
Cigarette Smoke-Induced Respiratory Response: Insights into Cellular Processes and Biomarkers
Abstract
:1. Introduction
2. Cellular Responses to Cigarette Smoke
2.1. Inflammation
2.2. Cigarette Smoke-Induced Cell Death
2.3. Effects of Cigarette Smoke on Cellular Senescence in the Lungs
2.4. Understanding the Link between Cigarette Smoke and Autophagy
3. CS-Induced Lung and Other Diseases
3.1. Chronic Obstructive Pulmonary Disease (COPD)
3.2. Pulmonary Fibrosis
3.3. Cancers
3.4. Acute Respiratory Distress Syndrome and Acute Exacerbations of COPD
3.5. Cardiovascular Disease
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Definitions |
| CS | cigarette smoke |
| ROS | reactive oxygen species |
| COPD | chronic obstructive pulmonary disease |
| PF | pulmonary fibrosis |
| WHO | World Health Organization |
| FCTC | Framework Convention on Tobacco Control |
| NOX | NADPH oxidase |
| NETs | neutrophil extracellular traps |
| DAMP | damage-associated molecular patterns |
| RAGE | receptor for advanced glycation end products |
| HMGB1 | high mobility group box 1 |
| sRAGE | soluble form RAGE |
| mRAGE | membrane-bound form RAGE |
| TLRs | toll-like receptors |
| H1R | histamine receptor-1 |
| COX2 | cyclooxygenase2 |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor kappa-light chain-enhancer of activated B cells |
| IKK | IκB kinase |
| ERK1/2 | extracellular signal-regulated kinases |
| JNK1/2 | c-Jun N-terminal Kinase |
| STAT | signal transducer and activator of transcription |
| IRF9 | interferon regulatory factor 9 |
| CBP | CREB-binding protein |
| CSE | cigarette smoke extract |
| BAL | bronchoalveolar lavage |
| BCL-2 | B-cell lymphoma 2 |
| TP53 | tumor protein p53 |
| IAPs | inhibitor of apoptosis proteins |
| TSSK4 | testis-specific serine/threonine kinase 4 |
| NQO1 | NAD(P)H dehydrogenase [quinone] 1 |
| AKR1C3 | aldo-keto reductase family 1 member C3 |
| GPX2 | glutathione peroxidase 2 |
| ILD | interstitial lung disease |
| MMPs | matrix metalloproteinases |
| SIRT1 | sirtuin-1 |
| SASP | senescence-associated secretory phenotype |
| OSGIN | oxidative stress-induced growth inhibitor |
| Nrf2 | nuclear factor erythroid 2-related factor |
| Keap1 | Kelch-like ECH-associated protein 1 |
| ARE | antioxidant response elements |
| HO1 | heme oxygenase 1 |
| GST | glutathione S-transferase |
| FEV1 | forced expiratory volume |
| SOD1 | superoxide dismutase type 1 |
| TXNRD1 | thioredoxin reductase 1 |
| GOLD | Global Initiative for Obstructive Lung Disease |
| HUC-MSC | Human umbilical cord mesenchymal system cell |
| LPHNPs | lipid-polymer hybrid nanoparticles |
| EMT | epithelial-mesenchymal transition |
| SNPs | single nucleotide polymorphisms |
| CYP1A1 | cytochrome P450 Family 1 Subfamily A Member 1 |
| CYP1B1 | cytochrome P450 Family 1 Subfamily B Member 1 |
| CCL18 | chemokine (C-C motif) ligand 18 |
| CXCR3 | chemokine (C-X-C motif) receptor 3 |
| TIMPs | tissue inhibitors of metalloproteinases |
| miRNAs | microRNAs |
| PDE4B | phosphodiesterase 4B |
| iNOS | inducible nitric oxide synthase |
| HIF | hypoxia-inducible factor |
| NSCLC | non-small-cell lung cancer |
| mAbs | monoclonal antibodies |
| MSCs | mesenchymal stem cells |
| PAHs | polycyclic aromatic hydrocarbons |
| ARDS | acute respiratory distress syndrome |
| NE | neutrophil elastase |
| ACE2 | angiotensin-converting enzyme 2 |
| TMPRSS2/4 | transmembrane protease, serine 2 and 4 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| AE-COPD | acute exacerbations of chronic obstructive pulmonary disease |
| FVC | forced vital capacity |
| CVD | cardiovascular disease |
| GMP | guanosine monophosphate |
| eNOS | endothelial NOS |
| NLR | NOD-like receptor |
| NLRP3 | NLR family pyrin domain containing 3 |
| sVCAM-1 | soluble vascular cell adhesion molecule 1 |
| LDL | low-density lipoprotein |
| oxLDL | oxidized LDLs |
| LOX-1 | lectin-like oxidized LDL receptor-1 |
References
- WHO. Global Report on Trends in Prevalence of Tobacco Smoking 2000–2025, 2nd ed.; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Dai, X.; Gakidou, E.; Lopez, A.D. Evolution of the global smoking epidemic over the past half century: Strengthening the evidence base for policy action. Tob. Control 2022, 31, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Hiilamo, H.; Glantz, S. Global Implementation of Tobacco Demand Reduction Measures Specified in Framework Convention on Tobacco Control. Nicotine Tob. Res. 2022, 24, 503–510. [Google Scholar] [CrossRef]
- Fong, G.T.; Chung-Hall, J.; Craig, L.; Group, W.F.I.A.E. Impact assessment of the WHO FCTC over its first decade: Methodology of the expert group. Tob. Control 2019, 28, s84–s88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung-Hall, J.; Craig, L.; Gravely, S.; Sansone, N.; Fong, G.T. Impact of the WHO FCTC over the first decade: A global evidence review prepared for the Impact Assessment Expert Group. Tob. Control 2019, 28, s119–s128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamceva, G.; Arsova-Sarafinovska, Z.; Ruskovska, T.; Zdravkovska, M.; Kamceva-Panova, L.; Stikova, E. Cigarette Smoking and Oxidative Stress in Patients with Coronary Artery Disease. Open Access Maced. J. Med. Sci. 2016, 4, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.C.; Byron, M.J.; Baig, S.A.; Stepanov, I.; Brewer, N.T. How people think about the chemicals in cigarette smoke: A systematic review. J. Behav. Med. 2017, 40, 553–564. [Google Scholar] [CrossRef]
