
Mammalian Animal and Human Retinal Organ Culture as Pre-Clinical Model to Evaluate Oxidative Stress and Antioxidant Intraocular Therapeutics

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Cells
2.2. Tissue Isolation and Culture Conditions
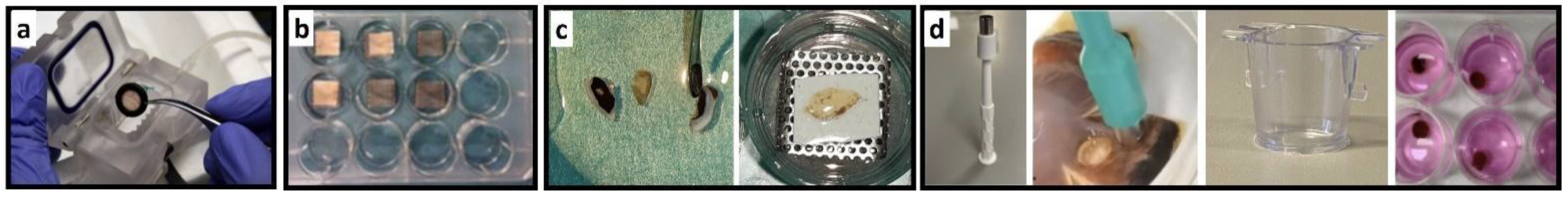
2.2.1. Bovine Retina–RPE Culture (Model 1)
2.2.2. Dynamic Porcine Retina Culture (Model 2)
2.2.3. Static Porcine Retina Culture (Model 3)
2.2.4. Rat Retina Culture (Model 4)
2.2.5. Semi-Long Porcine and Human Retina Culture (Model 5a and 5b)
2.2.6. Summary of Retina Models
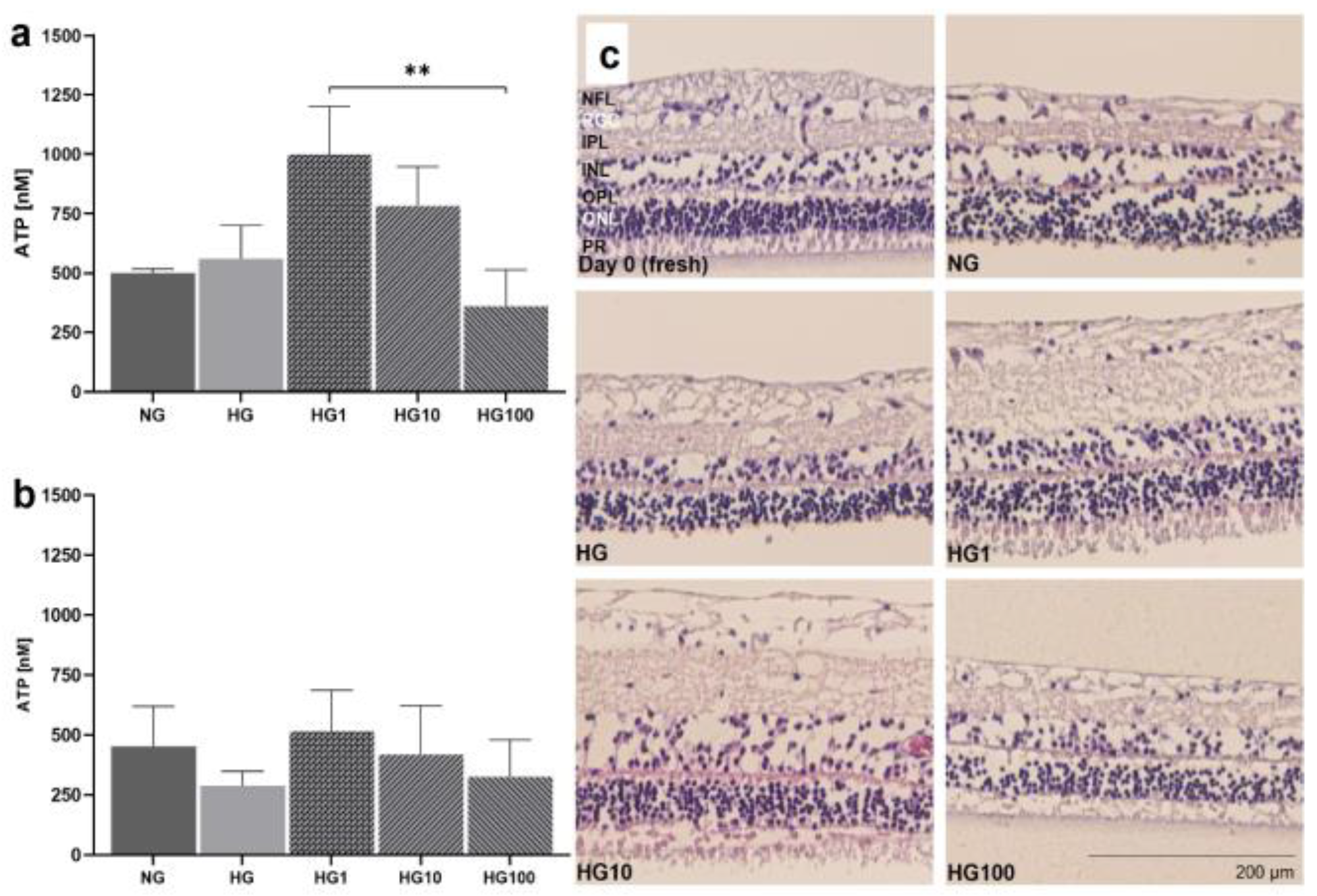
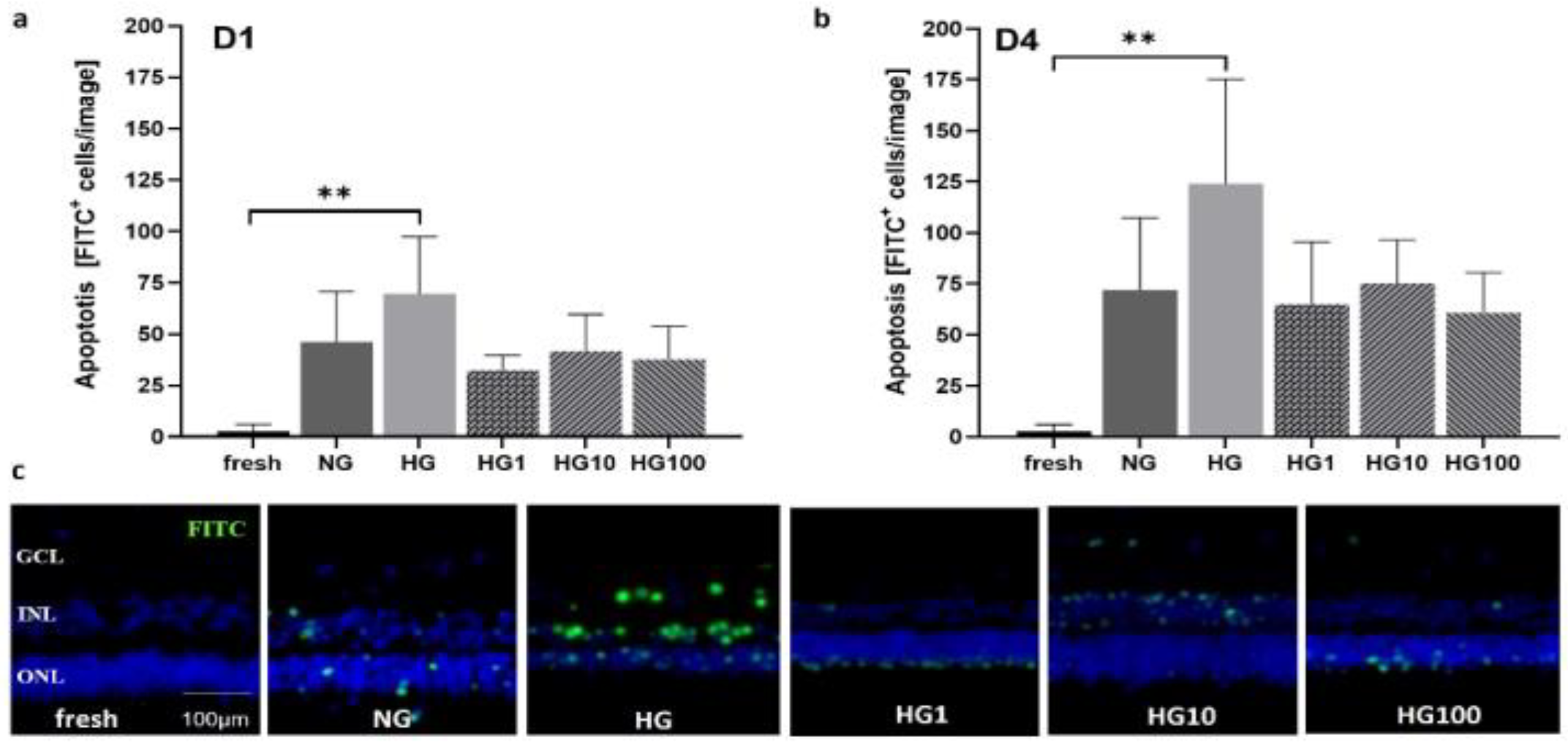
2.2.7. Induction of OS by High-Glucose Conditions and Scutellarin Treatment
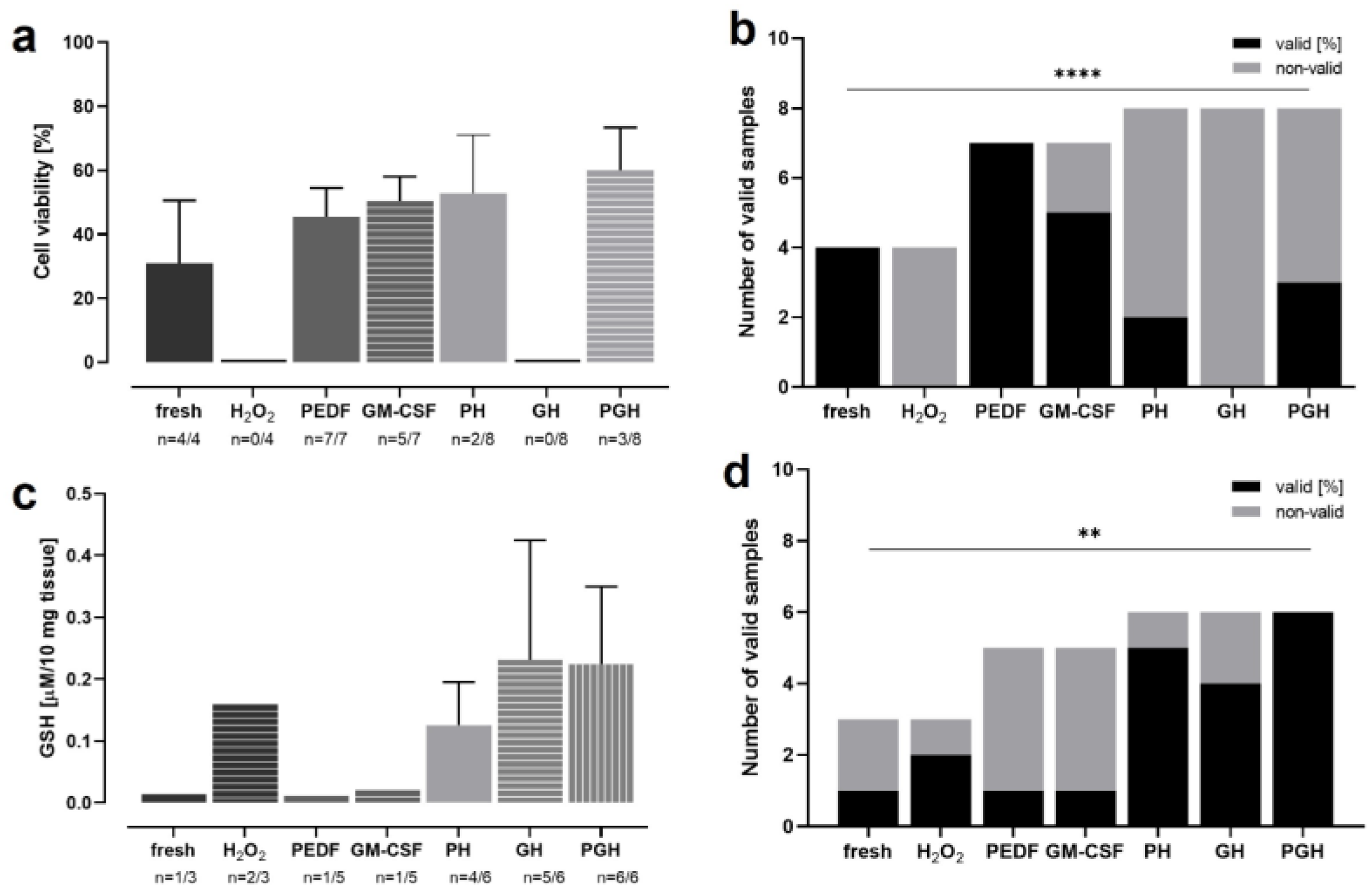
2.2.8. Induction of OS by H2O2 Incubation and PEDF/GM-CSF Treatment
2.3. Brightfield Microscopy of Flatmount Preparations and Degeneration Score
2.4. Hemalum and Eosin (H&E) Staining
2.5. Cell death and Viability Determination
2.5.1. CytoTox GloTM Assay
2.5.2. CellTiter Glo® Viability Assay
2.5.3. Propidium Iodide (PI) Staining
2.5.4. TUNEL Assay
2.6. GSH Assay
2.7. Immunohistology
2.8. Statistics
3. Results
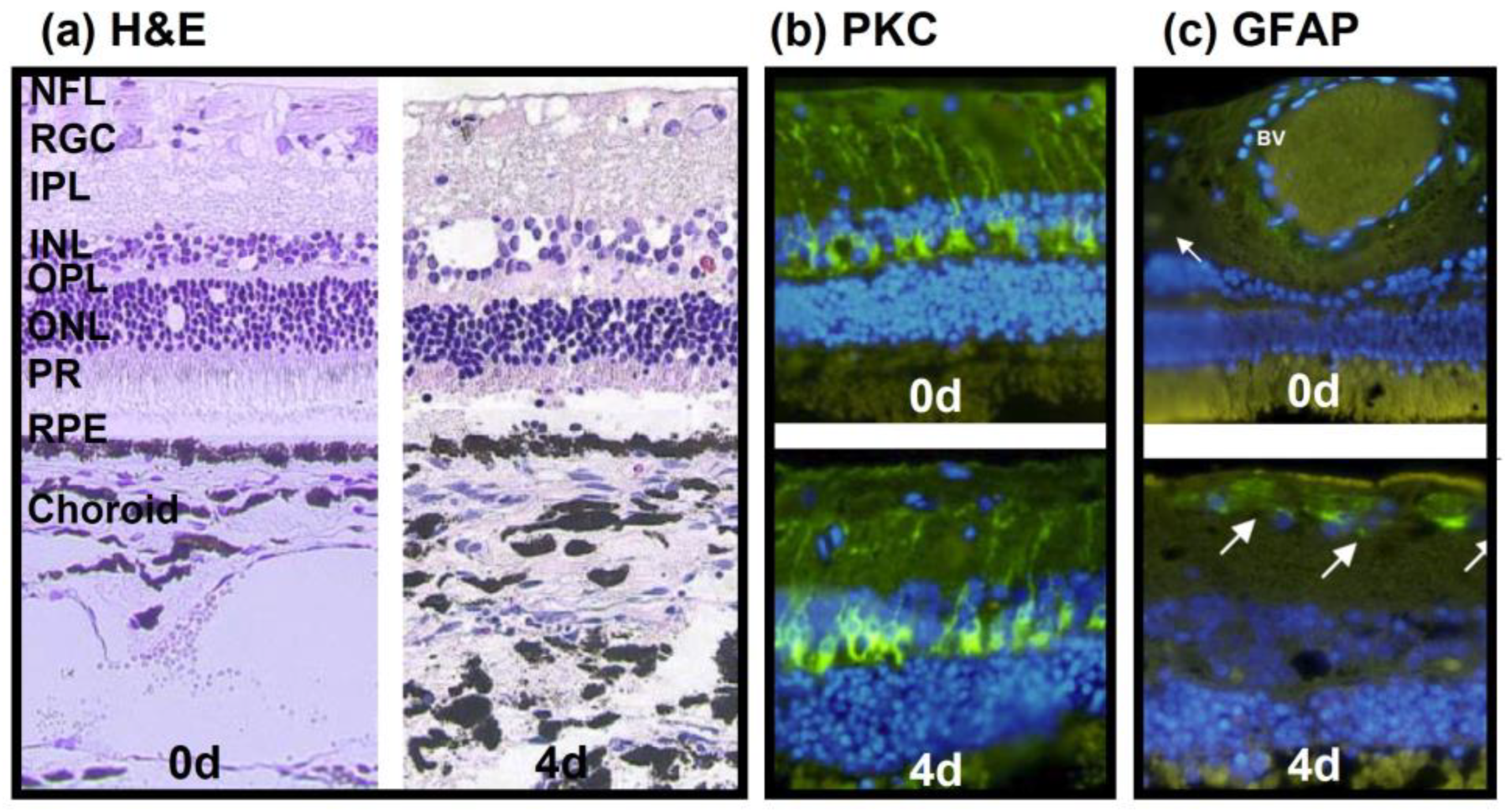
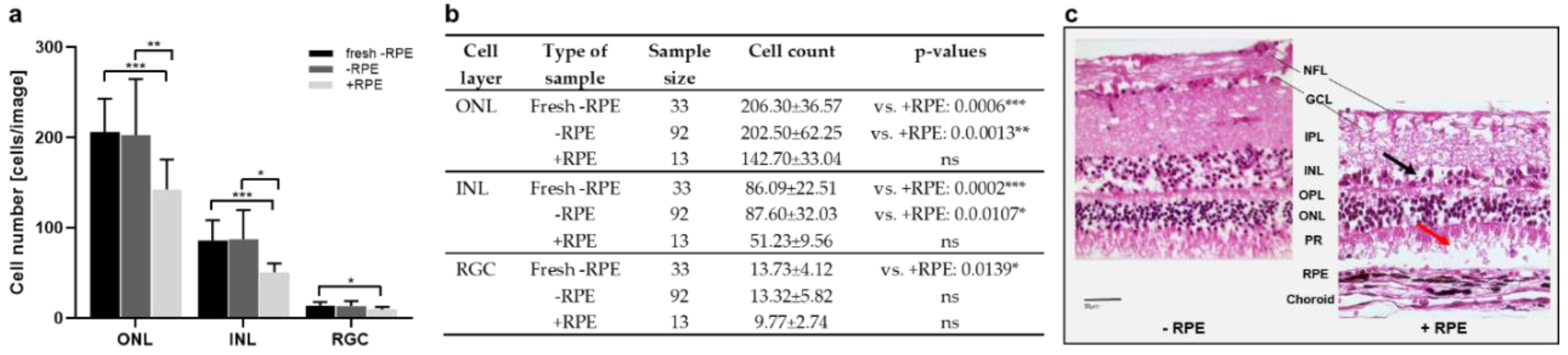
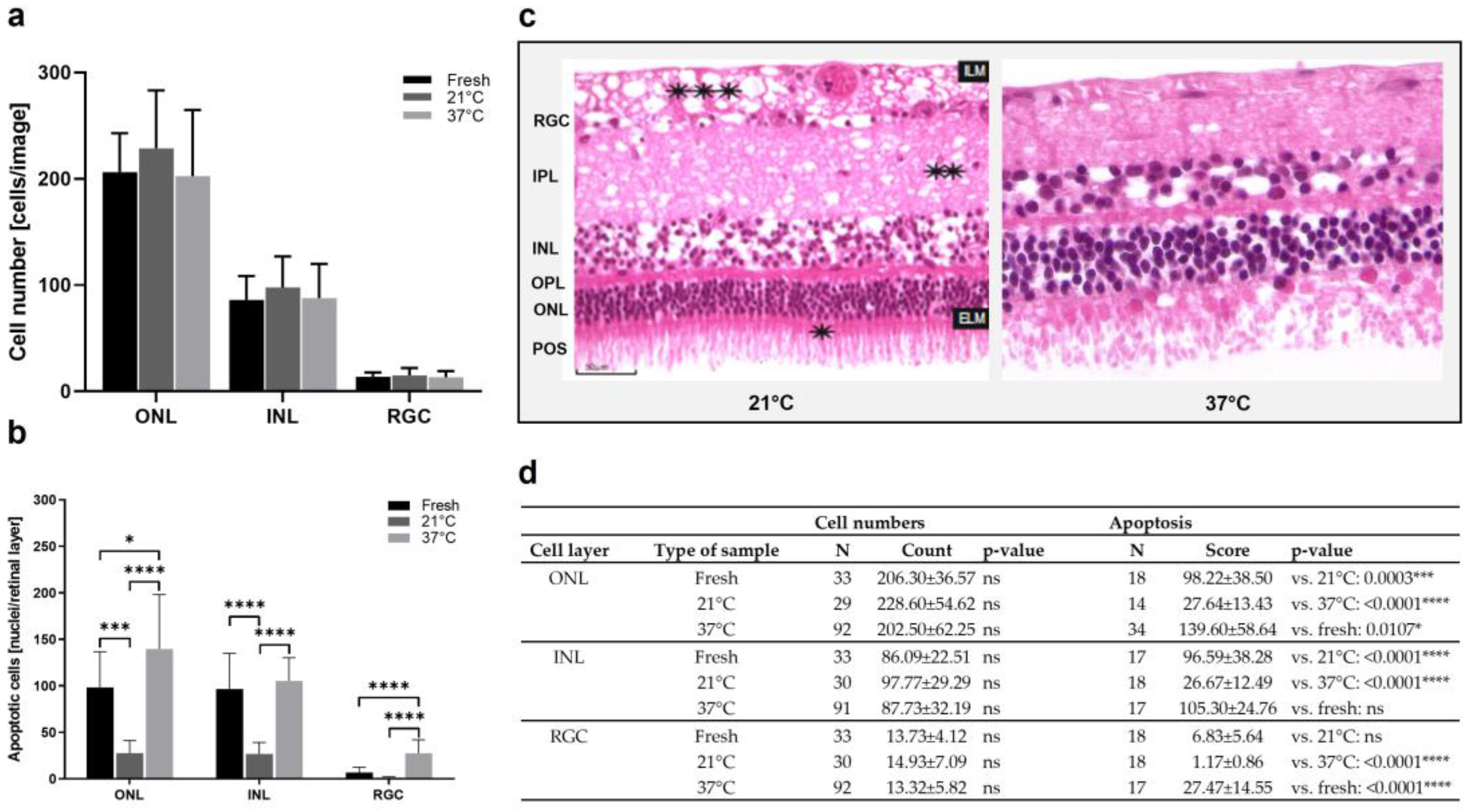
3.1. Model 1: Static Bovine Retina-RPE Culture, 4 Days
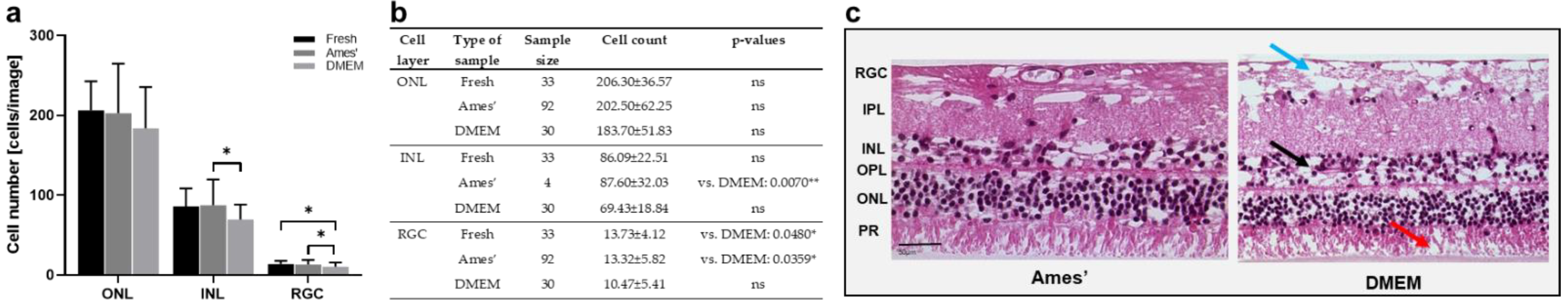
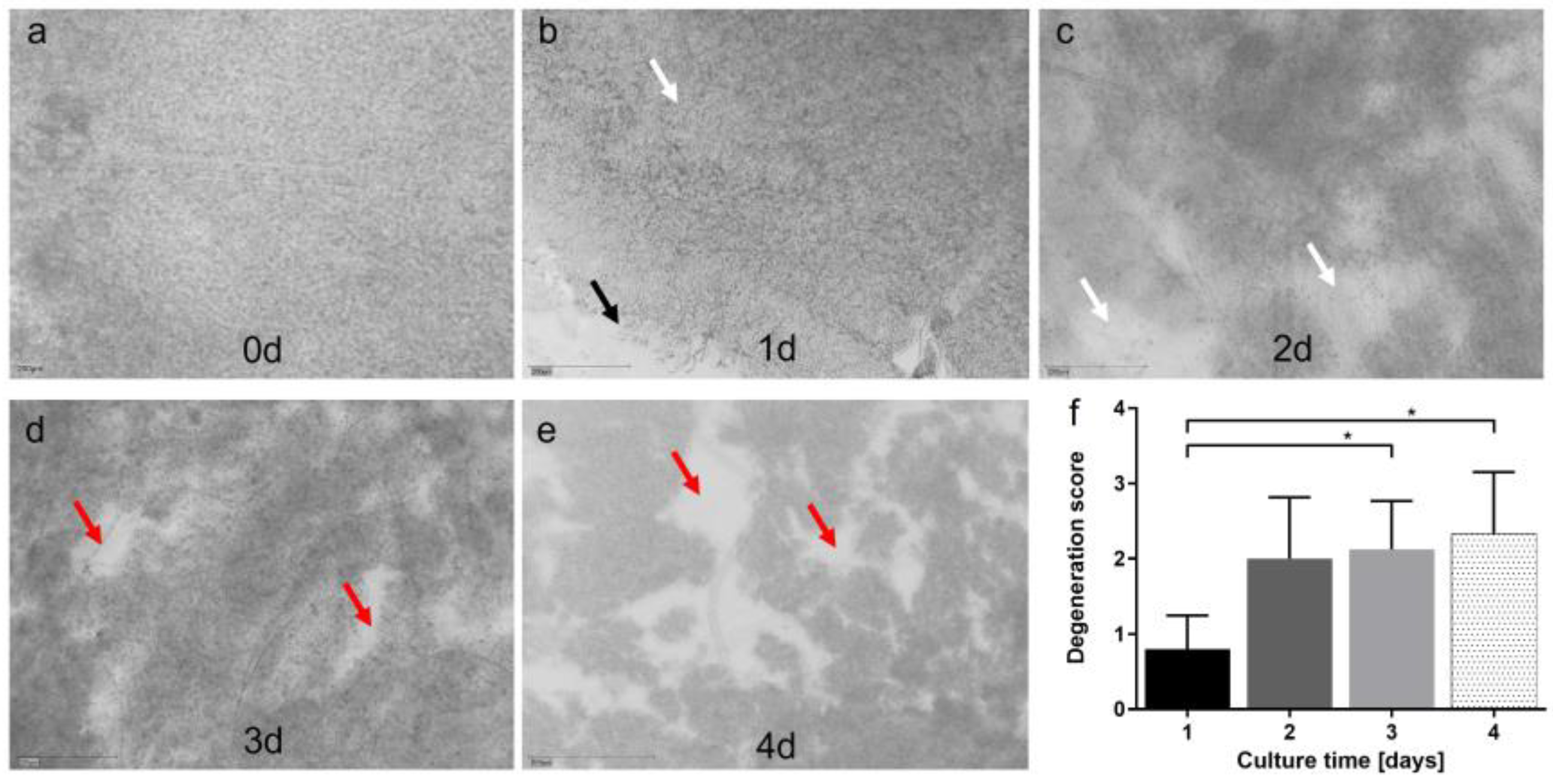
3.2. Model 2: Dynamic Porcine Retina Culture, 3 Days
3.3. Model 3: Static Porcine Retina Culture, 4 Days
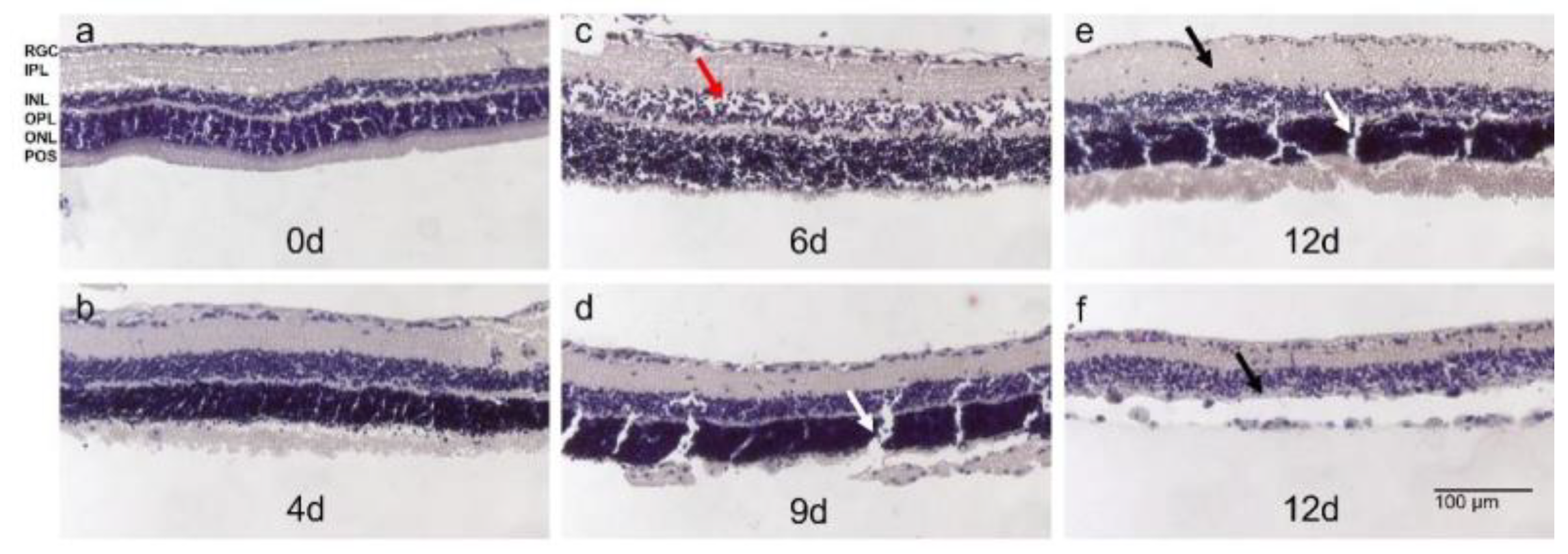
3.4. Model 4: Static Rat Retina Culture, 9–13 Days
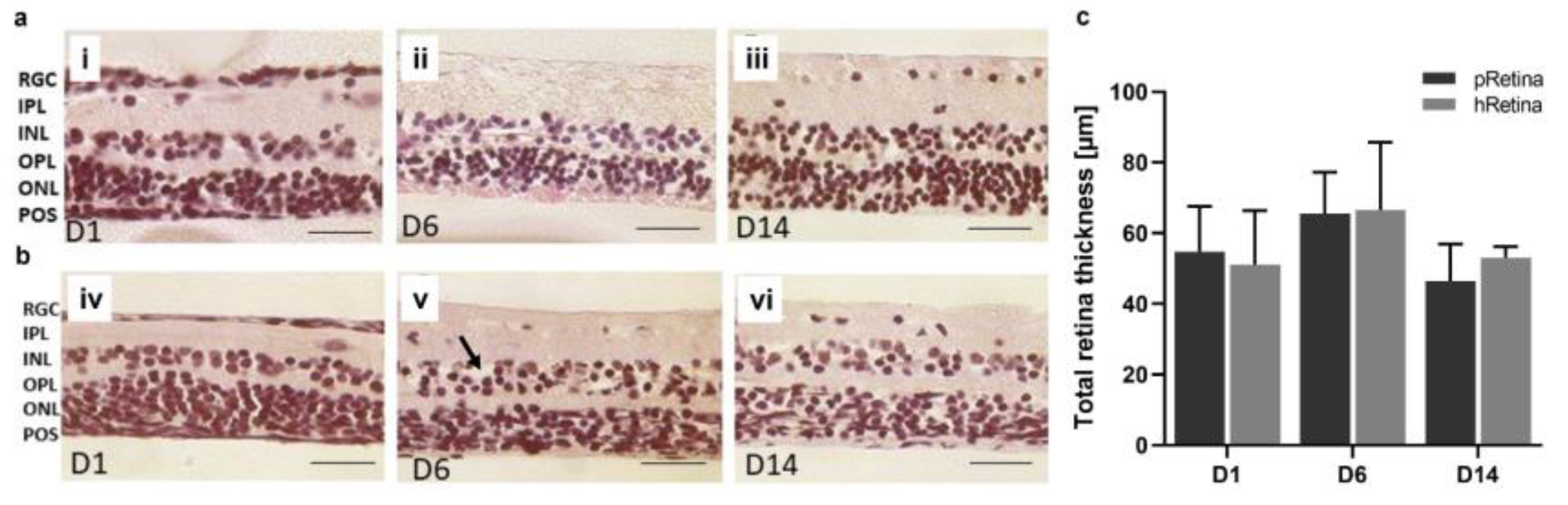
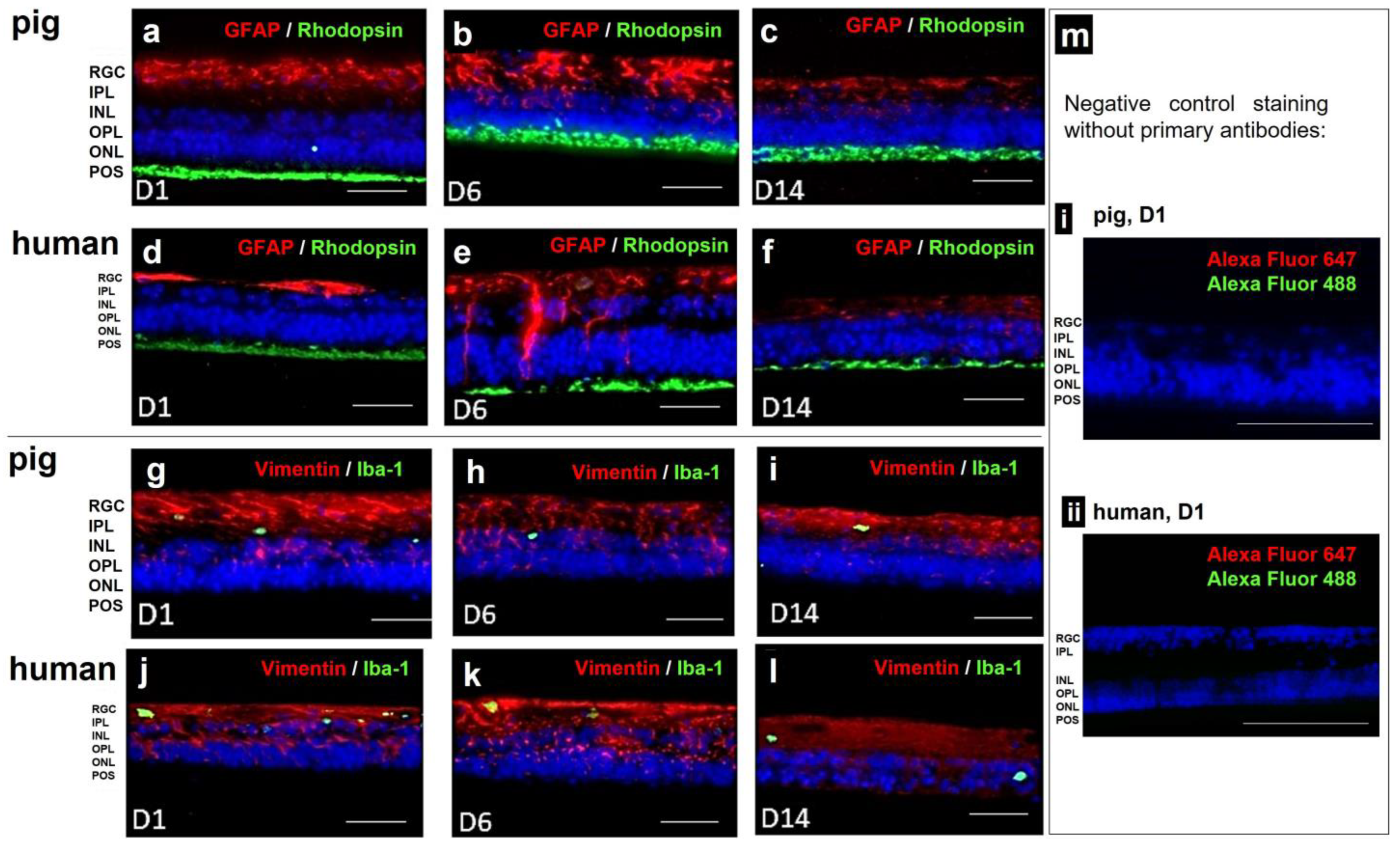
3.5. Model 5 and 5b: Static Porcine and Human Retina Culture, 14 Days
3.6. Antioxidant Scutellarin to Treat OS in High-Glucose Retina
3.7. OS Reduction and Cell Protection by PEDF and GM-CSF in H2O2-Treated Retina
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, V.; Bishayee, K.; Park, S.; Lee, U.; Kim, J. Oxidative stress in cerebrovascular disease and associated diseases. Front. Endocrinol. 2023, 14, 1124419. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, R.F.; Nagano, S. Crosstalk between Oxidative Stress and Aging in Neurodegeneration Disorders. Cells 2023, 12, 753. [Google Scholar] [CrossRef]
