
Bio-Hacking Better Health—Leveraging Metabolic Biochemistry to Maximise Healthspan

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular Ageing
3. Oxidative Stress Management; Reactive Oxygen Species Are Intimately Tied to Healthspan
4. Mitochondrial Hormesis
5. ROS on OXPHOS via Cardiolipin Upregulate Aerobic Glycolysis/Pseudo-Hypoxia-Signalling Cascades, a Cancer Phenotype
6. Excess ROS Alters the Mitochondrial Structure, Which Dictates Function
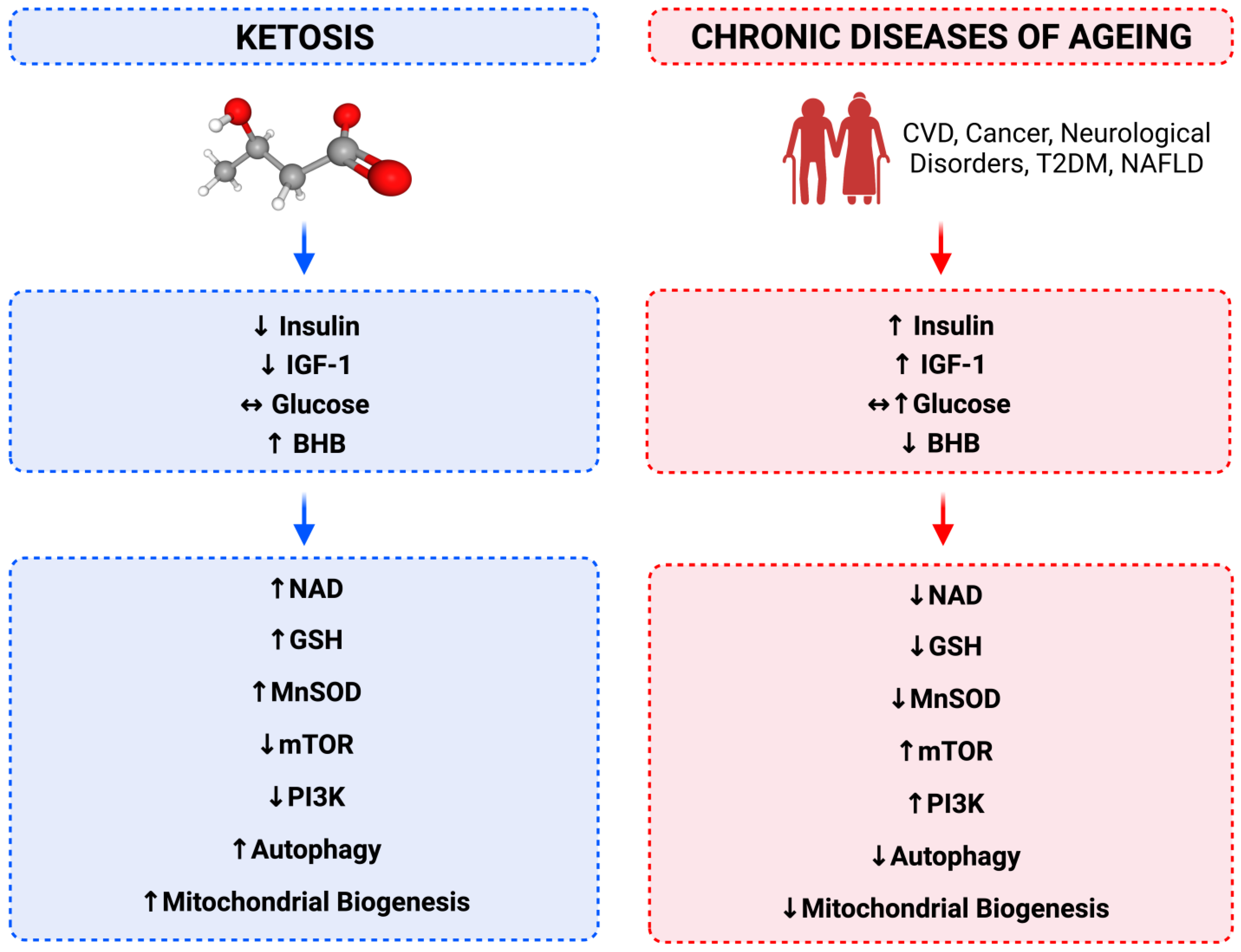
7. Relationship between Calorie Restriction, Beta-Hydroxybutyrate, Low-Insulin and Longevity
8. Cellular Energy Status Sensing
