
Full-Length Transcriptome Sequencing Reveals the Molecular Mechanism of Metasequoia glyptostroboides Seed Responding to Aging

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Artificial Aging Treatment
2.2. Germination Parameters Tests
2.3. Physiological Analysis
2.4. RNA Quantification and Qualification
2.5. cDNA Construction and PacBio Iso-Seq
2.6. Iso-Seq Data Processing with Standard Bioinformatics Pipeline
2.7. Illumina Library Construction and Sequencing
2.8. Weighted Correlation Network Analysis
2.9. Quantitative (q)RT-PCR Validation
3. Results
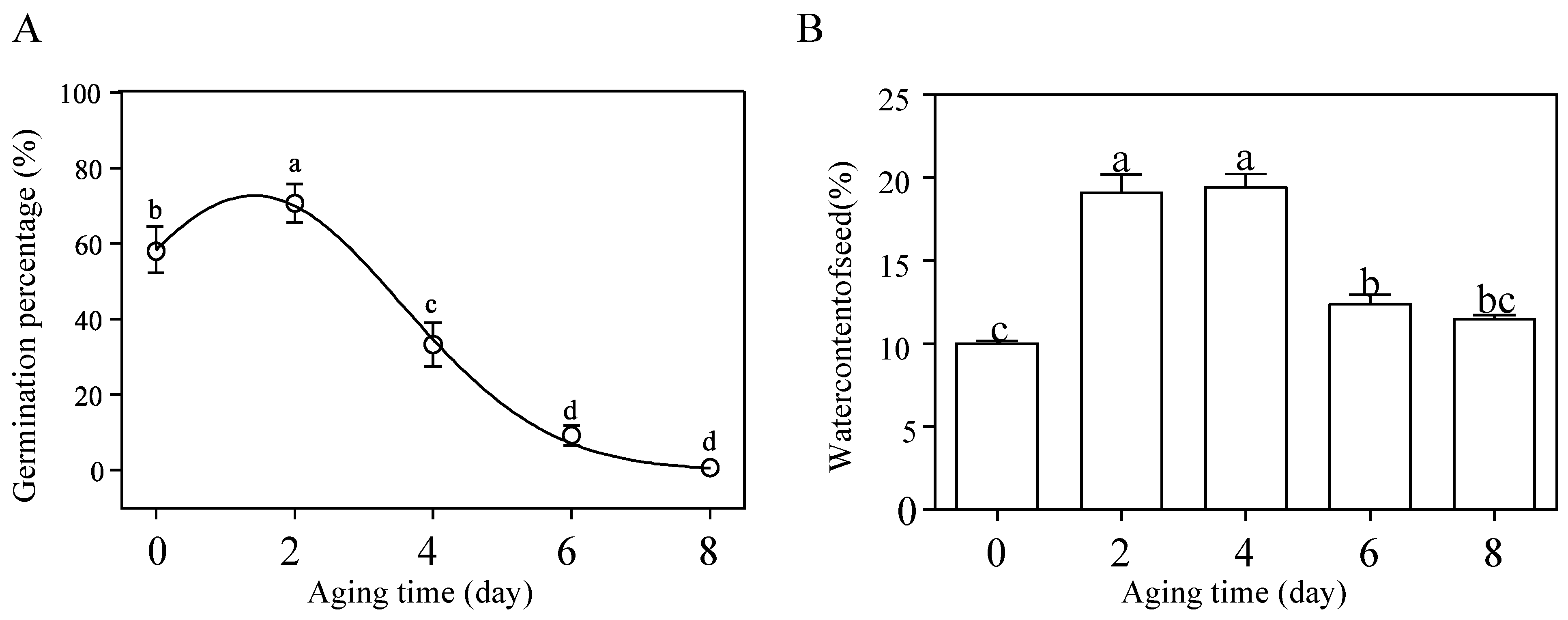
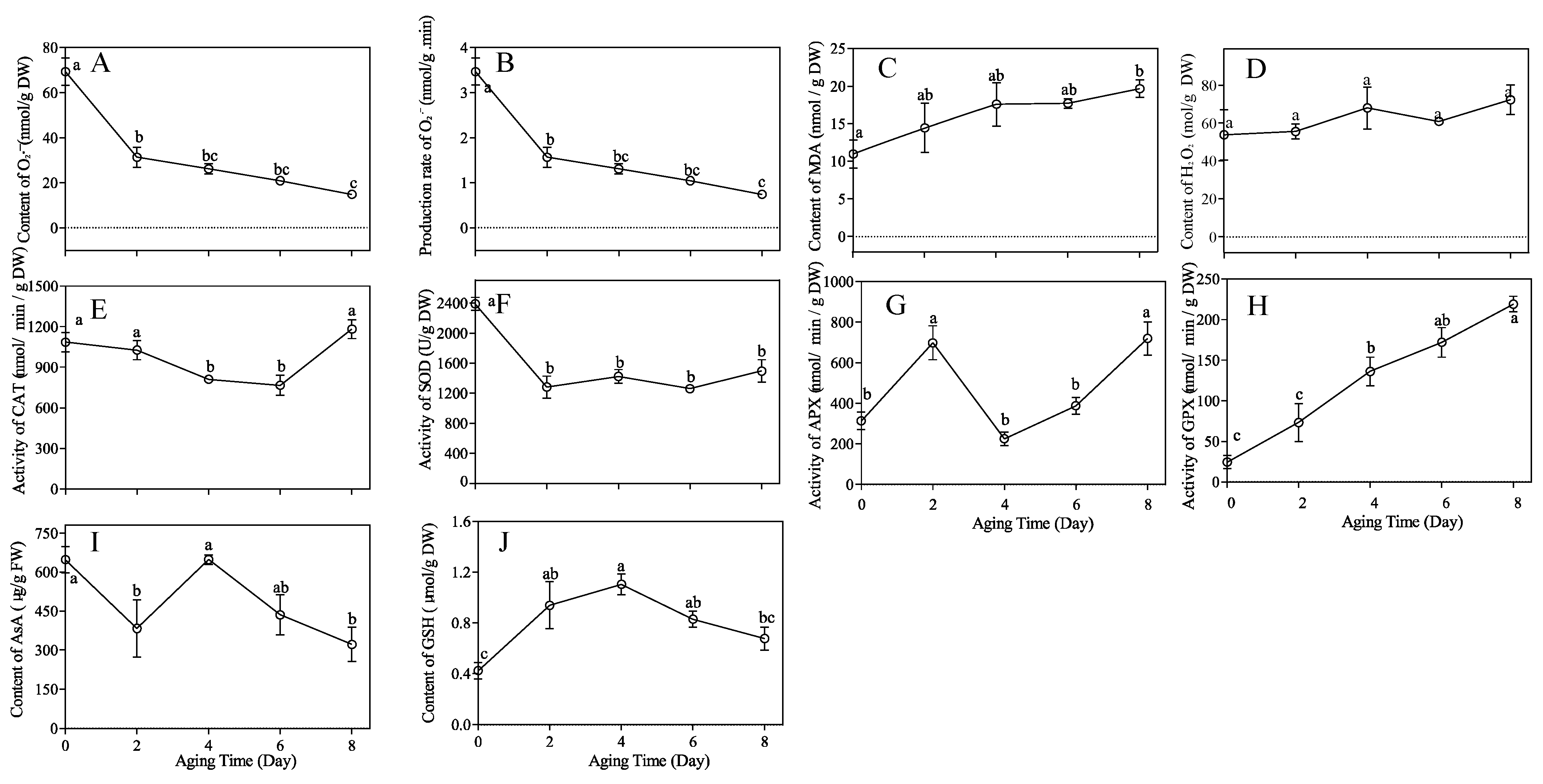
3.1. Germination Percentage and Physiological Changes during Artificial Seed Aging
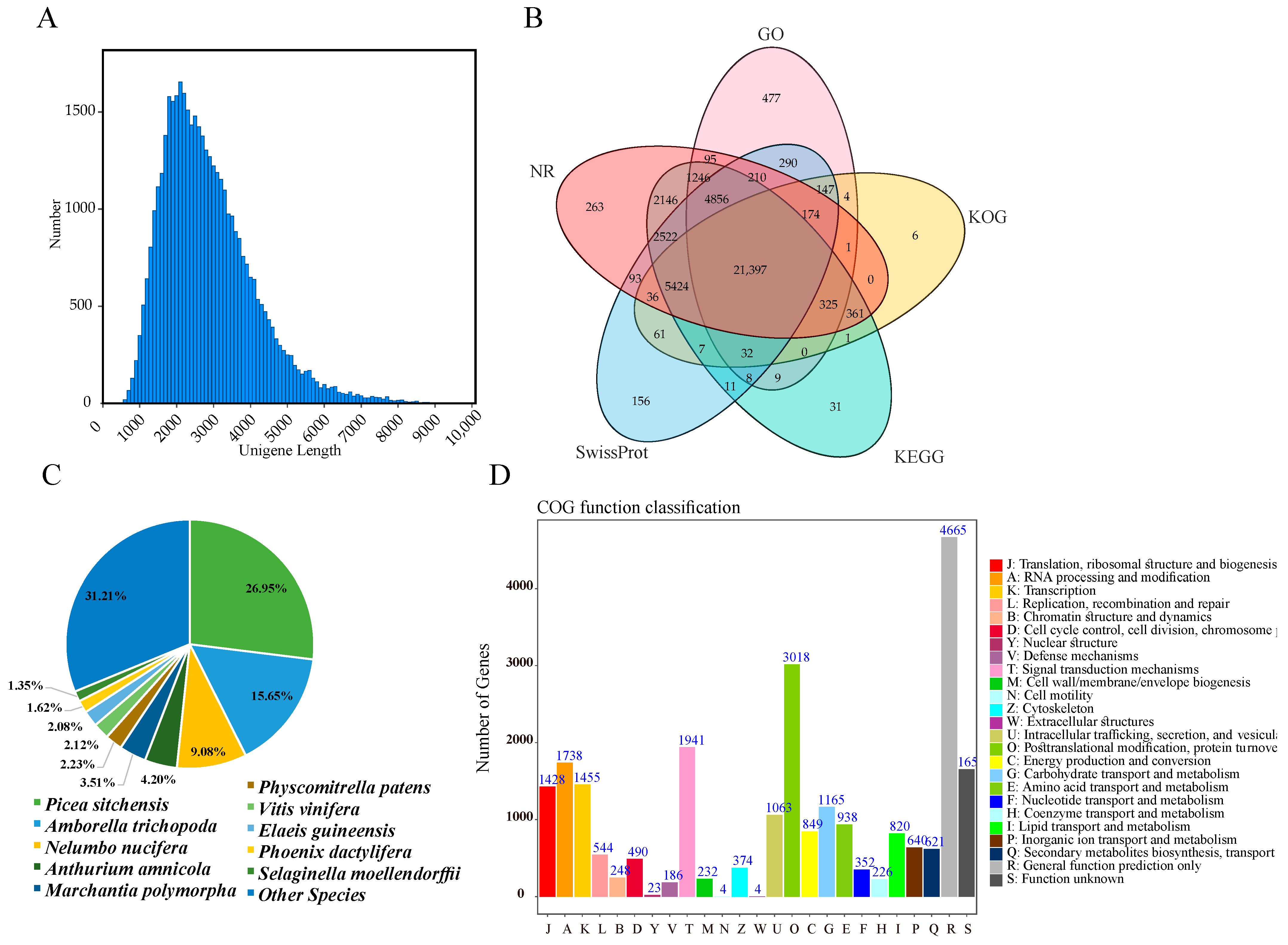
3.2. Functional Annotation of M. glyptostroboides Transcriptome
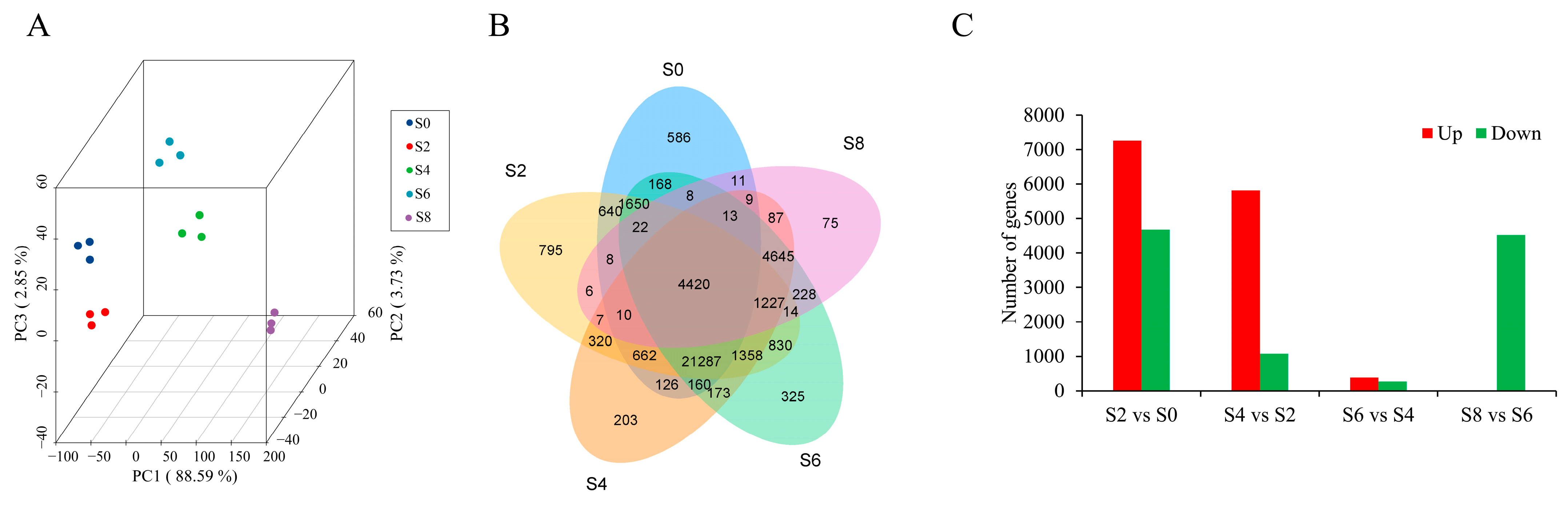
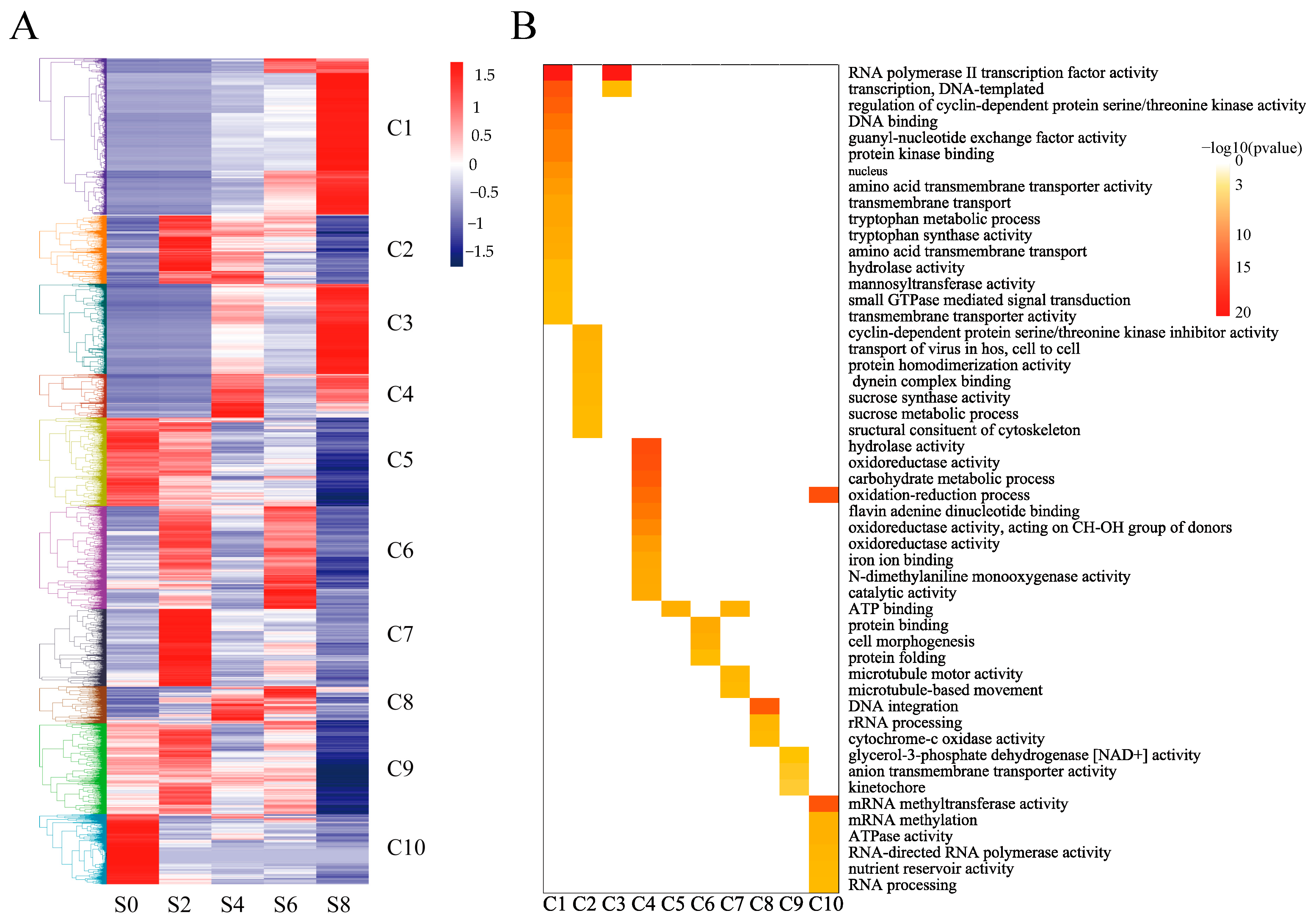
3.3. Global Analysis of the Time-Course Transcriptome Data from Different Samples
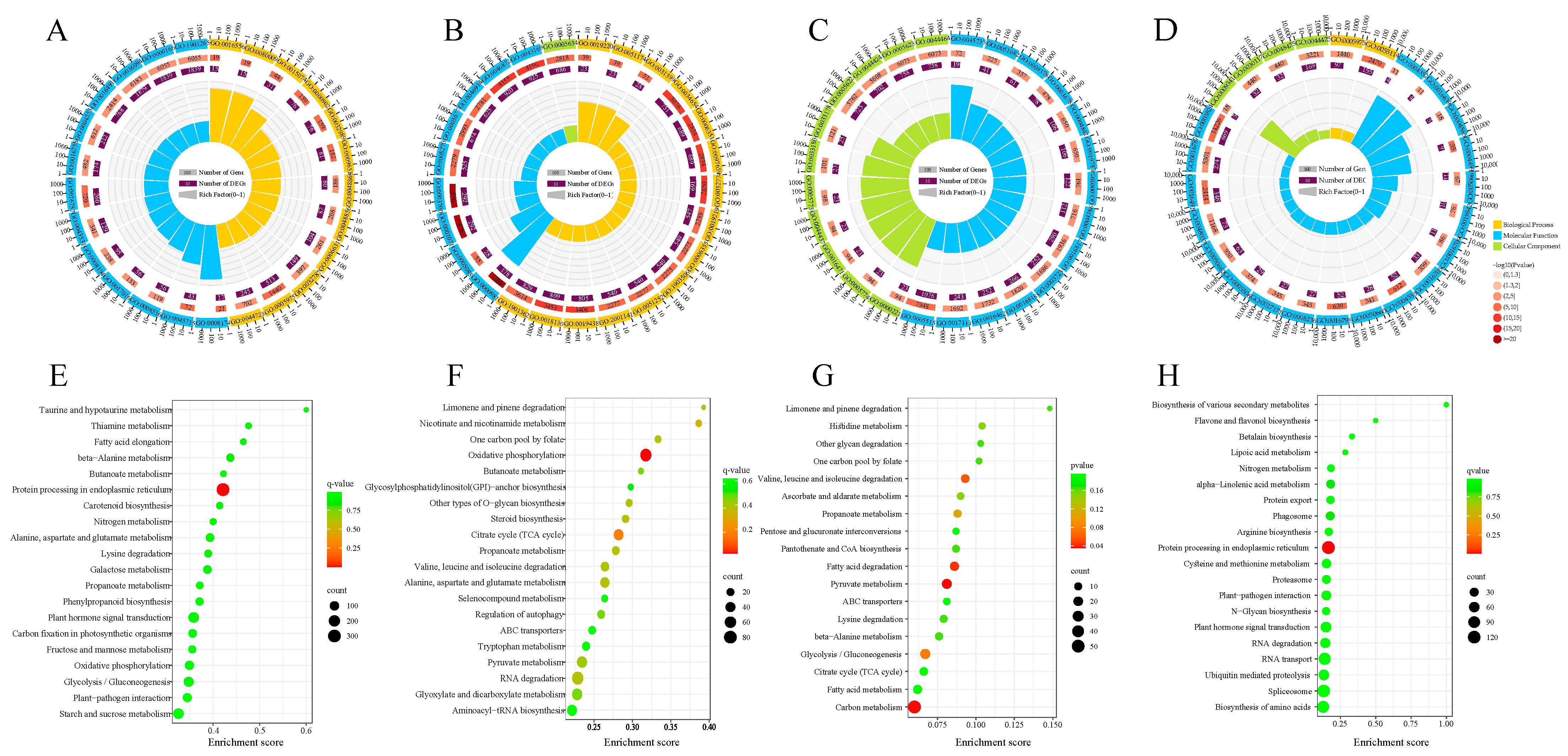
3.4. GO and KEGG Functional Enrichment Analysis
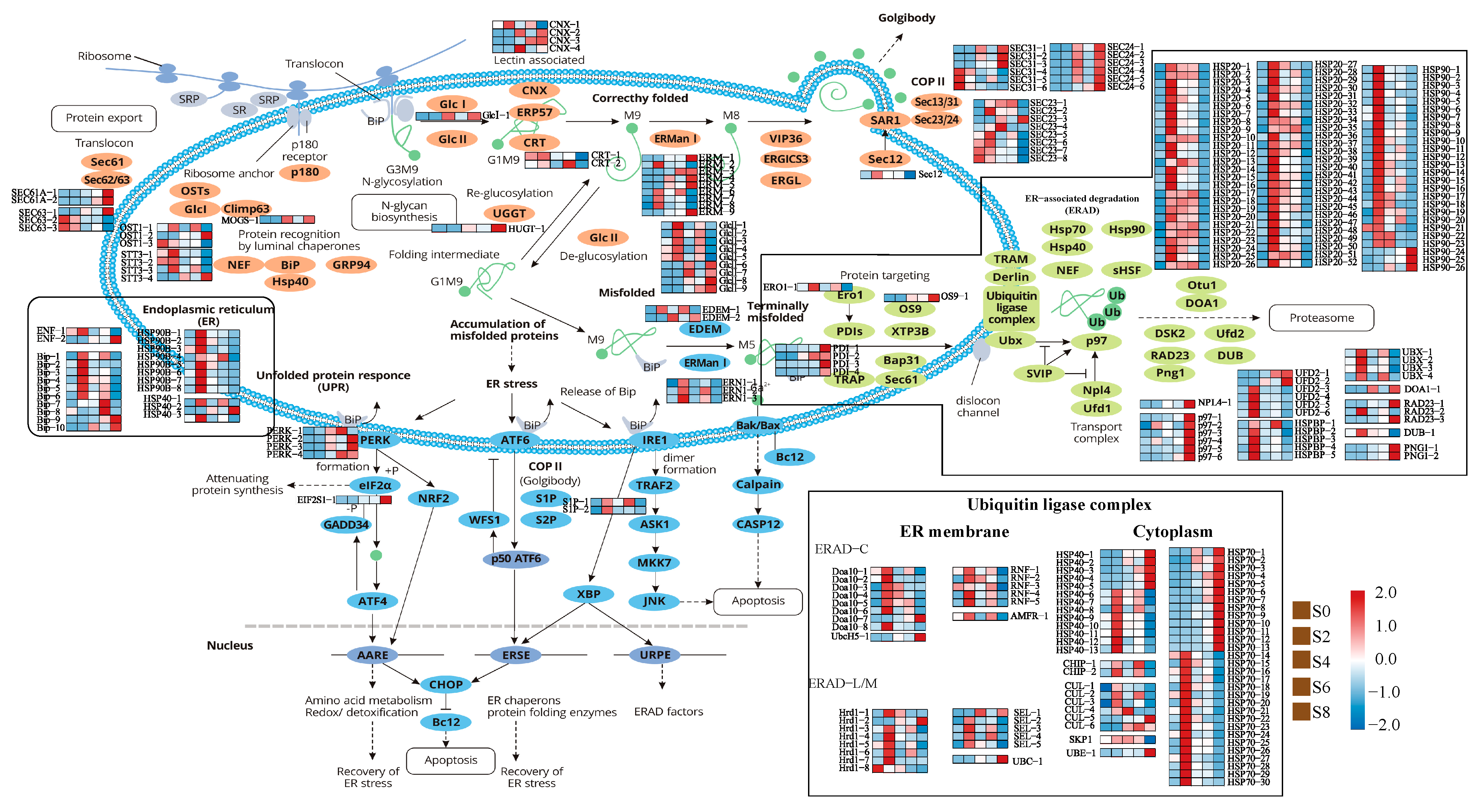
3.5. Expression Analysis of Genes Associated with the Protein Processing in Endoplasmic Reticulum Pathway
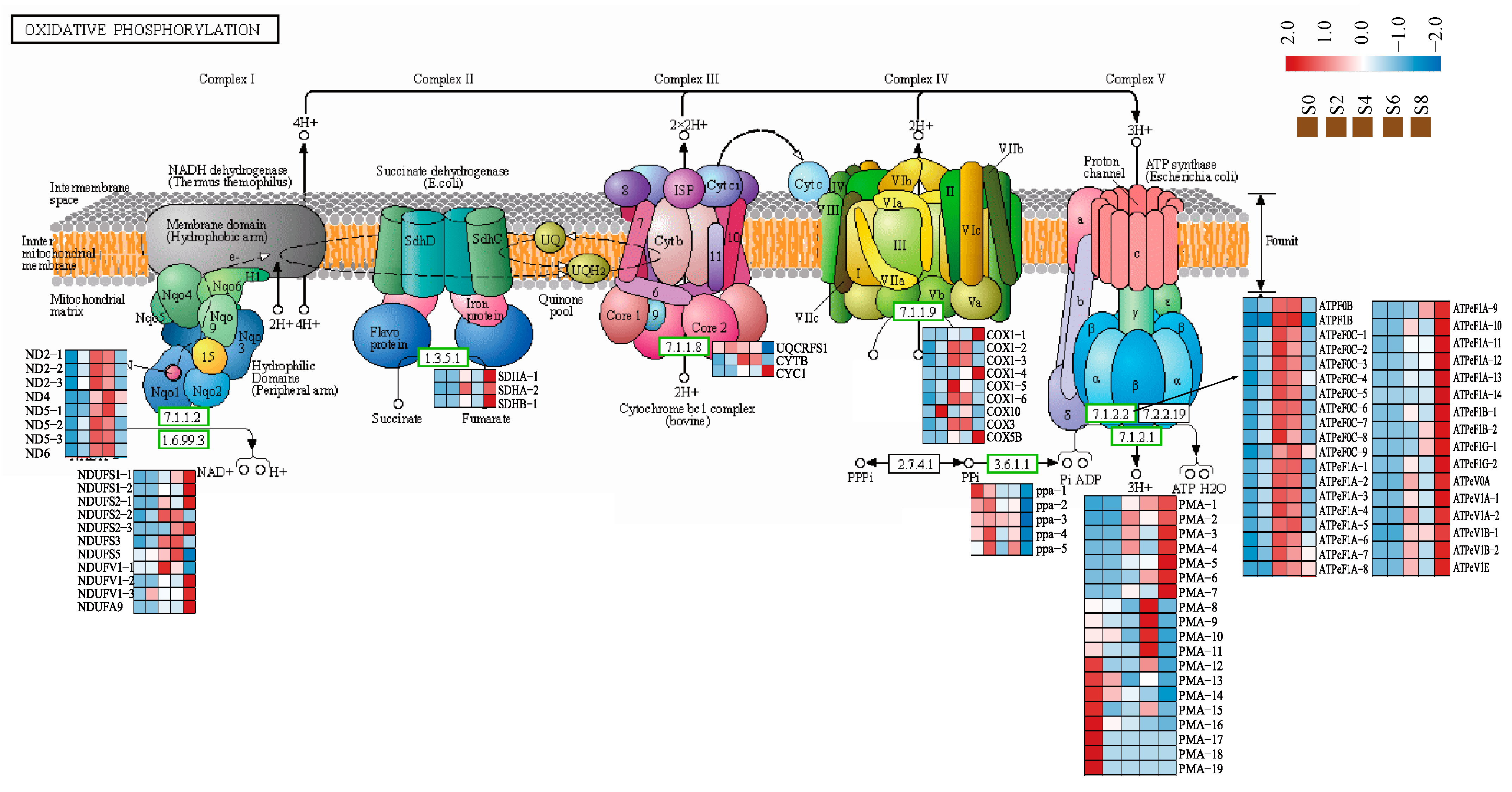
3.6. Expression Analysis of Genes Associated with the Oxidative Phosphorylation Pathway
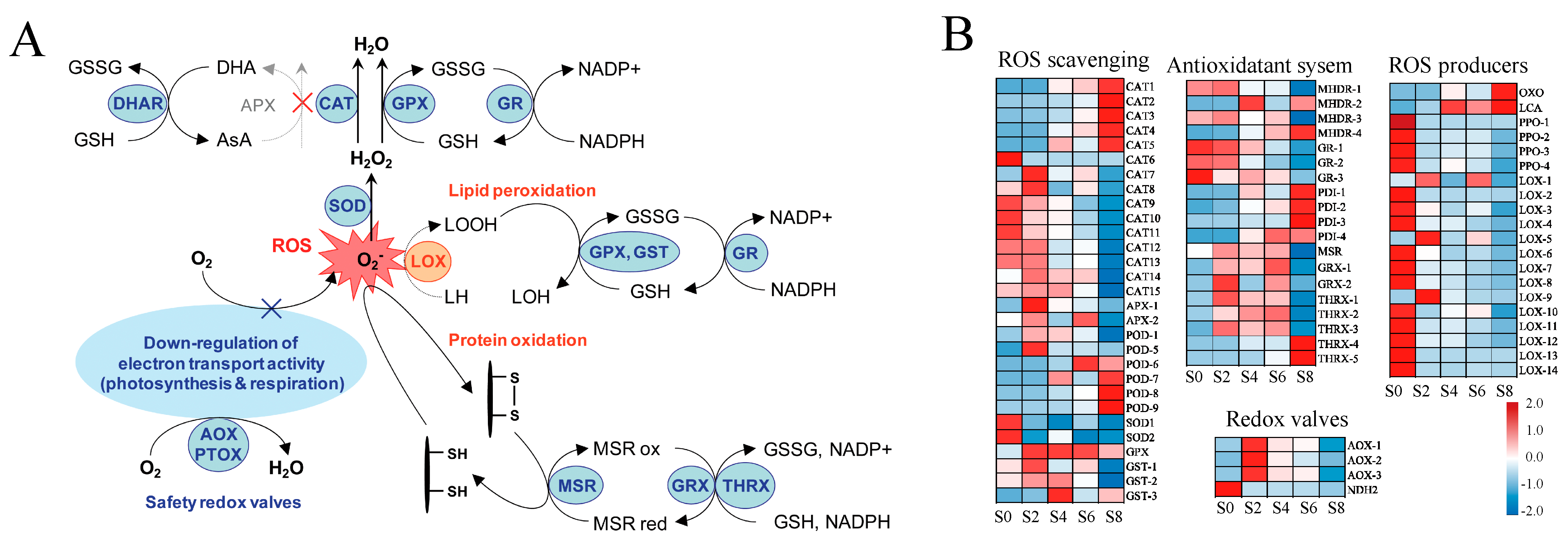