- Fantini, C.; Corinaldesi, C.; Lenzi, A.; Migliaccio, S.; Crescioli, C. Vitamin D as a Shield against Aging. Int. J. Mol. Sci. 2023, 24, 4546. [Google Scholar] [CrossRef]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, J.-Y.; Xu, Y.; Li, D.-L.; Zhou, M.; Wang, P.; Pan, C.-W. Impact of major age-related eye disorders on health-related quality of life assessed by EQ-5D: A systematic review and meta-analysis. Graefes Arch. Clin. Exp. Ophthalmol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.-Y.; Kim, B.-S.; Cho, K.-H.; Kim, I.-H.; Kim, J.-H.; Kim, J.-H. 10-year follow-up study on medical expenses and medical care use according to biological age: National Health Insurance Service Health Screening Cohort (NHIS-HealS 2002~2019). PLoS ONE 2023, 18, e0282466. [Google Scholar] [CrossRef]
- Tolentino, M.J.; Tolentino, A.J. Investigational drugs in clinical trials for macular degeneration. Expert Opin. Investig. Drugs 2022, 31, 1067–1085. [Google Scholar] [CrossRef]
- Li, Q.; Wang, M.; Li, X.; Shao, Y. Aging and diabetic retinopathy: Inherently intertwined pathophysiological processes. Exp. Gerontol. 2023, 175, 112138. [Google Scholar] [CrossRef]
- Cabral de Guimaraes, T.A.; Daich Varela, M.; Georgiou, M.; Michaelides, M. Treatments for dry age-related macular degeneration: Therapeutic avenues, clinical trials and future directions. Br. J. Ophthalmol. 2022, 106, 297–304. [Google Scholar] [CrossRef]
- Steinmetz, J.D.; Bourne, R.R.A.; Briant, P.S.; Flaxman, S.R.; Taylor, H.R.B.; Jonas, J.B.; Abdoli, A.A.; Abrha, W.A.; Abualhasan, A.; Abu-Gharbieh, E.G.; et al. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Mounirou, B.A.M.; Adam, N.D.; Yakoura, A.K.H.; Aminou, M.S.M.; Liu, Y.T.; Tan, L.Y. Diabetic Retinopathy: An Overview of Treatments. Indian J. Endocrinol. Metab. 2022, 26, 111–118. [Google Scholar] [CrossRef]
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarone, R. The Impact of Oxidative Stress on Blood-Retinal Barrier Physiology in Age-Related Macular Degeneration. Cells 2021, 10, 64. [Google Scholar] [CrossRef]
- Kiani, A.K.; Pheby, D.; Henehan, G.; Brown, R.; Sieving, P.; Sykora, P.; Marks, R.; Falsini, B.; Capodicasa, N.; Miertus, S.; et al. Ethical considerations regarding animal experimentation. J. Prev. Med. Hyg. 2022, 63, E255–E266. [Google Scholar] [CrossRef]
- Pastor, J.C.; Coco, R.M.; Fernandez-Bueno, I.; Alonso-Alonso, M.L.; Medina, J.; Sanz-Arranz, A.; Rull, F.; Gayoso, M.J.; Dueñas, A.; Garcia-Gutierrez, M.T.; et al. Acute retinal damage after using a toxic perfluoro-octane for vitreo-retinal surgery. Retina 2017, 37, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.; Fietz, A.; Tsai, T.; Joachim, S.C.; Schnichels, S. Organ Cultures for Retinal Diseases. Front. Neurosci. 2020, 14, 583392. [Google Scholar] [CrossRef]
- Strangeways, T.; Fell, B. Experimental studies on the differentiation of embryonic tissues growing in vivo and in vitro.—II. The development of the isolated early embryonic eye of the fowl when cultivated in vitro. Proc. R Soc. B. 1926, 100, 273–281. [Google Scholar]