- Koul, A.; Bhatia, V.; Bansal, M.P. Effect of alpha-tocopherol on pulmonary antioxidant defence system and lipid peroxidation in cigarette smoke inhaling mice. BMC Biochem. 2001, 2, 14. [Google Scholar] [CrossRef]
- Liu, X.Y.; Zhu, M.X.; Xie, J.P. Mutagenicity of acrolein and acrolein-induced DNA adducts. Toxicol. Mech. Methods 2010, 20, 36–44. [Google Scholar] [CrossRef]
- Pu, X.; Kamendulis, L.M.; Klaunig, J.E. Acrylonitrile-induced oxidative stress and oxidative DNA damage in male Sprague-Dawley rats. Toxicol. Sci. 2009, 111, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; MacNee, W. Oxidative stress and regulation of glutathione in lung inflammation. Eur. Respir. J. 2000, 16, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Almyroudis, N.G.; Grimm, M.J.; Davidson, B.A.; Rohm, M.; Urban, C.F.; Segal, B.H. NETosis and NADPH oxidase: At the intersection of host defense, inflammation, and injury. Front. Immunol. 2013, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Dusting, G.J.; Selemidis, S.; Jiang, F. Mechanisms for suppressing NADPH oxidase in the vascular wall. Mem. Inst. Oswaldo Cruz 2005, 100 (Suppl. S1), 97–103. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; Biswas, S.K.; Jimenez, L.A.; Torres, M.; Forman, H.J. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox Signal 2005, 7, 42–59. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef]
- Comhair, S.A.; Erzurum, S.C. Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal 2010, 12, 93–124. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [Green Version]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef]
- Ferrero-Miliani, L.; Nielsen, O.H.; Andersen, P.S.; Girardin, S.E. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1beta generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Oberdorster, G.; Finkelstein, J.N.; Johnston, C.; Gelein, R.; Cox, C.; Baggs, R.; Elder, A.C. Acute pulmonary effects of ultrafine particles in rats and mice. Res. Rep. Health Eff. Inst. 2000, 5–74, 75–86. [Google Scholar]
- Salvi, S.; Holgate, S.T. Mechanisms of particulate matter toxicity. Clin. Exp. Allergy 1999, 29, 1187–1194. [Google Scholar] [CrossRef]
- Pappas, R.S.; Halstead, M.M.; Watson, C.H. Electron Microscopic Analysis of Silicate and Calcium Particles in Cigarette Smoke Tar. Int. J. Respir. Pulm. Med. 2016, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Borm, P.J.; Schins, R.P.; Albrecht, C. Inhaled particles and lung cancer, part B: Paradigms and risk assessment. Int. J. Cancer 2004, 110, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Caliri, A.W.; Tommasi, S.; Besaratinia, A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108365. [Google Scholar] [CrossRef]
- Carta, S.; Castellani, P.; Delfino, L.; Tassi, S.; Vene, R.; Rubartelli, A. DAMPs and inflammatory processes: The role of redox in the different outcomes. J. Leukoc. Biol. 2009, 86, 549–555. [Google Scholar] [CrossRef]
- Logan, S.M.; Storey, K.B. Pro-inflammatory AGE-RAGE signaling is activated during arousal from hibernation in ground squirrel adipose. PeerJ 2018, 6, e4911. [Google Scholar] [CrossRef]
- Shirasawa, M.; Fujiwara, N.; Hirabayashi, S.; Ohno, H.; Iida, J.; Makita, K.; Hata, Y. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9, 165–174. [Google Scholar] [CrossRef]
- Hirschi-Budge, K.M.; Tsai, K.Y.F.; Curtis, K.L.; Davis, G.S.; Theurer, B.K.; Kruyer, A.M.M.; Homer, K.W.; Chang, A.; Van Ry, P.M.; Arroyo, J.A.; et al. RAGE signaling during tobacco smoke-induced lung inflammation and potential therapeutic utility of SAGEs. BMC Pulm. Med. 2022, 22, 160. [Google Scholar] [CrossRef]
- Robinson, A.B.; Stogsdill, J.A.; Lewis, J.B.; Wood, T.T.; Reynolds, P.R. RAGE and tobacco smoke: Insights into modeling chronic obstructive pulmonary disease. Front. Physiol. 2012, 3, 301. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lee, J.; Hong, S.H.; Rahman, I.; Yang, S.R. Inhibition of RAGE Attenuates Cigarette Smoke-Induced Lung Epithelial Cell Damage via RAGE-Mediated Nrf2/DAMP Signaling. Front. Pharmacol. 2018, 9, 684. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.A.; Delker, D.A.; Huecksteadt, T.; Beck, E.; Wuren, T.; Chen, Y.; Zhang, Y.; Hazel, M.W.; Hoidal, J.R. RAGE is a Critical Mediator of Pulmonary Oxidative Stress, Alveolar Macrophage Activation and Emphysema in Response to Cigarette Smoke. Sci. Rep. 2019, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Krutzik, S.R.; Tan, B.; Li, H.; Ochoa, M.T.; Liu, P.T.; Sharfstein, S.E.; Graeber, T.G.; Sieling, P.A.; Liu, Y.J.; Rea, T.H.; et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat. Med. 2005, 11, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, R.S.; Sharma, M.; Dileepan, K.N. Cigarette Smoke Amplifies Inflammatory Response and Atherosclerosis Progression Through Activation of the H1R-TLR2/4-COX2 Axis. Front. Immunol. 2015, 6, 572. [Google Scholar] [CrossRef] [Green Version]