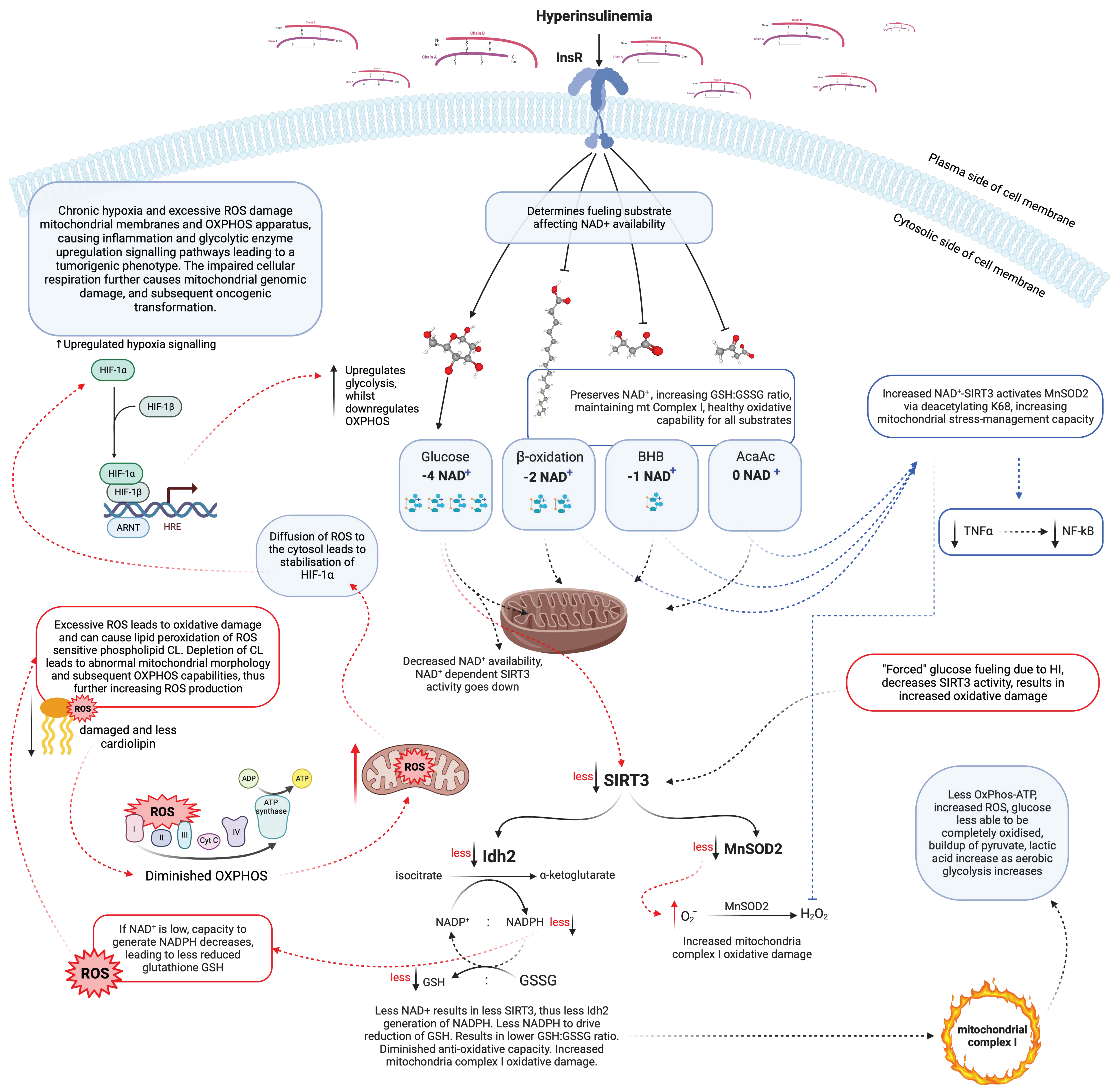
9. Hyperinsulinaemia and Insulin Resistance
10. Applications of Biochemistry into Behaviours
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poulain, M. On the age validation of supercentenarians. In Supercentenarians; Springer: Berlin/Heidelberg, Germany, 2010; pp. 3–30. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- NS C 21. National Life Tables—Life Expectancy in the UK—Office for National Statistics. 2021. Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/bulletins/nationallifetablesunitedkingdom/2018to2020 (accessed on 3 July 2023).
