M.; Evans, A.H.; Crewther, S.G. Escalating Bi-Directional Feedback Loops between Proinflammatory Microglia and Mitochondria in Ageing and Post-Diagnosis of Parkinson’s Disease. Antioxidants 2023, 12, 1117. https://doi.org/10.3390/antiox12051117
Ravenhill SM, Evans AH, Crewther SG. Escalating Bi-Directional Feedback Loops between Proinflammatory Microglia and Mitochondria in Ageing and Post-Diagnosis of Parkinson’s Disease. Antioxidants. 2023; 12(5):1117. https://doi.org/10.3390/antiox12051117
Chicago/Turabian StyleRavenhill, Shane Michael, Andrew Howard Evans, and Sheila Gillard Crewther. 2023. "Escalating Bi-Directional Feedback Loops between Proinflammatory Microglia and Mitochondria in Ageing and Post-Diagnosis of Parkinson’s Disease" Antioxidants 12, no. 5: 1117. https://doi.org/10.3390/antiox12051117