- Nadigel, J.; Prefontaine, D.; Baglole, C.J.; Maltais, F.; Bourbeau, J.; Eidelman, D.H.; Hamid, Q. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8+ T cells in chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Girard, S.; Kadhim, H.; Roy, M.; Lavoie, K.; Brochu, M.E.; Larouche, A.; Sebire, G. Role of perinatal inflammation in cerebral palsy. Pediatr. Neurol. 2009, 40, 168–174. [Google Scholar] [CrossRef]
- Moynagh, P.N. The NF-kappaB pathway. J. Cell Sci. 2005, 118, 4589–4592. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Wojciak, J.M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009, 28, 948–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesoriere, A.; Dinarello, A.; Argenton, F. The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions. Biomedicines 2021, 9, 956. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.K.; Lee, H.F.; Lin, A.H.; Liu, M.H.; Liu, C.I.; Lee, T.S.; Kou, Y.R. Regulation of Cigarette Smoke Induction of IL-8 in Macrophages by AMP-activated Protein Kinase Signaling. J. Cell Physiol. 2015, 230, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: Implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L567–L576. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, H.J.; Lea, S.; Hughes, D.; Khalaf, R.; Abbott-Banner, K.; Singh, D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin. Exp. Immunol. 2014, 176, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, T.; Ning, Q.; Li, F.; Chen, T.; Yao, Y.; Sun, Z. Cigarette smoke extract-treated mast cells promote alveolar macrophage infiltration and polarization in experimental chronic obstructive pulmonary disease. Inhal. Toxicol. 2015, 27, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, E.; Turato, G.; Tine, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, K.R.; Panth, N.; Manandhar, B.; Singh, S.K.; Gupta, G.; Wich, P.R.; Nammi, S.; MacLoughlin, R.; Adams, J.; Warkiani, M.E.; et al. Attenuation of Cigarette-Smoke-Induced Oxidative Stress, Senescence, and Inflammation by Berberine-Loaded Liquid Crystalline Nanoparticles: In Vitro Study in 16HBE and RAW264. 7 cells. Antioxidants 2022, 11, 873. [Google Scholar] [CrossRef]
- Ermert, D.; Niemiec, M.J.; Rohm, M.; Glenthoj, A.; Borregaard, N.; Urban, C.F. Candida albicans escapes from mouse neutrophils. J. Leukoc. Biol. 2013, 94, 223–236. [Google Scholar] [CrossRef]
- Hoenderdos, K.; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef]
- Thulborn, S.J.; Mistry, V.; Brightling, C.E.; Moffitt, K.L.; Ribeiro, D.; Bafadhel, M. Neutrophil elastase as a biomarker for bacterial infection in COPD. Respir. Res. 2019, 20, 170. [Google Scholar] [CrossRef] [Green Version]
- Voynow, J.A.; Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Ho, S.C.; Lin, H.C.; Lin, S.M.; Liu, C.Y.; Huang, C.D.; Wang, C.H.; Chung, K.F.; Kuo, H.P. Neutrophil-derived elastase induces TGF-beta1 secretion in human airway smooth muscle via NF-kappaB pathway. Am. J. Respir. Cell Mol. Biol. 2006, 35, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, K.; Skret, J.; Smagur, J.; Bzowska, M.; Gajkowska, B.; Scott, D.A.; Potempa, J.S. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death Dis. 2011, 2, e131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, N.; Matsumoto, K.; Fukuyama, S.; Asai, Y.; Kitajima, H.; Seki, N.; Matsunaga, Y.; Kan, O.K.; Moriwaki, A.; Morimoto, K.; et al. Cigarette smoke impairs phagocytosis of apoptotic neutrophils by alveolar macrophages via inhibition of the histone deacetylase/Rac/CD9 pathways. Int. Immunol. 2013, 25, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.; Krysko, D.V.; Vandenabeele, P.; de Vries, M.; van Oosterhout, A.J.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L377–L386. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- Wang, H.; Peng, W.; Weng, Y.; Ying, H.; Li, H.; Xia, D.; Yu, W. Imbalance of Th17/Treg cells in mice with chronic cigarette smoke exposure. Int. Immunopharmacol. 2012, 14, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Foley, J.; Bolognese, B.J.; Long, E., 3rd; Podolin, P.L.; Walsh, P.T. Airway infiltration of CD4+ CCR6+ Th17 type cells associated with chronic cigarette smoke induced airspace enlargement. Immunol. Lett. 2008, 121, 13–21. [Google Scholar] [CrossRef]
- Van Hove, C.L.; Moerloose, K.; Maes, T.; Joos, G.F.; Tournoy, K.G. Cigarette smoke enhances Th-2 driven airway inflammation and delays inhalational tolerance. Respir. Res. 2008, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Miyata, M.; Ohba, T.; Ando, T.; Hatsushika, K.; Suenaga, F.; Shimokawa, N.; Ohnuma, Y.; Katoh, R.; Ogawa, H.; et al. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. J. Allergy Clin. Immunol. 2008, 122, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Maeno, T.; Houghton, A.M.; Quintero, P.A.; Grumelli, S.; Owen, C.A.; Shapiro, S.D. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J. Immunol. 2007, 178, 8090–8096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasen, C.; Turkkila, M.; Bossios, A.; Erlandsson, M.; Andersson, K.M.; Ekerljung, L.; Malmhall, C.; Brisslert, M.; Toyra Silfversward, S.; Lundback, B.; et al. Smoking activates cytotoxic CD8+ T cells and causes survivin release in rheumatoid arthritis. J. Autoimmun. 2017, 78, 101–110. [Google Scholar] [CrossRef]