- Murali, A.; Ramlogan-Steel, C.A.; Steel, J.C.; Layton, C.J. Characterisation and validation of the 8-fold quadrant dissected human retinal explant culture model for pre-clinical toxicology investigation. Toxicol. Vitr. 2020, 63, 104716. [Google Scholar] [CrossRef]
- Schnichels, S.; Paquet-Durand, F.; Löscher, M.; Tsai, T.; Hurst, J.; Joachim, S.C.; Klettner, A. Retina in a dish: Cell cultures, retinal explants and animal models for common diseases of the retina. Prog. Retin. Eye Res. 2021, 81, 100880. [Google Scholar] [CrossRef]
- Koizumi, A.; Zeck, G.; Ben, Y.; Masland, R.H.; Jakobs, T.C. Organotypic culture of physiologically functional adult mammalian retinas. PLoS ONE 2007, 2, e221. [Google Scholar] [CrossRef] [Green Version]
- Ingensiep, C.; Schaffrath, K.; Denecke, B.; Walter, P.; Johnen, S. A multielectrode array-based hypoxia model for the analysis of electrical activity in murine retinae. J. Neurosci. Res. 2021, 99, 2172–2187. [Google Scholar] [CrossRef] [PubMed]
- Ingensiep, C.; Schaffrath, K.; Walter, P.; Johnen, S. Effects of Hydrostatic Pressure on Electrical Retinal Activity in a Multielectrode Array-Based ex vivo Glaucoma Acute Model. Front. Neurosci. 2022, 16, 831392. [Google Scholar] [CrossRef]
- Peynshaert, K.; Devoldere, J.; Forster, V.; Picaud, S.; Vanhove, C.; de Smedt, S.C.; Remaut, K. Toward smart design of retinal drug carriers: A novel bovine retinal explant model to study the barrier role of the vitreoretinal interface. Drug Deliv. 2017, 24, 1384–1394. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, S.; Hurst, J.; Rensinghoff, F.; Tsai, T.; Grauthoff, S.; Satgunarajah, Y.; Dick, H.B.; Schnichels, S.; Joachim, S.C. Degenerative effects of cobalt-chloride treatment on neurons and microglia in a porcine retina organ culture model. Exp. Eye Res. 2017, 155, 107–120. [Google Scholar] [CrossRef]
- Calbiague, V.M.; Vielma, A.H.; Cadiz, B.; Paquet-Durand, F.; Schmachtenberg, O. Physiological assessment of high glucose neurotoxicity in mouse and rat retinal explants. J. Comp. Neurol. 2020, 528, 989–1002. [Google Scholar] [CrossRef]
- Masri, R.A.; Lee, S.C.S.; Madigan, M.C.; Grünert, U. Particle-Mediated Gene Transfection and Organotypic Culture of Postmortem Human Retina. Transl. Vis. Sci. Technol. 2019, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Mueller-Buehl, A.M.; Doepper, H.; Grauthoff, S.; Kiebler, T.; Peters, L.; Hurst, J.; Kuehn, S.; Bartz-Schmidt, K.U.; Dick, H.B.; Joachim, S.C.; et al. Oxidative stress-induced retinal damage is prevented by mild hypothermia in an ex vivo model of cultivated porcine retinas. Clin. Experiment. Ophthalmol. 2020, 48, 666–681. [Google Scholar] [CrossRef]