3.7. Effects of Aging Stress on the Antioxidant Responses
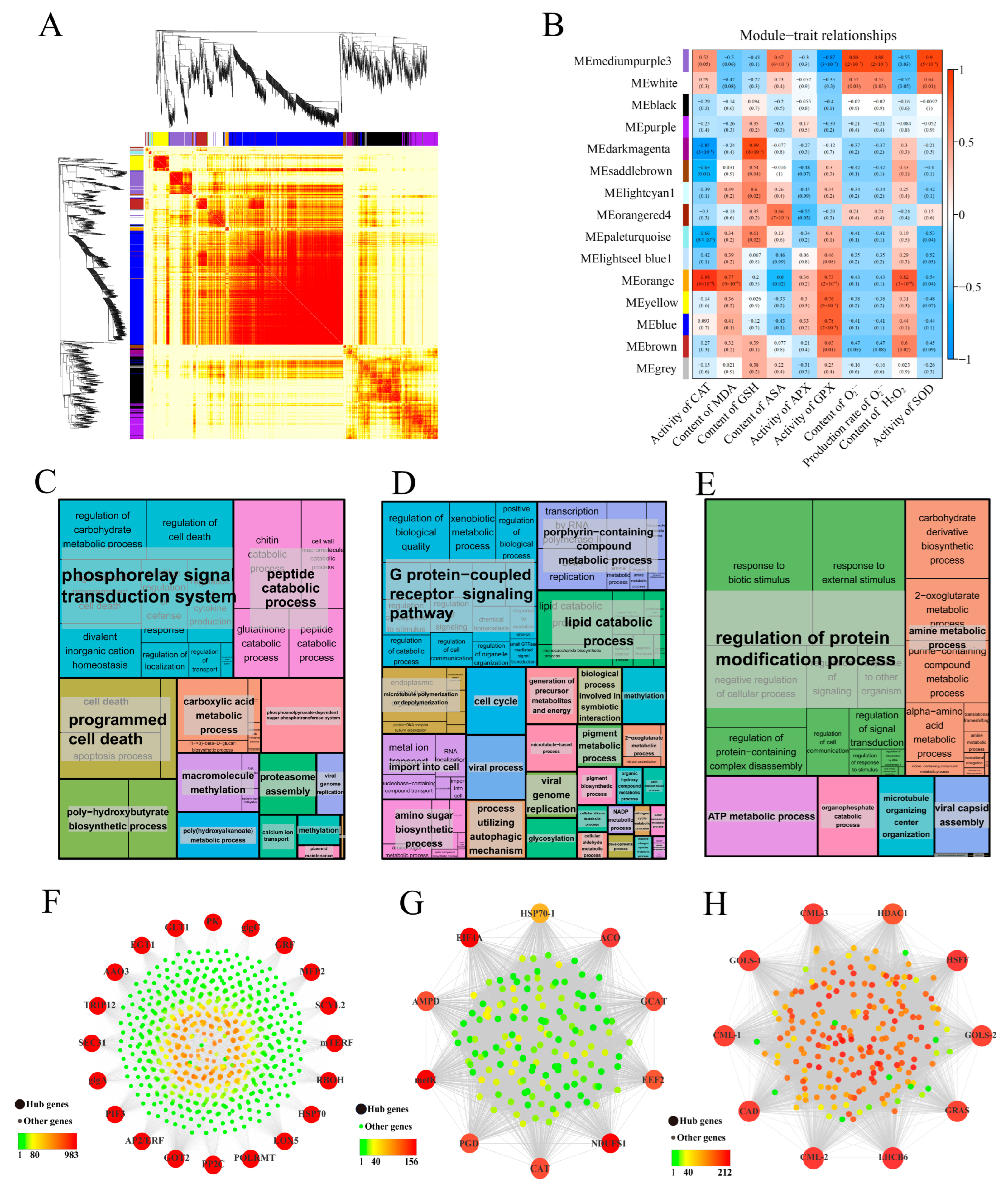
3.8. Co-Expression Network Analysis of DEGs by WGCNA
4. Discussion
4.1. Physiological Parameters of Seed Aging in M. glyptostroboides
4.2. Full-Length Sequences Identified by SMRT Sequencing in M. glyptostroboides Provided Resources for Studies of the Aging Stress Response
4.3. DEGs in Response to Aging Stress
4.4. Identification of Hub-Genes Associated with Scavenging ROS in Seeds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, D.; Hou, H.; Meng, A.; Meng, J.; Xie, L.; Zhang, C. Rapid Evaluation of Seed Vigor by the Absolute Content of Protein in Seed within the Same Crop. Sci. Rep. 2018, 8, 5569. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Audigier, C.; Ladonne, F.; Wagner, M.H.; Coste, F.; Corbineau, F.; Côme, D. Changes in Oligosaccharide Content and Antioxidant Enzyme Activities in Developing Bean Seeds as Related to Acquisition of Drying Tolerance and Seed Quality. J. Exp. Bot. 2001, 52, 701–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Baki, A.A. Biochemical Aspects of Seed Vigor. HortScience 1980, 15, 765–771. [Google Scholar] [CrossRef]
- Bewley, J.D.; Black, M. Seeds: Physiology of Development and Germination; Springer: Boston, MA, USA, 1985; ISBN 978-1-4613-5703-2. [Google Scholar]