- Pearce, M.; Raftery, A.E. Probabilistic forecasting of maximum human lifespan by 2100 using Bayesian population projections. Demogr. Res. 2021, 44, 1271–1294. [Google Scholar] [CrossRef]
- Endsjø, D.Ø. Flesh and Bones Forever. The History of Immortality from Heracles and Jesus to Vampires and Cyborgs; Apocryphile Press: Berkeley, CA, USA, 2023; ISBN 978-1958061367. [Google Scholar]
- Versele, R.; Corsi, M.; Fuso, A.; Sevin, E.; Businaro, R.; Gosselet, F.; Fenart, L.; Candela, P. Ketone bodies promote amyloid-β1–40 clearance in a human in vitro blood–brain barrier model. Int. J. Mol. Sci. 2020, 21, 934. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Jane Henley, S. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and trends in diabetes among adults in the united states, 1988-2012. JAMA J. Am. Med. Assoc. 2015, 314, 1021–1029. [Google Scholar] [CrossRef]
- Araújo, J.; Cai, J.; Stevens, J. Prevalence of optimal metabolic ealth in american adults: National health and nutrition examination survey 2009-2016. Metab. Syndr. Relat. Disord. 2018, 17, 46–52. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson pandemic—A call to action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Taylor, M.K.; Swerdlow, R.H.; Sullivan, D.K. Dietary neuroketotherapeutics for Alzheimer’s disease: An evidence update and the potential role for diet quality. Nutrients 2019, 11, 1910. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Hernlund, E.; Svedbom, A.; Ivergard, M.; Compston, J.; Cooper, C.; Stenmark, J.; McCloskey, E.V.; Jonsson, B.; Kanis, J.A. Osteoporosis in the European Union: Medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch. Osteoporos. 2013, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.N.; Lie, J.D.; Wan, V.; Cameron, M.; Austel, A.G.; Nguyen, J.K.; Van, K.; Hyun, D. Osteoporosis: A review of treatment options. P T 2018, 43, 92–104. [Google Scholar] [PubMed]
- Cooper, I.D.; Brookler, K.H.; Crofts, C.A.P. Rethinking fragility fractures in type 2 diabetes: The link between hyperinsulinaemia and osteofragilitas. Biomedicines 2021, 9, 1165. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry; United States Atomic Energy Commission: Washington, DC, USA, 1955. [Google Scholar]
- Harman, D. The aging process: Major risk factor for disease and death. Proc. Natl. Acad. Sci. USA 1991, 88, 5360–5363. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Free radical theory of aging: An update: Increasing the functional life span. Ann. N. Y. Acad. Sci. 2006, 1067, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Carboneau, B.A.; Krzysik-Walker, S.M.; De Cabo, R. Of mice and men: The benefits of caloric restriction, exercise, and mimetics. Ageing Res. Rev. 2012, 11, 390–398. [Google Scholar] [CrossRef]
- Konopka, A.R.; Sreekumaran Nair, K. Mitochondrial and skeletal muscle health with advancing age. Mol. Cell. Endocrinol. 2013, 379, 19–29. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Davies, P.C.W.W.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model. BioEssays 2014, 36, 827–835. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Soto, A.M. Carcinogenesis explained within the context of a theory of organisms. Prog. Biophys. Mol. Biol. 2016, 122, 70–76. [Google Scholar] [CrossRef]
- Khokhlov, A.N. Evolution of the term “Cellular Senescence” and its impact on the current cytogerontological research. Moscow Univ. Biol. Sci. Bull. 2013, 68, 158–161. [Google Scholar] [CrossRef]