- Thumann, G.; Harmening, N.; Prat-Souteyrand, C.; Marie, C.; Pastor, M.; Sebe, A.; Miskey, C.; Hurst, L.D.; Diarra, S.; Kropp, M.; et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol. Ther. Nucleic Acids 2017, 6, 302–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bascuas, T.; Zedira, H.; Kropp, M.; Harmening, N.; Asrih, M.; Prat-Souteyrand, C.; Tian, S.; Thumann, G. Human Retinal Pigment Epithelial Cells Overexpressing the Neuroprotective Proteins PEDF and GM-CSF to Treat Degeneration of the Neural Retina. CGT 2022, 21, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, M.L.; Dass, C.R.; Choong, P.F.M. Systemically administered PEDF against primary and secondary tumours in a clinically relevant osteosarcoma model. Br. J. Cancer 2011, 105, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Michelis, G.; German, O.L.; Villasmil, R.; Soto, T.; Rotstein, P.N.; Politi, L.; Becerra, S.P. Pigment Epithelium-derived Factor (PEDF) and Derived Peptides Promote Survival and Differentiation of Photoreceptors and Induce Neurite-outgrowth in Amacrine Neurons. J. Neurochem. 2021, 159, 791–931. [Google Scholar] [CrossRef]
- Craword, S.E.; Fitchev, P.; Veliceasa, D.; Volpert, O.V. The many facets of PEDF in drug discovery and disease: A diamond in the rough or split personality disorder? Expert Opin. Drug Discov. 2013, 8, 769–792. [Google Scholar] [CrossRef]
- Crane, I.J.; Wallace, C.A.; Forrester, J.V. Regulation of granulocyte-macrophage colony-stimulating factor in human retinal pigment epithelial cells by IL-1beta and IFN-gamma. Cell. Immunol. 2001, 209, 132–139. [Google Scholar] [CrossRef]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2020, 217, e20190945. [Google Scholar] [CrossRef] [Green Version]
- Chitu, V.; Biundo, F.; Stanley, E.R. Colony stimulating factors in the nervous system. Semin. Immunol. 2021, 54, 101511. [Google Scholar] [CrossRef] [PubMed]
- Schallenberg, M.; Charalambous, P.; Thanos, S. GM-CSF protects rat photoreceptors from death by activating the SRC-dependent signalling and elevating anti-apoptotic factors and neurotrophins. Graefes Arch. Clin. Exp. Ophthalmol. 2012, 250, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Leroy-Ciocanea, C. Establishment of an In Vitro Organotypic Retinopathy Model to Test the Neuroprotective Impact of Scutellarin. Master’s Thesis, Sorbonne Universités, Université Pierre et Marie Curie Paris, Paris, France, 2016. [Google Scholar]
- Wang, L.; Ma, Q. Clinical benefits and pharmacology of scutellarin: A comprehensive review. Pharmacol. Ther. 2018, 190, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Minuth, W.W.; Rudolph, U. A compatible support system for cell culture in biomedical research. Cytotechnology 1990, 4, 181–189. [Google Scholar] [CrossRef]