- Wojtyla, Ł.; Lechowska, K.; Kubala, S.; Garnczarska, M. Different Modes of Hydrogen Peroxide Action During Seed Germination. Front. Plant Sci. 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kijowska-Oberc, J.; Staszak, A.M.; Ratajczak, E. Climate Change Affects Seed Aging? Initiation Mechanism and Consequences of Loss of Forest Tree Seed Viability. Trees 2021, 35, 1099–1108. [Google Scholar] [CrossRef]
- Ciacka, K.; Krasuska, U.; Staszek, P.; Wal, A.; Zak, J.; Gniazdowska, A. Effect of Nitrogen Reactive Compounds on Aging in Seed. Front. Plant Sci. 2020, 11, 1011. [Google Scholar] [CrossRef]
- Luo, Y.; Le, J.; Zhang, Y.; Wang, R.; Li, Q.; Lu, X.; Liu, J.; Deng, Z. Identification and Functional Analysis of LncRNAs in Response to Seed Aging in Metasequoia Glyptostroboides by Third Generation Sequencing Technology. Forests 2022, 13, 1579. [Google Scholar] [CrossRef]
- Gerna, D.; Ballesteros, D.; Arc, E.; Stöggl, W.; Seal, C.E.; Marami-Zonouz, N.; Na, C.S.; Kranner, I.; Roach, T. Does Oxygen Affect Ageing Mechanisms of Pinus Densiflora Seeds? A Matter of Cytoplasmic Physical State. J. Exp. Bot. 2022, 73, 2631–2649. [Google Scholar] [CrossRef]
- Roach, T.; Nagel, M.; Börner, A.; Eberle, C.; Kranner, I. Changes in Tocochromanols and Glutathione Reveal Differences in the Mechanisms of Seed Ageing under Seedbank Conditions and Controlled Deterioration in Barley. Environ. Exp. Bot. 2018, 156, 8–15. [Google Scholar] [CrossRef]