- Xia, C.; Møller, A.P. An explanation for negligible senescence in animals. Ecol. Evol. 2022, 12, e8970. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [CrossRef]
- Crofts, C.A.P.; Zinn, C.; Wheldon, M.; Schofield, M. Hyperinsulinemia: A unifying theory of chronic disease? Diabesity 2015, 1, 34. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.D.; Yen, K.; Tissenbaum, H.A. Converging pathways in lifespan regulation. Curr. Biol. 2009, 19, R657. [Google Scholar] [CrossRef] [PubMed]
- Draznin, B. Mitogenic action of insulin: Friend, foe or “frenemy”? Diabetologia 2010, 53, 229–233. [Google Scholar] [CrossRef]
- Draznin, B. Mechanism of the mitogenic influence of hyperinsulinemia. Diabetol. Metab. Syndr. 2011, 3, 1–3. [Google Scholar] [CrossRef]
- Jin, K. Modern biological theories of aging. Aging Dis. 2010, 1, 72–74. [Google Scholar]
- Nourshahi, M.; Damirchi, A.; Babaei, P.; Gholamali, M.; Salehpour, M. Mitochondrial biogenesis in skeletal muscle: Exercise and aging. In Skeletal Muscle—From Myogenesis to Clinical Relations; IntechOpen: Rijeka, CO, USA, 2012. [Google Scholar] [CrossRef]
- Schiavi, A.; Torgovnick, A.; Kell, A.; Megalou, E.; Castelein, N.; Guccini, I.; Marzocchella, L.; Gelino, S.; Hansen, M.; Malisan, F.; et al. Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp. Gerontol. 2013, 48, 191–201. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. Fed. Eur. Biochem. Soc. J. 2016, 284, 183–195. [Google Scholar] [CrossRef]
- Moloudizargari, M.; Asghari, M.H.; Ghobadi, E.; Fallah, M.; Rasouli, S.; Abdollahi, M. Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res. Rev. 2017, 40, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. Autophagy and longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, C.; Devin, A. Mitochondrial biogenesis and mitochondrial reactive oxygen species (ROS): A complex relationship regulated by the cAMP/PKA signaling pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C. The Tissue Organization Field Theory of Cancer: A testable replacement for the Somatic Mutation Theory. BioEssays 2011, 33, 332–340. [Google Scholar] [CrossRef]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional mitochondria in health and disease. Front. Endocrinol. (Lausanne) 2017, 8, 296. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-Ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707. [Google Scholar] [CrossRef]
- Vassilopoulos, A.; Fritz, K.S.; Petersen, D.R.; Gius, D. The human sirtuin family: Evolutionary divergences and functions. Hum. Genomics 2011, 5, 485–496. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef]
- Muoio, D.M.; Newgard, C.B. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional ketosis and mitohormesis: Potential implications for mitochondrial function and human health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: Molecular and pharmacological aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Harman, D. Origin and evolution of the free radical theory of ageing: A brief personal history, 1954–2009. Biogerontology 2009, 10, 773–781. [Google Scholar] [CrossRef]
- Frezza, C.; Gottlieb, E. Mitochondria in cancer: Not just innocent bystanders. Semin. Cancer Biol. 2009, 19, 4–11. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 48. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Milder, J.; Patel, M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012, 100, 295–303. [Google Scholar] [CrossRef]
- Hill, S.; Van Remmen, H. Mitochondrial stress signalling in longevity: A new role for mitochondrial function in ageing. Redox Biol. 2014, 2, 936–944. [Google Scholar] [CrossRef]