- Zhong, L.; Qin, L.; Ding, X.; Ma, L.; Wang, Y.; Liu, M.; Chen, H.; Yan, H.; Song, L. The regulatory effect of fermented black barley on the gut microbiota and metabolic dysbiosis in mice exposed to cigarette smoke. Food Res. Int. 2022, 157, 111465. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Targeting apoptosis pathways in cancer therapy. Curr. Cancer Drug Targets 2004, 4, 569–576. [Google Scholar] [CrossRef]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384, 201–232. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, 8730. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Chen, H.; He, A.; Li, H.; Chen, H.; Xie, H.; Luo, L.; Huang, Y.; Chen, J.; Guan, J.; He, Q.; et al. TSSK4 upregulation in alveolar epithelial type-II cells facilitates pulmonary fibrosis through HSP90-AKT signaling restriction and AT-II apoptosis. Cell Death Dis. 2021, 12, 938. [Google Scholar] [CrossRef]
- Dong, W.; Zhu, X.; Liu, X.; Zhao, X.; Lei, X.; Kang, L.; Liu, L. Role of the SENP1-SIRT1 pathway in hyperoxia-induced alveolar epithelial cell injury. Free Radic. Biol. Med. 2021, 173, 142–150. [Google Scholar] [CrossRef]
- Bao, T.; Zhu, H.; Zheng, Y.; Hu, J.; Wang, H.; Cheng, H.; Zhang, Y.; Tian, Z. Expression of long noncoding RNA uc.375 in bronchopulmonary dysplasia and its function in the proliferation and apoptosis of mouse alveolar epithelial cell line MLE 12. Front. Physiol. 2022, 13, 971732. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; MacNee, W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic. Biol. Med. 1996, 21, 669–681. [Google Scholar] [CrossRef]
- Ma, T.L.; Zhou, Y.; Wang, C.; Wang, L.; Chen, J.X.; Yang, H.H.; Zhang, C.Y.; Zhou, Y.; Guan, C.X. Targeting Ferroptosis for Lung Diseases: Exploring Novel Strategies in Ferroptosis-Associated Mechanisms. Oxid. Med. Cell Longev. 2021, 2021, 1098970. [Google Scholar] [CrossRef] [PubMed]
- Batista Napotnik, T.; Polajzer, T.; Miklavcic, D. Cell death due to electroporation-A review. Bioelectrochemistry 2021, 141, 107871. [Google Scholar] [CrossRef]
- Tojo, K.; Yamamoto, N.; Tamada, N.; Mihara, T.; Abe, M.; Nishii, M.; Takeuchi, I.; Goto, T. Early alveolar epithelial cell necrosis is a potential driver of COVID-19-induced acute respiratory distress syndrome. iScience 2023, 26, 105748. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Lin, Z.; Xu, Y.; Guan, L.; Qin, L.; Ding, J.; Zhang, Q.; Zhou, L. Seven ferroptosis-specific expressed genes are considered as potential biomarkers for the diagnosis and treatment of cigarette smoke-induced chronic obstructive pulmonary disease. Ann. Transl. Med. 2022, 10, 331. [Google Scholar] [CrossRef]
- Mercado, N.; Ito, K.; Barnes, P.J. Accelerated ageing of the lung in COPD: New concepts. Thorax 2015, 70, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Brereton, C.F.; Blander, J.M. Responding to infection and apoptosis—A task for TH17 cells. Ann. N. Y. Acad. Sci. 2010, 1209, 56–67. [Google Scholar] [CrossRef]
- Gaillat, J. Should patients with chronic obstructive pulmonary disease be vaccinated against pneumococcal diseases? Expert Rev. Respir. Med. 2009, 3, 585–596. [Google Scholar] [CrossRef]
- Haegele, J.A.; Yessick, A.; Zhu, X. Females with Visual Impairments in Physical Education: Exploring the Intersection between Disability and Gender Identities. Res. Q. Exerc. Sport 2018, 89, 298–308. [Google Scholar] [CrossRef]
- Muradian, K.; Schachtschabel, D.O. The role of apoptosis in aging and age-related disease: Update. Z. Gerontol. Geriatr. 2001, 34, 441–446. [Google Scholar] [CrossRef]
- Yao, H.; Yang, S.R.; Kode, A.; Rajendrasozhan, S.; Caito, S.; Adenuga, D.; Henry, R.; Edirisinghe, I.; Rahman, I. Redox regulation of lung inflammation: Role of NADPH oxidase and NF-kappaB signalling. Biochem. Soc. Trans. 2007, 35, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef] [PubMed]
- Morsch, A.; Wisniewski, E.; Luciano, T.F.; Comin, V.H.; Silveira, G.B.; Marques, S.O.; Thirupathi, A.; Silveira Lock, P.C.; De Souza, C.T. Cigarette smoke exposure induces ROS-mediated autophagy by regulating sestrin, AMPK, and mTOR level in mice. Redox Rep. 2019, 24, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Vella, G.; Ritzmann, F.; Wolf, L.; Kamyschnikov, A.; Stodden, H.; Herr, C.; Slevogt, H.; Bals, R.; Beisswenger, C. IL-17C contributes to NTHi-induced inflammation and lung damage in experimental COPD and is present in sputum during acute exacerbations. PLoS ONE 2021, 16, e0243484. [Google Scholar] [CrossRef] [PubMed]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.E. The pulmonary surfactant: Impact of tobacco smoke and related compounds on surfactant and lung development. Tob. Induc. Dis. 2004, 2, 3–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Sundar, I.K.; Ahmad, T.; Lerner, C.; Gerloff, J.; Friedman, A.E.; Phipps, R.P.; Sime, P.J.; McBurney, M.W.; Guarente, L.; et al. SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L816–L828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopoulou, M.E.; Papakonstantinou, E.; Stolz, D. Matrix Metalloproteinases in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2023, 24, 3786. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Ma, Z.; Hu, Y.; Chen, J.; Shetty, S.; Fu, J. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L847–L853. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Zhou, T.Y.; Zhang, Z.D.; Liu, H.Y.; Zheng, Z.Y.; Xie, H.Q. Current therapeutic strategies for respiratory diseases using mesenchymal stem cells. MedComm 2021, 2, 351–380. [Google Scholar] [CrossRef]