- Wang, W.-Q.; Xu, D.-Y.; Sui, Y.-P.; Ding, X.-H.; Song, X.-J. A Multiomic Study Uncovers a BZIP23-PER1A-Mediated Detoxification Pathway to Enhance Seed Vigor in Rice. Proc. Natl. Acad. Sci. USA 2022, 119, e2026355119. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, F.; Zhang, Q.; Luo, Y.; Liu, Q.; Gao, J.; Liu, J.; Chen, G.; Zhang, H. Identification and Functional Analysis of Long Non-Coding RNA (LncRNA) in Response to Seed Aging in Rice. Plants 2022, 11, 3223. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hou, D.; Li, Y.; Chao, H.; Zhang, K.; Wang, H.; Xiang, J.; Raboanatahiry, N.; Wang, B.; Li, M. Integration of Proteomic and Genomic Approaches to Dissect Seed Germination Vigor in Brassica Napus Seeds Differing in Oil Content. BMC Plant Biol. 2019, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Raquid, R.; Kohli, A.; Reinke, R.; Dionisio-Sese, M.; Kwak, J.; Chebotarov, D.; Mo, Y.; Lee, J.-S. Genetic Factors Enhancing Seed Longevity in Tropical Japonica Rice. Curr. Plant Biol. 2021, 26, 100196. [Google Scholar] [CrossRef]
- Shin, J.-H.; Kim, S.-R.; An, G. Rice Aldehyde Dehydrogenase7 Is Needed for Seed Maturation and Viability. Plant Physiol. 2009, 149, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wei, Y.; Zhu, Y.; Lian, L.; Xie, H.; Cai, Q.; Chen, Q.; Lin, Z.; Wang, Z.; Xie, H.; et al. Antisense Suppression of LOX3 Gene Expression in Rice Endosperm Enhances Seed Longevity. Plant Biotechnol. J. 2015, 13, 526–539. [Google Scholar] [CrossRef]