- Framme, C.; Kobuch, K.; Eckert, E.; Monzer, J.; Roininen, E. RPE in Perfusion Tissue Culture and Its Response to Laser Application: Preliminary Report. Ophthalmologica 2002, 216, 320–328. [Google Scholar] [CrossRef]
- Bull, N.; Johnson, T.; Martin, K.R. Organotypic explant culture of adult rat retina for in vitro investigations of neurodegeneration, neuroprotection and cell transplantation. Res. Sq. 2011. preprint. Available online: https://protocolexchange.researchsquare.com/article/nprot-2042/v1 (accessed on 30 May 2023).
- Kaempf, S.; Walter, P.; Salz, A.K.; Thumann, G. Novel organotypic culture model of adult mammalian neurosensory retina in co-culture with retinal pigment epithelium. J. Neurosci. Methods 2008, 173, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Zedira, H. Optimization of an In Vitro Retinal Organotypic Culture Model of Retinal Degeneration to Assess the Neuroprotective Effect of the Recombinant Proteins PEDF and GM-CSF. Master’s Thesis, UNIGE, Geneva, Switzerland, Université Grenoble Alpes, Lyon, France, 2019. [Google Scholar]
- Mohit, M. Up-Scaling and Humanization of Retinal Organotypic Culture as a Model for Intraocular EFFICIENCY and Toxicity Analyses. Master’s Thesis, University of Geneva, Geneva, Switzerland, 2023. [Google Scholar]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon Olga, L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci. 2021, 22, 1296. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.G.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergen, A.A.; Arya, S.; Koster, C.; Pilgrim, M.G.; Wiatrek-Moumoulidis, D.; van der Spek, P.J.; Hauck, S.M.; Boon, C.J.F.; Emri, E.; Stewart, A.J.; et al. On the origin of proteins in human drusen: The meet, greet and stick hypothesis. Prog. Retin. Eye Res. 2019, 70, 55–84. [Google Scholar] [CrossRef] [Green Version]
- Al-Dosari, D.I.; Ahmed, M.M.; Al-Rejaie, S.S.; Alhomida, A.S.; Ola, M.S. Flavonoid Naringenin Attenuates Oxidative Stress, Apoptosis and Improves Neurotrophic Effects in the Diabetic Rat Retina. Nutrients 2017, 9, 1161. [Google Scholar] [CrossRef] [Green Version]
- Kropp, M.; Golubnitschaja, O.; Mazurakova, A.; Koklesova, L.; Sargheini, N.; Vo, T.-T.K.S.; de Clerck, E.; Polivka Jr., J.; Potuznik, P.; Polivka, J.; et al. Diabetic retinopathy as the leading cause of blindness and early predictor of cascading complications—Risks and mitigation. EMPA J. 2023, 14, 21–42. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [Green Version]