- Babu, K.S.; Kastelik, J.A.; Morjaria, J.B. Inhaled corticosteroids in chronic obstructive pulmonary disease: A pro-con perspective. Br. J. Clin. Pharmacol. 2014, 78, 282–300. [Google Scholar] [CrossRef] [Green Version]
- Jerkic, M.; Szaszi, K.; Laffey, J.G.; Rotstein, O.; Zhang, H. Key Role of Mesenchymal Stromal Cell Interaction with Macrophages in Promoting Repair of Lung Injury. Int. J. Mol. Sci. 2023, 24, 3376. [Google Scholar] [CrossRef]
- van Eerd, E.A.; van der Meer, R.M.; van Schayck, O.C.; Kotz, D. Smoking cessation for people with chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2016, 2016, CD010744. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, M.; Yoshida, Y.; Ohtani, N. Cellular senescence and the tumour microenvironment. Mol. Oncol. 2022, 16, 3333–3351. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Muthumalage, T.; Rahman, I. Clearance of senescent cells reverts the cigarette smoke-induced lung senescence and airspace enlargement in p16-3MR mice. Aging Cell 2023, e13850. [Google Scholar] [CrossRef]
- Lord, J.M. The effect of ageing of the immune system on vaccination responses. Hum. Vaccin Immunother. 2013, 9, 1364–1367. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Lee, K.A.; Flores, R.R.; Jang, I.H.; Saathoff, A.; Robbins, P.D. Immune Senescence, Immunosenescence and Aging. Front. Aging 2022, 3, 900028. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S. Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy. Biol. Pharm. Bull. 2015, 38, 1098–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation and role in disease. Crit. Rev. Clin. Lab. Sci. 2009, 46, 210–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Ndoye, A.; Weeraratna, A.T. Autophagy—An emerging target for melanoma therapy. F1000Res 2016, 5, 8347. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, D.D.; Yan, F.; Jing, Y.Y.; Han, Z.P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Loos, B.; du Toit, A.; Hofmeyr, J.H. Defining and measuring autophagosome flux-concept and reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoubridge, A.P.; Fourrier, C.; Choo, J.M.; Proud, C.G.; Sargeant, T.J.; Rogers, G.B. Gut Microbiome Regulation of Autophagic Flux and Neurodegenerative Disease Risks. Front. Microbiol. 2021, 12, 817433. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, H.; Strulovici-Barel, Y.; Al-Hijji, M.; Ou, X.; Salit, J.; Walters, M.S.; Staudt, M.R.; Kaner, R.J.; Crystal, R.G. Role of OSGIN1 in mediating smoking-induced autophagy in the human airway epithelium. Autophagy 2017, 13, 1205–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szoka, P.; Lachowicz, J.; Cwiklinska, M.; Lukaszewicz, A.; Rybak, A.; Baranowska, U.; Holownia, A. Cigarette Smoke-Induced Oxidative Stress and Autophagy in Human Alveolar Epithelial Cell Line (A549 Cells). Adv. Exp. Med. Biol. 2019, 1176, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kono, Y.; Colley, T.; To, M.; Papaioannou, A.I.; Mercado, N.; Baker, J.R.; To, Y.; Abe, S.; Haruki, K.; Ito, K.; et al. Cigarette smoke-induced impairment of autophagy in macrophages increases galectin-8 and inflammation. Sci. Rep. 2021, 11, 335. [Google Scholar] [CrossRef]
- Tribulatti, M.V.; Carabelli, J.; Prato, C.A.; Campetella, O. Galectin-8 in the onset of the immune response and inflammation. Glycobiology 2020, 30, 134–142. [Google Scholar] [CrossRef]
- Sampson, J.F.; Suryawanshi, A.; Chen, W.S.; Rabinovich, G.A.; Panjwani, N. Galectin-8 promotes regulatory T-cell differentiation by modulating IL-2 and TGFbeta signaling. Immunol. Cell Biol. 2016, 94, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Eckardt, V.; Miller, M.C.; Blanchet, X.; Duan, R.; Leberzammer, J.; Duchene, J.; Soehnlein, O.; Megens, R.T.; Ludwig, A.K.; Dregni, A.; et al. Chemokines and galectins form heterodimers to modulate inflammation. EMBO Rep. 2020, 21, e47852. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dong, X.; Zhao, R.; Zhang, R.; Xu, C.; Wang, X.; Liu, C.; Hu, X.; Huang, S.; Chen, L. Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell Signal 2019, 55, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Button, R.W.; Roberts, S.L.; Willis, T.L.; Hanemann, C.O.; Luo, S. Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.V.; Klionsky, D.J. Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr. Opin. Cell Biol. 1998, 10, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, W.; Zheng, Z.; Wang, W.; Yuan, Y.; Hong, Q.; Lin, J.; Li, X.; Meng, Y. Cigarette smoke-inactivated SIRT1 promotes autophagy-dependent senescence of alveolar epithelial type 2 cells to induce pulmonary fibrosis. Free Radic. Biol. Med. 2021, 166, 116–127. [Google Scholar] [CrossRef]