- Yuan, Z.; Fan, K.; Wang, Y.; Tian, L.; Zhang, C.; Sun, W.; He, H.; Yu, S. OsGRETCHENHAGEN3-2 Modulates Rice Seed Storability via Accumulation of Abscisic Acid and Protective Substances. Plant Physiol. 2021, 186, 469–482. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, H.; Diao, L.; Zhu, Y.; Xie, H.; Cai, Q.; Wu, F.; Wang, Z.; Zhang, J.; Xie, H. Protein Repair L-Isoaspartyl Methyltransferase 1 (PIMT1) in Rice Improves Seed Longevity by Preserving Embryo Vigor and Viability. Plant Mol. Biol. 2015, 89, 475–492. [Google Scholar] [CrossRef]
- Kaur, H.; Petla, B.; Kamble, N.; Singh, A.; Rao, V.; Salvi, P.; Ghosh, S.; Majee, M. Differentially Expressed Seed Aging Responsive Heat Shock Protein OsHSP18.2 Implicates in Seed Vigor, Longevity and Improves Germination and Seedling Establishment under Abiotic Stress. Front. Plant Sci. 2015, 6, 713. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.-J.; Tao, Y.; Wang, S.; Ma, H.-Y.; Song, L.-R.; Yu, X.-W.; Ma, H. Research Progress on Seed Vigor Biology of Higher Plant. Acta Bot. Boreali Occident. Sin. 2013, 33, 1709–1716. [Google Scholar]
- Ma, J.-S. A Survey of Metasequoia Cultivated Worldwide (1947–2007). J. Wuhan Bot. Res. 2008, 186–196. Available online: https://en.cnki.com.cn/Article_en/CJFDTOTAL-WZXY200802015.htm (accessed on 15 February 2023).