- Murali, A.; Ramlogan-Steel, C.A.; Andrzejewski, S.; Steel, J.C.; Layton, C.J. Retinal explant culture: A platform to investigate human neuro-retina. Clin. Exp. Ophthalmol. 2019, 47, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Schnichels, S.; Dorfi, T.; Schultheiss, M.; Arango-Gonzalez, B.; Bartz-Schmidt, K.-U.; Januschowski, K.; Spitzer, M.S.; Ziemssen, F. Ex-vivo-examination of ultrastructural changes in organotypic retina culture using near-infrared imaging and optical coherence tomography. Exp. Eye Res. 2016, 147, 31–36. [Google Scholar] [CrossRef]
- Kobuch, K.; Herrmann, W.A.; Framme, C.; Sachs, H.G.; Gabel, V.-P.; Hillenkamp, J. Maintenance of adult porcine retina and retinal pigment epithelium in perfusion culture: Characterisation of an organotypic in vitro model. Exp. Eye Res. 2008, 86, 661–668. [Google Scholar] [CrossRef]
- Narayanamurthy, R.; Armstrong, E.A.; Yang, J.-L.J.; Yager, J.Y.; Unsworth, L.D. Administration of Selective Brain Hypothermia Using a Simple Cooling Device in Neonatal Rats. J. Neurosci. Methods 2023, 390, 109838. [Google Scholar] [CrossRef]
- Choi, D.E.; Jeong, J.Y.; Choi, H.; Chang, Y.K.; Ahn, M.S.; Ham, Y.R.; Na, K.R.; Lee, K.W. ERK phosphorylation plays an important role in the protection afforded by hypothermia against renal ischemia-reperfusion injury. Surgery 2017, 161, 444–452. [Google Scholar] [CrossRef]
- Xi, L. Research Progress of the Application of Hypothermia in the Eye. Oxid. Med. Cell. Longev. 2020, 2020, 3897168. [Google Scholar] [CrossRef] [PubMed]
- Bilkhu, P.S.; Wolffsohn, J.S.; Naroo, S.A.; Robertson, L.; Kennedy, R. Effectiveness of nonpharmacologic treatments for acute seasonal allergic conjunctivitis. Ophthalmology 2014, 121, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Ames, A., III; Gurian, B.S. Effects of glucose and oxygen deprivation on function of isolated mammalian retina. J. Neurophysiol. 1963, 26, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Ames, A., III; Nesbett, F.B. In vitro retina as an experimental model of the central nervous system. J. Neurochem. 1981, 37, 867–877. [Google Scholar] [CrossRef]
- Kropp, M.; Harmening, N.; Bascuas, T.; Johnen, S.; de Clerck, E.; Fernández, V.; Ronchetti, M.; Cadossi, R.; Zanini, C.; Scherman, D.; et al. GMP-Grade Manufacturing and Quality Control of a Non-Virally Engineered Advanced Therapy Medicinal Product for Personalized Treatment of Age-Related Macular Degeneration. Biomedicines 2022, 10, 2777. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, F.; Wu, L. NLRX1 increases human retinal pigment epithelial autophagy and reduces H2O2-induced oxidative stress and inflammation by suppressing FUNDC1 phosphorylation and NLRP3 activation. Allergol. Immunopathol. 2023, 51, 177–186. [Google Scholar] [CrossRef]