- Margaritelis, N.V.; Cobley, J.N.; Paschalis, V.; Veskoukis, A.S.; Theodorou, A.A.; Kyparos, A.; Nikolaidis, M.G. Going retro: Oxidative stress biomarkers in modern redox biology. Free Radic. Biol. Med. 2016, 98, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Tan, T.M.C.; Takao, I.; Tang, B.L. The biochemistry and cell biology of ageing: Metabolic regulation through mitochondrial signalling. Am. J. Physiol. Metab. 2014, 306, E581–E591. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. [Google Scholar] [CrossRef]
- Li, S.; Fasipe, B.; Laher, I. Potential harms of supplementation with high doses of antioxidants in athletes. J. Exerc. Sci. Fit. 2022, 20, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, E.; Robinson, N.C. Phospholipase A2 digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure. Biochemistry 1999, 38, 14966–14972. [Google Scholar] [CrossRef]
- Schild, L.; Lendeckel, U.; Gardemann, A.; Wiswedel, I.; Schmidt, C.A.; Wolke, C.; Walther, R.; Grabarczyk, P.; Busemann, C. Composition of molecular cardiolipin species correlates with proliferation of lymphocytes. Exp. Biol. Med. 2012, 237, 372–379. [Google Scholar] [CrossRef]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.W.; Li, C.Y. Regulation of HIF-1α stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Source Sci. New Ser. 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.; Arismendi-Morillo, G.; Kalamian, M.; Elsakka, A.; Maroon, J.; Mukherjee, P. Provocative question: Should ketogenic metabolic therapy become the standard of care for glioblastoma? Neurochem. Res. 2019, 44, 2392–2404. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 2009, 19, 12–16. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Meyskens, F.L. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.; Poff, A.M.; D’Agostino, D.P.; Mukherjee, P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015, 356, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Arismendi-Morillo, G.J.; Castellano-Ramirez, A.V. Ultrastructural mitochondrial pathology in human astrocytic tumors: Potentials implications pro-therapeutics strategies. J. Electron Microsc. 2008, 57, 33–39. [Google Scholar] [CrossRef]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Cooper, I.D.; Crofts, C.A.P.; DiNicolantonio, J.J.; Malhotra, A.; Elliott, B.; Kyriakidou, Y.; Brookler, K.H. Relationships between hyperinsulinaemia, magnesium, vitamin D, thrombosis and COVID-19: Rationale for clinical management. Open Hear. 2020, 7, e001356. [Google Scholar] [CrossRef] [PubMed]
- Fine, E.J.; Segal-Isaacson, C.J.; Feinman, R.D.; Herszkopf, S.; Romano, M.C.; Tomuta, N.; Bontempo, A.F.; Negassa, A.; Sparano, J.A. Targeting insulin inhibition as a metabolic therapy in advanced cancer: A pilot safety and feasibility dietary trial in 10 patients. Nutrition 2012, 28, 1028–1035. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int. J. Cancer 2017, 141, 102–111. [Google Scholar] [CrossRef]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef]
- Hansen, M.E.; Tippetts, T.S.; Anderson, M.C.; Holub, Z.E.; Moulton, E.R.; Swensen, A.C.; Prince, J.T.; Bikman, B.T. Insulin increases ceramide synthesis in skeletal muscle. J. Diabetes Res. 2014, 2014, 765784. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.L.; Letian, A.; Dlugos, T.; Leveau, C.; Dixit, V.D. Innate immune cell-intrinsic ketogenesis is dispensable for organismal metabolism and age-related inflammation. J. Biol. Chem. 2023, 299, 103005. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F.; Herrera, M.G.; Morgan, A.P.; Soeldner, J.S.; Steinke, J.; Levy, P.L.; Reichard, G.A.; Kipnis, D.M. Hormone-fuel interrelationships during fasting. J. Clin. Investig. 1966, 45, 1751–1769. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; Tapia, E.; Pedraza-Chaverri, J. β-hydroxybutyrate: A signaling metabolite in starvation response? Cell. Signal. 2016, 28, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.; Hayat, F.; Cunningham, R.; Li, J.; Garavaglia, S.; Zamani, L.; Ferraris, D.M.; Sykora, P.; Andrews, J.; Clark, J.; et al. Extracellular NAD+ enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 2020, 10, 651. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kim, O.Y.; Song, J. Role of ketone bodies in diabetes-induced dementia: Sirtuins, insulin resistance, synaptic plasticity, mitochondrial dysfunction, and neurotransmitter. Nutr. Rev. 2022, 80, 774–785. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: A signaling metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-hydroxybutyrate in the brain: One molecule, multiple mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31, S262–S268. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, S.-Y.; Soo Choi, C. Insulin resistance: From mechanisms to therapeutic strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Cooper, I.D.; Brookler, K.H.; Kyriakidou, Y.; Elliott, B.T.; Crofts, C.A.P. Metabolic phenotypes and step by step evolution of type 2 diabetes: A new paradigm. Biomedicines 2021, 9, 800. [Google Scholar] [CrossRef]
- Leitner, J.W.; Kline, T.; Carel, K.; Goalstone, M.; Draznin, B. Hyperinsulinemia potentiates activation of p21Ras by growth factors. Endocrinology 1997, 138, 2211–2214. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, R.W. Does weight cycling present a health risk? Am. J. Clin. Nutr. 1996, 63, 452S–455S. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schünemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Bosco, G.; Camporesi, E.M.; Mangar, D. Ketosis, ketogenic diet and food intake control: A complex relationship. Front. Psychol. 2015, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Freidenreich, D.J.; Saenz, C.; Kunces, L.J.; Creighton, B.C.; Bartley, J.M.; Davitt, P.M.; Munoz, C.X.; Anderson, J.M.; Maresh, C.M.; et al. Metabolic characteristics of keto-adapted ultra-endurance runners. Metabolism. 2015, 65, 100–110. [Google Scholar] [CrossRef]
- Kinzig, K.P.; Honors, M.A.; Hargrave, S.L. Insulin sensitivity and glucose tolerance are altered by maintenance on a ketogenic diet. Endocrinology 2010, 151, 3105–3114. [Google Scholar] [CrossRef]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-oxidant and anti-inflammatory activity of ketogenic diet: New perspectives for neuroprotection in Alzheimer’s disease. Antioxidants 2018, 7, 63. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, I.D.; Kyriakidou, Y.; Petagine, L.; Edwards, K.; Elliott, B.T. Bio-Hacking Better Health—Leveraging Metabolic Biochemistry to Maximise Healthspan. Antioxidants 2023, 12, 1749. https://doi.org/10.3390/antiox12091749
Cooper ID, Kyriakidou Y, Petagine L, Edwards K, Elliott BT. Bio-Hacking Better Health—Leveraging Metabolic Biochemistry to Maximise Healthspan. Antioxidants. 2023; 12(9):1749. https://doi.org/10.3390/antiox12091749
Chicago/Turabian StyleCooper, Isabella D., Yvoni Kyriakidou, Lucy Petagine, Kurtis Edwards, and Bradley T. Elliott. 2023. "Bio-Hacking Better Health—Leveraging Metabolic Biochemistry to Maximise Healthspan" Antioxidants 12, no. 9: 1749. https://doi.org/10.3390/antiox12091749