- Lin, Y.; Ai, X.-R.; Yao, L.; Guo, Q.-J.; Zhang, M.-X.; Chne, J. Population structure and dynamics of Metasequoia glyptostroboides parent trees. Chin. J. Ecol. 2017, 36, 1531–1538. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Kang, S.C. Antifungal Activity of Leaf Essential Oil and Extracts of Metasequoia Glyptostroboides Miki Ex Hu. J. Am. Oil Chem. Soc. 2010, 87, 327–336. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Tsang, E.P.K.; Cui, M.-Y.; Chen, X.-Y. Too Early to Call It Success: An Evaluation of the Natural Regeneration of the Endangered Metasequoia Glyptostroboides. Biol. Conserv. 2012, 150, 1–4. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Y.; Liu, X.; Jiang, Y.; Deng, S.; Ai, X.; Deng, Z. Effect of Artificially Accelerated Aging on the Vigor of Metasequoia Glyptostroboides Seeds. J. For. Res. 2020, 31, 769–779. [Google Scholar] [CrossRef]
- Wang, B.; Kumar, V.; Olson, A.; Ware, D. Reviving the Transcriptome Studies: An Insight Into the Emergence of Single-Molecule Transcriptome Sequencing. Front. Genet. 2019, 10, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, L.C.; Garcia, Q.S. Accelerated Ageing and Subsequent Imbibition Affect Seed Viability and the Efficiency of Antioxidant System in Macaw Palm Seeds. Acta Physiol. Plant. 2017, 39, 72. [Google Scholar] [CrossRef]
- Sun, M.; Sun, S.; Mao, C.; Zhang, H.; Ou, C.; Jia, Z.; Wang, Y.; Ma, W.; Li, M.; Jia, S.; et al. Dynamic Responses of Antioxidant and Glyoxalase Systems to Seed Aging Based on Full-Length Transcriptome in Oat (Avena Sativa L.). Antioxidants 2022, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.J.; Cheng, H.Y.; Song, S.Q. Effects of Temperature, Scarification, Dry Storage, Stratification, Phytohormone and Light on Dormancy-Breaking and Germination of Cotinus coggygria Var. cinerea (Anacardiaceae) Seeds. Seed Sci. Technol. 2010, 38, 572–584. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ratajczak, E.; Małecka, A.; Ciereszko, I.; Staszak, A. Mitochondria Are Important Determinants of the Aging of Seeds. Int. J. Mol. Sci. 2019, 20, 1568. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.-N.; Wang, F.-Z.; Yang, C.-H.; Yuan, J.-Z.; Guo, H.; Zhang, J.-L.; Wang, S.-M.; Ma, Q. Transcriptomic Profiling Identifies Candidate Genes Involved in the Salt Tolerance of the Xerophyte Pugionium Cornutum. Genes 2019, 10, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Cheng, L.-L.; Xia, F.-S.; Yan, H.-F.; Mao, P.-S. Advances in the molecular biology study of seed aging. Pratacultural Sci. 2017, 34, 129–137. [Google Scholar]
- Wu, M.-L. Study on Barriers and Regulation of Natural Regeneration Seed Reproduction in Native Population of Metasequoia glyptostroboides. Master’s Thesis, Hubei University for Nationalities, Enshi, China, 2020. Available online: https://wap.cnki.net/touch/web/Dissertation/Article/10517-1020632069.nh.html (accessed on 15 February 2023).
- Li, S.-X.; Gao, Y.-Y.; Li, Y.-H.; Tian, S.-X. Methold of Measuring Seed Water Content and It’s Expectation. Seed 2010, 29, 57–59+61. [Google Scholar] [CrossRef]
- Liu, H. Effects of Accelerated Aging and Simulated Drought and Salt Stress on Seed Viability of Metasequoia glyptostroboides. Master’s Thesis, Hubei University for Nationalities, Enshi, China, 2019. Available online: https://kns.cnki.net/kns8/defaultresult/index (accessed on 15 February 2023).
- Han, Q.; Zhou, Y.-F.; Gao, Y.-M.; Zhang, Z.; Huang, R.-D. Effect of Artificial Accelerated Aging on Germination and Physiological and Biochemical Characteristics of Sorghum Seeds. Seed 2015, 34, 9–12+16. [Google Scholar] [CrossRef]
- Fang, J.-Y.; Zhu, Y.; Wang, C.-Y.; Ye, K.-K.; Gao, W.-D.; Zhang, Z.-H.; Yan, J.-J.; Li, Q.-M. Physiological and Biochemical Changes of Toona sinensis Seeds During Artificial Aging. For. Res. 2020, 33, 163–169. [Google Scholar] [CrossRef]
- Yu, P.-L. Effects of Artificial Aging and Induction on Physiological Characteristics of Seeds of Pyrus betulaefoia Bge. Master’s Thesis, Hubei University for Nationalities, Enshi, China, 2019. Available online: https://kns.cnki.net/kns8/defaultresult/index (accessed on 15 February 2023).