- Zheng, H.-L.; Li, M.-T.; Zhou, T.; Wang, Y.-Y.; Shang, E.-X.; Hua, Y.-Q.; Duan, J.-A.; Zhu, Y. Protective effects of Lycium barbarum L. berry extracts against oxidative stress-induced damage of the retina of aging mouse and ARPE-19 cells. Food Funct. 2023, 14, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Jüttner, J.; Szabo, A.; Gross-Scherf, B.; Morikawa, R.K.; Rompani, S.B.; Hantz, P.; Szikra, T.; Esposti, F.; Cowan, C.S.; Bharioke, A.; et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nat. Neurosci. 2019, 22, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Schnichels, S.; Kiebler, T.; Hurst, J.; Maliha, A.M.; Löscher, M.; Dick, H.B.; Bartz-Schmidt, K.-U.; Joachim, S.C. Retinal Organ Cultures as Alternative Research Models. Altern. Lab. Anim. 2019, 47, 19–29. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Species | Tissue | Type of culture | Processing | Medium | Temperature | Culture duration | Advantages | Disadvantages | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | bRetina | cattle | Choroid-RPE-retina | Static, customized grids | Scalpel | DMEM; Neurobasal, 2% B27 | 37 °C | 4 d | Supporting tissue; easy available tissue | Complex isolation |
| 2 | pRetina, dynamic | pig | RPE-retina; retina | Dynamic, Minuth chamber | Scalpel | DMEM; Ames’ | 37 °C; 21 °C | 3 d | Continuous medium flow; easy available tissue | More complex set up; only neural retina (+RPE) |
| 3 | pRetina, static | pig | Retina | Static, customized grids | Scalpel | Ames’ | 21 °C | 4 d | Simple isolation; simple set-up; easy available tissue | Only neural retina |
| 4 | rRetina | rat | Retina | Static, inserts | Scalpel | Neurobasal, 2% B-27 & 1% N-2; Ames’; Ames’, 2% B-27 & 1% N-2 | 21 °C | 9 d | Fresh tissue | Ethical concerns; only neural retina |
| 5a | pRetina | pig | Retina | Static, inserts | Punch | Ames’ with 1% N2 & 1% B-27 | 21 °C | 14 d | Low tissue damage; long culture; easy available | only neural retina |
| 5b | hRetina | human | Retina | Static, inserts | Punch | Ames’, 1% N2 & 1% B-27 | 21 °C | 14 d | Transferability; low tissue damage; long culture | Limited tissue availability; only neural retina |
| Process | Reagents | Time | Temperature |
|---|---|---|---|
| Deparaffinization | Xylol | 3 × 3 min | RT |
| Rehydration | EtOH 2× 100%, 96%, 90%, 80%, 70%, 50%, dd H2O | 30 s each 3 min | RT |
| Staining | Hemalum | 4 min | RT |
| Tap H2O | 3 × 2 min | ||
| Acid alcohol | 10 s | ||
| dd H2O | 2 × 1 min | ||
| Eosin | 3 min | ||
| dd H2O | 1 min | ||
| Dehydration | 50%, 70%, 80%, 90%, 96% 2 × 100% EtOH | 30 s each 2 × 2 min | RT |
| Mounting | Mounting medium | 1 min | RT |
| Grade | Tissue Quality |
|---|---|
| 0 | Healthy tissue, integral, no damage; |
| 1 | Irregular borders, small holes; |
| 2 | Big holes, patches of cell loss; |