- Myc, L.A.; Shim, Y.M.; Laubach, V.E.; Dimastromatteo, J. Role of medical and molecular imaging in COPD. Clin. Transl. Med. 2019, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Devine, J.F. Chronic obstructive pulmonary disease: An overview. Am. Health Drug Benefits 2008, 1, 34–42. [Google Scholar]
- Oh, C.M.; Oh, I.H.; Lee, J.K.; Park, Y.H.; Choe, B.K.; Yoon, T.Y.; Choi, J.M. Blood cadmium levels are associated with a decline in lung function in males. Environ. Res. 2014, 132, 119–125. [Google Scholar] [CrossRef]
- Girod, C.E.; King, T.E., Jr. COPD: A dust-induced disease? Chest 2005, 128, 3055–3064. [Google Scholar] [CrossRef]
- Devillier, P. Limitations of drug prescriptions in patients with chronic obstructive pulmonary disease. Rev. Pneumol. Clin. 2004, 60, 203–208. [Google Scholar] [CrossRef]
- Laniado-Laborin, R. Smoking and chronic obstructive pulmonary disease (COPD). Parallel epidemics of the 21 century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharib, S.A.; Manicone, A.M.; Parks, W.C. Matrix metalloproteinases in emphysema. Matrix Biol. 2018, 73, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.A.; Cox, J.H.; Bellac, C.L.; Doucet, A.; Starr, A.E.; Overall, C.M. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008, 112, 3455–3464. [Google Scholar] [CrossRef] [Green Version]
- Gueders, M.M.; Foidart, J.M.; Noel, A.; Cataldo, D.D. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: Potential implications in asthma and other lung diseases. Eur. J. Pharmacol. 2006, 533, 133–144. [Google Scholar] [CrossRef]
- Churg, A.; Wang, R.D.; Tai, H.; Wang, X.; Xie, C.; Wright, J.L. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 2004, 170, 492–498. [Google Scholar] [CrossRef]
- March, T.H.; Wilder, J.A.; Esparza, D.C.; Cossey, P.Y.; Blair, L.F.; Herrera, L.K.; McDonald, J.D.; Campen, M.J.; Mauderly, J.L.; Seagrave, J. Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. Toxicol. Sci. 2006, 92, 545–559. [Google Scholar] [CrossRef]
- Gosselink, J.V.; Hayashi, S.; Elliott, W.M.; Xing, L.; Chan, B.; Yang, L.; Wright, C.; Sin, D.; Pare, P.D.; Pierce, J.A.; et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 1329–1335. [Google Scholar] [CrossRef] [Green Version]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- Pelkonen, M. Smoking: Relationship to chronic bronchitis, chronic obstructive pulmonary disease and mortality. Curr. Opin. Pulm. Med. 2008, 14, 105–109. [Google Scholar] [CrossRef]
- Kesimer, M.; Ford, A.A.; Ceppe, A.; Radicioni, G.; Cao, R.; Davis, C.W.; Doerschuk, C.M.; Alexis, N.E.; Anderson, W.H.; Henderson, A.G.; et al. Airway Mucin Concentration as a Marker of Chronic Bronchitis. N. Engl. J. Med. 2017, 377, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Radicioni, G.; Ceppe, A.; Ford, A.A.; Alexis, N.E.; Barr, R.G.; Bleecker, E.R.; Christenson, S.A.; Cooper, C.B.; Han, M.K.; Hansel, N.N.; et al. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: An analysis of the SPIROMICS cohort. Lancet Respir. Med. 2021, 9, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ye, Z. The Potential Role and Regulatory Mechanisms of MUC5AC in Chronic Obstructive Pulmonary Disease. Molecules 2020, 25, 4437. [Google Scholar] [CrossRef] [PubMed]
- Ghorani, V.; Boskabady, M.H.; Khazdair, M.R.; Kianmeher, M. Experimental animal models for COPD: A methodological review. Tob. Induc. Dis. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.W. Canine models of asthma and COPD. Pulm. Pharmacol. Ther. 2008, 21, 731–742. [Google Scholar] [CrossRef]
- Park, S.S.; Kikkawa, Y.; Goldring, I.P.; Daly, M.M.; Zelefsky, M.; Shim, C.; Spierer, M.; Morita, T. An animal model of cigarette smoking in beagle dogs: Correlative evaluation of effects on pulmonary function, defense, and morphology. Am. Rev. Respir. Dis. 1977, 115, 971–979. [Google Scholar] [CrossRef]
- Plopper, C.G.; Hyde, D.M. The non-human primate as a model for studying COPD and asthma. Pulm. Pharmacol. Ther. 2008, 21, 755–766. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. The Role of HO-1 and Its Crosstalk with Oxidative Stress in Cancer Cell Survival. Cells 2021, 10, 2401. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H.; Asai, K.; Kojima, K.; Sugitani, A.; Kyomoto, Y.; Okamoto, A.; Yamada, K.; Ijiri, N.; Watanabe, T.; Hirata, K.; et al. Astaxanthin Suppresses Cigarette Smoke-Induced Emphysema through Nrf2 Activation in Mice. Mar. Drugs 2019, 17, 673. [Google Scholar] [CrossRef] [Green Version]
- Fratta Pasini, A.M.; Stranieri, C.; Ferrari, M.; Garbin, U.; Cazzoletti, L.; Mozzini, C.; Spelta, F.; Peserico, D.; Cominacini, L. Oxidative stress and Nrf2 expression in peripheral blood mononuclear cells derived from COPD patients: An observational longitudinal study. Respir. Res. 2020, 21, 37. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Baker, J.; Higham, A.; Shah, R.; Montero-Fernandez, A.; Murray, C.; Cooper, N.; Lucas, C.; Fox, C.; Singh, D.; et al. COPD lung studies of Nrf2 expression and the effects of Nrf2 activators. Inflammopharmacology 2022, 30, 1431–1443. [Google Scholar] [CrossRef]