- Zhang, Z.; Jin, H.; Suo, J.; Yu, W.; Zhou, M.; Dai, W.; Song, L.; Hu, Y.; Wu, J. Effect of Temperature and Humidity on Oil Quality of Harvested Torreya grandis Cv. Merrillii Nuts During the After-Ripening Stage. Front. Plant Sci. 2020, 11, 573681. [Google Scholar] [CrossRef] [PubMed]
- Morscher, F.; Kranner, I.; Arc, E.; Bailly, C.; Roach, T. Glutathione Redox State, Tocochromanols, Fatty Acids, Antioxidant Enzymes and Protein Carbonylation in Sunflower Seed Embryos Associated with after-Ripening and Ageing. Ann. Bot. 2015, 116, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Jisha, K.C.; Vijayakumari, K.; Puthur, J.T. Seed Priming for Abiotic Stress Tolerance: An Overview. Acta Physiol. Plant. 2013, 35, 1381–1396. [Google Scholar] [CrossRef]
- Reed, R.C.; Bradford, K.J.; Khanday, I. Seed Germination and Vigor: Ensuring Crop Sustainability in a Changing Climate. Heredity 2022, 128, 450–459. [Google Scholar] [CrossRef]
- Kurek, K.; Plitta-Michalak, B.; Ratajczak, E. Reactive Oxygen Species as Potential Drivers of the Seed Aging Process. Plants 2019, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Yin, G.; Xin, X.; Song, C.; Chen, X.; Zhang, J.; Wu, S.; Li, R.; Liu, X.; Lu, X. Activity Levels and Expression of Antioxidant Enzymes in the Ascorbate-Glutathione Cycle in Artificially Aged Rice Seed. Plant Physiol. Biochem. 2014, 80, 1–9. [Google Scholar] [CrossRef]
- Xia, F.; Wang, X.; Li, M.; Mao, P. Mitochondrial Structural and Antioxidant System Responses to Aging in Oat (Avena sativa L.) Seeds with Different Moisture Contents. Plant Physiol. Biochem. 2015, 94, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Ebone, L.A.; Caverzan, A.; Silveira, D.C.; de Oliveira Siqueira, L.; Lângaro, N.C.; Chiomento, J.L.T.; Chavarria, G. Biochemical Profile of the Soybean Seed Embryonic Axis and Its Changes during Accelerated Aging. Biology 2020, 9, 186. [Google Scholar] [CrossRef]
- Mittler, R. Oxidative Stress, Antioxidants and Stress Tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Ratajczak, E.; Małecka, A.; Bagniewska-Zadworna, A.; Kalemba, E.M. The Production, Localization and Spreading of Reactive Oxygen Species Contributes to the Low Vitality of Long-Term Stored Common Beech (Fagus sylvatica L.) Seeds. J. Plant Physiol. 2015, 174, 147–156. [Google Scholar] [CrossRef]
- Kong, L.; Huo, H.; Mao, P. Antioxidant Response and Related Gene Expression in Aged Oat Seed. Front. Plant Sci. 2015, 6, 158. [Google Scholar] [CrossRef]
- Rasheed, A.; Rasool, S.G.; Gul, B.; Ajmal Khan, M.; Hameed, A. Reactive Oxygen Species Production and Scavenging During Seed Germination of Halophytes. In Ecophysiology, Abiotic Stress Responses and Utilization of Halophytes; Hasanuzzaman, M., Nahar, K., Öztürk, M., Eds.; Springer: Singapore, 2019; pp. 63–81. ISBN 9789811337611. [Google Scholar]
- Fu, Y.-B.; Ahmed, Z.; Diederichsen, A. Towards a Better Monitoring of Seed Ageing under Ex Situ Seed Conservation. Conserv. Physiol. 2015, 3, cov026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaweł, S.; Wardas, M.; Niedworok, E.; Wardas, P. Malondialdehyde (MDA) as a lipid peroxidation marker. Wiad. Lek. 2004, 57, 453–455. [Google Scholar] [PubMed]
- Tsikas, D. Assessment of Lipid Peroxidation by Measuring Malondialdehyde (MDA) and Relatives in Biological Samples: Analytical and Biological Challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lei, B.; Zhai, M.-H.; Wang, L.; Zhang, J.-G.; Zhou, X.-Y.; Liang, J. Study on the Response Mechanism of the AsA-GSH Cycle in Cotton Seedling Under Low Temperature Stress. J. Nucl. Agric. Sci. 2021, 35, 221–228. [Google Scholar]
- Yuan, H.; Yu, H.; Huang, T.; Shen, X.; Xia, J.; Pang, F.; Wang, J.; Zhao, M. The Complexity of the Fragaria x Ananassa (Octoploid) Transcriptome by Single-Molecule Long-Read Sequencing. Hortic. Res. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; den Dunnen, J.T.; et al. Full-Length MRNA Sequencing Uncovers a Widespread Coupling between Transcription Initiation and MRNA Processing. Genome Biol. 2018, 19, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, C.D.; Springer, N.M.; Hirsch, C.N. Genomic Limitations to RNA Sequencing Expression Profiling. Plant J. 2015, 84, 491–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global Identification of Alternative Splicing via Comparative Analysis of SMRT- and Illumina-Based RNA-Seq in Strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Yuan, J.; Guo, T.; Xu, L.; Mu, Z.; Han, L. Analysis of Transcripts and Splice Isoforms in Medicago sativa L. by Single-Molecule Long-Read Sequencing. Plant Mol. Biol. 2019, 99, 219–235. [Google Scholar] [CrossRef]
- Jia, X.; Tang, L.; Mei, X.; Liu, H.; Luo, H.; Deng, Y.; Su, J. Single-Molecule Long-Read Sequencing of the Full-Length Transcriptome of Rhododendron lapponicum L. Sci. Rep. 2020, 10, 6755. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Luo, L.; Xu, X.-H.; Yang, K.; Li, Z.; Zhang, X.-Q. Senescence and heat shock protein in plants in response to abiotic stressSenescence and heat shock protein in plants in response to abiotic stress. Pratacultural Sci. 2020, 37, 2320–2333. [Google Scholar]
- Gao, J.; Fu, H.; Zhou, X.; Chen, Z.; Luo, Y.; Cui, B.; Chen, G.; Liu, J. Comparative Proteomic Analysis of Seed Embryo Proteins Associated with Seed Storability in Rice (Oryza sativa L.) during Natural Aging. Plant Physiol. Biochem. 2016, 103, 31–44. [Google Scholar] [CrossRef]
- Wang, T.; Hou, L.; Jian, H.; Di, F.; Li, J.; Liu, L. Combined QTL Mapping, Physiological and Transcriptomic Analyses to Identify Candidate Genes Involved in Brassica Napus Seed Aging. Mol. Genet. Genom. 2018, 293, 1421–1435. [Google Scholar] [CrossRef]
- Su, X.; Xin, L.; Li, Z.; Zheng, H.; Mao, J.; Yang, Q. Physiology and Transcriptome Analyses Reveal a Protective Effect of the Radical Scavenger Melatonin in Aging Maize Seeds. Free. Radic. Res. 2018, 52, 1094–1109. [Google Scholar] [CrossRef]
- Yang, Z.; Zheng, H.; Wei, X.; Song, J.; Wang, B.; Sui, N. Transcriptome Analysis of Sweet Sorghum Inbred Lines Differing in Salt Tolerance Provides Novel Insights into Salt Exclusion by Roots. Plant Soil 2018, 430, 423–439. [Google Scholar] [CrossRef]
- Han, G.; Yuan, F.; Guo, J.; Zhang, Y.; Sui, N.; Wang, B. AtSIZ1 Improves Salt Tolerance by Maintaining Ionic Homeostasis and Osmotic Balance in Arabidopsis. Plant Sci. 2019, 285, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Navrot, N.; Rouhier, N.; Gelhaye, E.; Jacquot, J.-P. Reactive Oxygen Species Generation and Antioxidant Systems in Plant Mitochondria. Physiol. Plant. 2007, 129, 185–195. [Google Scholar] [CrossRef]
- Jin, Z.-Y.; Xu, T.-Q.; Zhang, Y.; Zhang, K.-Y. Review of the Relationship between Mitochondria and Seed Aging. Mol. Plant Breed. 2021, 19, 1687–1691. [Google Scholar] [CrossRef]
- Talla, S.; Riazunnisa, K.; Padmavathi, L.; Sunil, B.; Rajsheel, P.; Raghavendra, A.S. Ascorbic Acid Is a Key Participant during the Interactions between Chloroplasts and Mitochondria to Optimize Photosynthesis and Protect against Photoinhibition. J. Biosci. 2011, 36, 163–173. [Google Scholar] [CrossRef]
- Schwarzländer, M.; Finkemeier, I. Mitochondrial Energy and Redox Signaling in Plants. Antioxid. Redox Signal. 2013, 18, 2122–2144. [Google Scholar] [CrossRef]
- Chew, O.; Whelan, J.; Millar, A.H. Molecular Definition of the Ascorbate-Glutathione Cycle in Arabidopsis Mitochondria Reveals Dual Targeting of Antioxidant Defenses in Plants. J. Biol. Chem. 2003, 278, 46869–46877. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox Regulation of Mitochondrial Function with Emphasis on Cysteine Oxidation Reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-Q.; Cheng, H.-Y.; Møller, I.M.; Song, S.-Q. The Role of Recovery of Mitochondrial Structure and Function in Desiccation Tolerance of Pea Seeds. Physiol. Plant. 2012, 144, 20–34. [Google Scholar] [CrossRef]