| 3 | Big holes, major cell loss. |
| Process | Reagents | Time | Temperature |
|---|---|---|---|
| Deparaffinization | Xylol | 3 × 3 min | RT |
| Rehydration | EtOH 100%, 96%, 90%, 80%, 70%, 50%, dd H2O | 3 min each 3 × 5 min | RT |
| Demasking | Citrate buffer, 0.1 M pH 6 (boiling) cooling | 15 min 30 min | 95 °C–100 °C RT |
| Washing | PBS | 3 × 10 min | RT |
| Blocking | PBS-BSA 3% | 2 h | 37 °C |
| Staining | Primary antibody (PBS-BSA 1%) | overnight | 4 °C |
| Washing | PBS | 3 × 10 min | RT |
| Staining | Secondary antibody (PBS) | 30 min | 37 °C |
| Washing | PBS | 3 × 10 min | RT |
| Mounting | Mounting medium with DAPI | 1 min | RT |
| Specificity | Cells Labelled | Host | Conjugate | Clonality | Company | ID | Dilution |
|---|---|---|---|---|---|---|---|
| Primary antibodies | |||||||
| Protein kinase C | Bipolar cells | Chicken | Unconjugated | Polyclonal | Abcam * ab14078 | n.a. | 1:50 |
| Rhodopsin | Rods | Rabbit | Unconjugated | Polyclonal | Novus Biological $ NLS1052 | AB_2178795 | 1:700 |
| GFAP | Astrocytes | Mouse | Unconjugated | Monoclonal | Millipore † MAB360 | AB_11212597 | 1:300 |
| Iba-1 | Microglia | Rabbit | Unconjugated | Polyclonal | Fujifilm Wako Pure Chemical Corporation § 01-1874 | AB_2314666 | 1:750 |
| Vimentin | Müller cells | Mouse | Unconjugated | Monoclonal | Merck† MAB3400 | AB_94843 | 1:120 |
| Secondary antibodies | |||||||
| Mouse | NA | Donkey | Alexa Fluor 647 | NA | Abcam ab150107 | AB_2890037 | 1:250 |
| Rabbit | NA | Donkey | Alexa Fluor 488 | NA | Jackson # 711546152 | AB_2340619 | 1:100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kropp, M.; Mohit, M.; Leroy-Ciocanea, C.I.; Schwerm, L.; Harmening, N.; Bascuas, T.; De Clerck, E.; Kreis, A.J.; Pajic, B.; Johnen, S.; et al. Mammalian Animal and Human Retinal Organ Culture as Pre-Clinical Model to Evaluate Oxidative Stress and Antioxidant Intraocular Therapeutics. Antioxidants 2023, 12, 1211. https://doi.org/10.3390/antiox12061211
Kropp M, Mohit M, Leroy-Ciocanea CI, Schwerm L, Harmening N, Bascuas T, De Clerck E, Kreis AJ, Pajic B, Johnen S, et al. Mammalian Animal and Human Retinal Organ Culture as Pre-Clinical Model to Evaluate Oxidative Stress and Antioxidant Intraocular Therapeutics. Antioxidants. 2023; 12(6):1211. https://doi.org/10.3390/antiox12061211
Chicago/Turabian StyleKropp, Martina, Mohit Mohit, Cristina Ioana Leroy-Ciocanea, Laura Schwerm, Nina Harmening, Thais Bascuas, Eline De Clerck, Andreas J. Kreis, Bojan Pajic, Sandra Johnen, and et al. 2023. "Mammalian Animal and Human Retinal Organ Culture as Pre-Clinical Model to Evaluate Oxidative Stress and Antioxidant Intraocular Therapeutics" Antioxidants 12, no. 6: 1211. https://doi.org/10.3390/antiox12061211