- Smith, D.J.; Yerkovich, S.T.; Towers, M.A.; Carroll, M.L.; Thomas, R.; Upham, J.W. Reduced soluble receptor for advanced glycation end-products in COPD. Eur. Respir. J. 2011, 37, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Hoonhorst, S.J.; Lo Tam Loi, A.T.; Pouwels, S.D.; Faiz, A.; Telenga, E.D.; van den Berge, M.; Koenderman, L.; Lammers, J.W.; Boezen, H.M.; van Oosterhout, A.J.; et al. Advanced glycation endproducts and their receptor in different body compartments in COPD. Respir. Res. 2016, 17, 46. [Google Scholar] [CrossRef] [Green Version]
- Ferhani, N.; Letuve, S.; Kozhich, A.; Thibaudeau, O.; Grandsaigne, M.; Maret, M.; Dombret, M.C.; Sims, G.P.; Kolbeck, R.; Coyle, A.J.; et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 917–927. [Google Scholar] [CrossRef]
- Lee, H.; Park, J.R.; Kim, W.J.; Sundar, I.K.; Rahman, I.; Park, S.M.; Yang, S.R. Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling. FASEB J. 2017, 31, 2076–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambamurthy, N.; Leme, A.S.; Oury, T.D.; Shapiro, S.D. The receptor for advanced glycation end products (RAGE) contributes to the progression of emphysema in mice. PLoS ONE 2015, 10, e0118979. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Henry, A.P.; Hodge, E.; Kheirallah, A.K.; Billington, C.K.; Rimington, T.L.; Bhaker, S.K.; Obeidat, M.; Melen, E.; Merid, S.K.; et al. The Ser82 RAGE Variant Affects Lung Function and Serum RAGE in Smokers and sRAGE Production In Vitro. PLoS ONE 2016, 11, e0164041. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.T.; Kim, D.K.; Cockayne, D.A.; Belousov, A.; Bitter, H.; Cho, M.H.; Duvoix, A.; Edwards, L.D.; Lomas, D.A.; Miller, B.E.; et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 188, 948–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaens, K.H.; Ferreira, I.; van der Kallen, C.J.; van Greevenbroek, M.M.; Blaak, E.E.; Feskens, E.J.; Dekker, J.M.; Nijpels, G.; Heine, R.J.; Hart, L.M.; et al. Association of polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating RAGE levels. J. Clin. Endocrinol. Metab. 2009, 94, 5174–5180. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, X.; Wang, H.; Xie, F.; Liu, H.; Xie, J. Cigarette smoke triggers inflammation mediated by autophagy in BEAS-2B cells. Ecotoxicol. Environ. Saf. 2019, 184, 109617. [Google Scholar] [CrossRef] [PubMed]
- Marumo, S.; Hoshino, Y.; Kiyokawa, H.; Tanabe, N.; Sato, A.; Ogawa, E.; Muro, S.; Hirai, T.; Mishima, M. p38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm. Med. 2014, 14, 79. [Google Scholar] [CrossRef] [Green Version]
- Renda, T.; Baraldo, S.; Pelaia, G.; Bazzan, E.; Turato, G.; Papi, A.; Maestrelli, P.; Maselli, R.; Vatrella, A.; Fabbri, L.M.; et al. Increased activation of p38 MAPK in COPD. Eur. Respir. J. 2008, 31, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Mercer, B.A.; Kolesnikova, N.; Sonett, J.; D’Armiento, J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J. Biol. Chem. 2004, 279, 17690–17696. [Google Scholar] [CrossRef] [Green Version]
- Zong, D.; Li, J.; Cai, S.; He, S.; Liu, Q.; Jiang, J.; Chen, S.; Long, Y.; Chen, Y.; Chen, P.; et al. Notch1 regulates endothelial apoptosis via the ERK pathway in chronic obstructive pulmonary disease. Am. J. Physiol. Cell Physiol. 2018, 315, C330–C340. [Google Scholar] [CrossRef]
- Deshmukh, H.S.; Case, L.M.; Wesselkamper, S.C.; Borchers, M.T.; Martin, L.D.; Shertzer, H.G.; Nadel, J.A.; Leikauf, G.D. Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activation. Am. J. Respir. Crit. Care Med. 2005, 171, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal 2014, 20, 2838–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Murugesan, P.; Zhang, P.; Xu, S.; Peng, L.; Wang, C.; Cai, H. NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation. Antioxidants 2022, 11, 1539. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.H.; Brassington, K.; Almerdasi, S.A.; Dobric, A.; De Luca, S.N.; Coward-Smith, M.; Wang, H.; Mou, K.; Akhtar, A.; Alateeq, R.A.; et al. Inhibition of oxidative stress by apocynin attenuated chronic obstructive pulmonary disease progression and vascular injury by cigarette smoke exposure. Br. J. Pharmacol. 2023, 16068. [Google Scholar] [CrossRef]
- Schiffers, C.; Reynaert, N.L.; Wouters, E.F.M.; van der Vliet, A. Redox Dysregulation in Aging and COPD: Role of NOX Enzymes and Implications for Antioxidant Strategies. Antioxidants 2021, 10, 11799. [Google Scholar] [CrossRef]
- Hollins, F.; Sutcliffe, A.; Gomez, E.; Berair, R.; Russell, R.; Szyndralewiez, C.; Saunders, R.; Brightling, C. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir. Res. 2016, 17, 84. [Google Scholar] [CrossRef] [Green Version]
- Seimetz, M.; Sommer, N.; Bednorz, M.; Pak, O.; Veith, C.; Hadzic, S.; Gredic, M.; Parajuli, N.; Kojonazarov, B.; Kraut, S.; et al. NADPH oxidase subunit NOXO1 is a target for emphysema treatment in COPD. Nat. Metab. 2020, 2, 532–546. [Google Scholar] [CrossRef]