- Hong, Y.; Zhao, J.; Guo, L.; Kim, S.-C.; Deng, X.; Wang, G.; Zhang, G.; Li, M.; Wang, X. Plant Phospholipases D and C and Their Diverse Functions in Stress Responses. Prog. Lipid Res. 2016, 62, 55–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Osuna, D.; Colville, L.; Lorenzo, O.; Graeber, K.; Küster, H.; Leubner-Metzger, G.; Kranner, I. Transcriptome-Wide Mapping of Pea Seed Ageing Reveals a Pivotal Role for Genes Related to Oxidative Stress and Programmed Cell Death. PLoS ONE 2013, 8, e78471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisa, S.; Domingo, B.; Martínez, J.; Gilch, S.; Llopis, J.F.; Schätzl, H.M.; Gasset, M. Failure of Prion Protein Oxidative Folding Guides the Formation of Toxic Transmembrane Forms. J. Biol. Chem. 2017, 292, 20045. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.P.; Weissman, J.S. Oxidative Protein Folding in Eukaryotes: Mechanisms and Consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics during Early Phases of ER Stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.-C.; Lin, G.; Xue, M.-J.; Xing, L.-M.; Lv, W.-Z.; Yang, W.-F.; Chen, J.-Y. Responses of Endoplasmic Reticulum Stress-Related Genes in Maize Embryo to Artificial Aging Treatment. Sci. Agric. Sin. 2016, 49, 429–442. [Google Scholar]
- Kamauchi, S.; Nakatani, H.; Nakano, C.; Urade, R. Gene Expression in Response to Endoplasmic Reticulum Stress in Arabidopsis Thaliana: Unfolded Protein Response Genes in Arabidopsis. FEBS J. 2005, 272, 3461–3476. [Google Scholar] [CrossRef]
- Liu, L.; Cui, F.; Li, Q.; Yin, B.; Zhang, H.; Lin, B.; Wu, Y.; Xia, R.; Tang, S.; Xie, Q. The Endoplasmic Reticulum-Associated Degradation Is Necessary for Plant Salt Tolerance. Cell Res. 2011, 21, 957–969. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Srivastava, R.; Howell, S. Endoplasmic Reticulum (ER) Stress Response and Its Physiological Roles in Plants. Int. J. Mol. Sci. 2013, 14, 8188–8212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, K.K.; Vago, R.; Calanca, V.; Galli, C.; Paganetti, P.; Molinari, M. EDEM Contributes to Maintenance of Protein Folding Efficiency and Secretory Capacity. J. Biol. Chem. 2004, 279, 44600–44605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Liu, Y.; Xia, Y.; Hong, Z.; Li, J. Conserved Endoplasmic Reticulum-Associated Degradation System to Eliminate Mutated Receptor-like Kinases in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Grice, G.L.; Nathan, J.A. The Recognition of Ubiquitinated Proteins by the Proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, X.; Qi, Y.; Lei, Y.; Wang, S.; Hu, H.; Wei, A. Transcriptome and Metabolome Dynamics Explain Aroma Differences between Green and Red Prickly Ash Fruit. Foods 2021, 10, 391. [Google Scholar] [CrossRef]
- Yu, B.; Liu, J.; Wu, D.; Liu, Y.; Cen, W.; Wang, S.; Li, R.; Luo, J. Weighted Gene Coexpression Network Analysis-Based Identification of Key Modules and Hub Genes Associated with Drought Sensitivity in Rice. BMC Plant Biol. 2020, 20, 478. [Google Scholar] [CrossRef]
- Long, W.; Yao, X.; Wang, K.; Sheng, Y.; Lv, L. De Novo Transcriptome Assembly of the Cotyledon of Camellia Oleifera for Discovery of Genes Regulating Seed Germination. BMC Plant Biol. 2022, 22, 265. [Google Scholar] [CrossRef]
- Chapman, J.M.; Muhlemann, J.K.; Gayomba, S.R.; Muday, G.K. RBOH-Dependent ROS Synthesis and ROS Scavenging by Plant Specialized Metabolites To Modulate Plant Development and Stress Responses. Chem. Res. Toxicol. 2019, 32, 370–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, L.; Cui, Z.; Jiang, J.; Chen, C.; Jiang, M. The NADPH Oxidase OsRbohA Increases Salt Tolerance by Modulating K+ Homeostasis in Rice. Crop J. 2022, 10, 1611–1622. [Google Scholar] [CrossRef]
- Takayanagi, K.; Harrington, J.F. Enhancement of Germination Rate of Aged Seeds by Ethylene. Plant Physiol. 1971, 47, 521–524. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.B.; Adams, D.O.; Yang, S.F. 1-Aminocyclopropanecarboxylate Synthase, a Key Enzyme in Ethylene Biosynthesis. Arch. Biochem. Biophys. 1979, 198, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, M.; Li, Z.; Niu, Y.; Jin, Q.; Zhu, B.; Xu, Y. Effects of Ethylene Biosynthesis and Signaling on Oxidative Stress and Antioxidant Defense System in Nelumbo Nucifera G. under Cadmium Exposure. Environ. Sci. Pollut. Res. Int. 2020, 27, 40156–40170. [Google Scholar] [CrossRef]
- Qi, Y.; Wang, H.; Zou, Y.; Liu, C.; Liu, Y.; Wang, Y.; Zhang, W. Over-Expression of Mitochondrial Heat Shock Protein 70 Suppresses Programmed Cell Death in Rice. FEBS Lett. 2011, 585, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Jiang, J. Research progress of small molecule heat shock protein gene family (sHSPs) in plants. Plant Physiol. J. 2017, 53, 943–948. [Google Scholar] [CrossRef]
- Garbuz, D.G. Regulation of heat shock gene expression in response to stress. Mol. Biol. 2017, 51, 400–417. [Google Scholar] [CrossRef]
- Zhang, Q.; Geng, J.; Du, Y.; Zhao, Q.; Zhang, W.; Fang, Q.; Yin, Z.; Li, J.; Yuan, X.; Fan, Y.; et al. Heat Shock Transcription Factor (Hsf) Gene Family in Common Bean (Phaseolus vulgaris): Genome-Wide Identification, Phylogeny, Evolutionary Expansion and Expression Analyses at the Sprout Stage under Abiotic Stress. BMC Plant Biol. 2022, 22, 33. [Google Scholar] [CrossRef]
- Prieto-Dapena, P.; Castaño, R.; Almoguera, C.; Jordano, J. Improved Resistance to Controlled Deterioration in Transgenic Seeds. Plant Physiol. 2006, 142, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-T.; Li, B.; Shang, Z.-L.; Li, X.-Z.; Mu, R.-L.; Sun, D.-Y.; Zhou, R.-G. Calmodulin Is Involved in Heat Shock Signal Transduction in Wheat. Plant Physiol. 2003, 132, 1186–1195. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aging Time (d) | Germination Percentage (%) | Germination Potential (%) | Germination Index (%) | Vigor Index (%) | Root Length (mm) | Seedling Height (mm) | Fresh Weight (mg) | Dry Weight (mg) |
|---|---|---|---|---|---|---|---|---|
| 0 | 58.00 ± 5.71 b | 54 ± 5.29 a | 12 ± 1.32 a | 59.54 ± 7.40 a | 12.79 ± 1.55 a | 36.63 ± 1.08 a | 18.55 ± 0.50 a | 2.01 ± 0.02 a |
| 2 | 70.67 ± 3.68 a | 48 ± 3.46 a | 11.46 ± 0.53 a | 63.16 ± 4.63 a | 17.90 ± 1.15 a | 37.09 ± 0.58 a | 20.13 ± 0.71 a | 2.04 ± 0.06 a |
| 4 | 33.33 ± 4.11 c | 10 ± 2.31 b | 4.64 ± 0.84 b | 22.86 ± 4.63 b | 12.59 ± 1.13 a | 36.39 ± 0.52 a | 20.59 ± 0.54 a | 1.94 ± 0.02 ab |
| 6 | 9.33 ± 1.88 d | 0 c | 1.23 ± 0.33 c | 5.55 ± 1.41 c | 12.03 ± 1.31 a | 33.84 ± 1.12 a | 21.51 ± 1.22 a | 1.82 ± 0.04 b |
| 8 | 0 e | — | — | — | — | — | — | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Zhang, Y.; Le, J.; Li, Q.; Mou, J.; Deng, S.; Li, J.; Wang, R.; Deng, Z.; Liu, J. Full-Length Transcriptome Sequencing Reveals the Molecular Mechanism of Metasequoia glyptostroboides Seed Responding to Aging. Antioxidants 2023, 12, 1353. https://doi.org/10.3390/antiox12071353
Luo Y, Zhang Y, Le J, Li Q, Mou J, Deng S, Li J, Wang R, Deng Z, Liu J. Full-Length Transcriptome Sequencing Reveals the Molecular Mechanism of Metasequoia glyptostroboides Seed Responding to Aging. Antioxidants. 2023; 12(7):1353. https://doi.org/10.3390/antiox12071353
Chicago/Turabian StyleLuo, Yongjian, Yixin Zhang, Jingyu Le, Qing Li, Jiaolin Mou, Shiming Deng, Jitao Li, Ru Wang, Zhijun Deng, and Jun Liu. 2023. "Full-Length Transcriptome Sequencing Reveals the Molecular Mechanism of Metasequoia glyptostroboides Seed Responding to Aging" Antioxidants 12, no. 7: 1353. https://doi.org/10.3390/antiox12071353