- Schiffers, C.; van de Wetering, C.; Bauer, R.A.; Habibovic, A.; Hristova, M.; Dustin, C.M.; Lambrichts, S.; Vacek, P.M.; Wouters, E.F.; Reynaert, N.L.; et al. Downregulation of epithelial DUOX1 in chronic obstructive pulmonary disease. JCI Insight 2021, 6, 142189. [Google Scholar] [CrossRef]
- Nagai, K.; Betsuyaku, T.; Suzuki, M.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid. Redox Signal 2008, 10, 705–714. [Google Scholar] [CrossRef]
- van der Vliet, A.; Danyal, K.; Heppner, D.E. Dual oxidase: A novel therapeutic target in allergic disease. Br. J. Pharmacol. 2018, 175, 1401–1418. [Google Scholar] [CrossRef] [Green Version]
- Wesley, U.V.; Bove, P.F.; Hristova, M.; McCarthy, S.; van der Vliet, A. Airway epithelial cell migration and wound repair by ATP-mediated activation of dual oxidase 1. J. Biol. Chem. 2007, 282, 3213–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, J.J.; Senior, R.M. Matrix metalloproteinase-9 in lung remodeling. Am. J. Respir. Cell Mol. Biol. 2003, 28, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, N.; Khan, M.; Paintlia, M.K.; Singh, I.; Hoda, M.N.; Giri, S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J. Immunol. 2009, 182, 8005–8014. [Google Scholar] [CrossRef] [Green Version]
- Polverino, F.; Wu, T.D.; Rojas-Quintero, J.; Wang, X.; Mayo, J.; Tomchaney, M.; Tram, J.; Packard, S.; Zhang, D.; Cleveland, K.H.; et al. Metformin: Experimental and Clinical Evidence for a Potential Role in Emphysema Treatment. Am. J. Respir. Crit. Care Med. 2021, 204, 651–666. [Google Scholar] [CrossRef]
- Oslan, S.N.H.; Tan, J.S.; Oslan, S.N.; Matanjun, P.; Mokhtar, R.A.M.; Shapawi, R.; Huda, N. Haematococcus pluvialis as a Potential Source of Astaxanthin with Diverse Applications in Industrial Sectors: Current Research and Future Directions. Molecules 2021, 26, 6470. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Yoshida, H.; Kondo, K. Potential Anti-Atherosclerotic Properties of Astaxanthin. Mar. Drugs 2016, 14, 35. [Google Scholar] [CrossRef]
- Piermarocchi, S.; Saviano, S.; Parisi, V.; Tedeschi, M.; Panozzo, G.; Scarpa, G.; Boschi, G.; Lo Giudice, G.; Carmis Study, G. Carotenoids in Age-related Maculopathy Italian Study (CARMIS): Two-year results of a randomized study. Eur. J. Ophthalmol. 2012, 22, 216–225. [Google Scholar] [CrossRef]
- Liu, S.Z.; Ali, A.S.; Campbell, M.D.; Kilroy, K.; Shankland, E.G.; Roshanravan, B.; Marcinek, D.J.; Conley, K.E. Building strength, endurance, and mobility using an astaxanthin formulation with functional training in elderly. J. Cachexia Sarcopenia Muscle 2018, 9, 826–833. [Google Scholar] [CrossRef]
- Deng, M.; Tong, R.; Bian, Y.; Hou, G. Astaxanthin attenuates cigarette smoking-induced oxidative stress and inflammation in a sirtuin 1-dependent manner. Biomed. Pharmacother. 2023, 159, 114230. [Google Scholar] [CrossRef]
- Ridzuan, N.; Zakaria, N.; Widera, D.; Sheard, J.; Morimoto, M.; Kiyokawa, H.; Mohd Isa, S.A.; Chatar Singh, G.K.; Then, K.Y.; Ooi, G.C.; et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles ameliorate airway inflammation in a rat model of chronic obstructive pulmonary disease (COPD). Stem Cell Res. Ther. 2021, 12, 54. [Google Scholar] [CrossRef]
- Craparo, E.F.; Cabibbo, M.; Scialabba, C.; Giammona, G.; Cavallaro, G. Inhalable Formulation Based on Lipid-Polymer Hybrid Nanoparticles for the Macrophage Targeted Delivery of Roflumilast. Biomacromolecules 2022, 23, 3439–3451. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, H.; Song, L. Novel drug delivery systems targeting oxidative stress in chronic obstructive pulmonary disease: A review. J. Nanobiotechnol. 2020, 18, 145. [Google Scholar] [CrossRef] [PubMed]
- Barjaktarevic, I.Z.; Arredondo, A.F.; Cooper, C.B. Positioning new pharmacotherapies for COPD. Int. J. Chron. Obstruct. Pulm. Dis. 2015, 10, 1427–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paranjpe, M.; Muller-Goymann, C.C. Nanoparticle-mediated pulmonary drug delivery: A review. Int. J. Mol. Sci. 2014, 15, 5852–5873. [Google Scholar] [CrossRef] [Green Version]
- Geiser, M.; Quaile, O.; Wenk, A.; Wigge, C.; Eigeldinger-Berthou, S.; Hirn, S.; Schaffler, M.; Schleh, C.; Moller, W.; Mall, M.A.; et al. Cellular uptake and localization of inhaled gold nanoparticles in lungs of mice with chronic obstructive pulmonary disease. Part Fibre Toxicol. 2013, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Barrera, L.; Mendoza, F.; Zuniga, J.; Estrada, A.; Zamora, A.C.; Melendro, E.I.; Ramirez, R.; Pardo, A.; Selman, M. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2008, 177, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Carcoforo, P.; Feo, C.; Sortini, D.; Pozza, E.; Carrella, G.; Sortini, A. Localization of pulmonary nodules. Chest 2004, 125, 796; author reply 796–797. [Google Scholar] [CrossRef]
- Kanoh, S.; Kobayashi, H.; Motoyoshi, K. Exhaled ethane: An in vivo biomarker of lipid peroxidation in interstitial lung diseases. Chest 2005, 128, 2387–2392. [Google Scholar] [CrossRef]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Jiang, W.H.; Tian, T.; Tan, H.S.; Chen, Y.; Qiao, G.L.; Han, J.; Huang, S.Y.; Yang, Y.; Li, S.; et al. CBX6 overexpression contributes to tumor progression and is predictive of a poor prognosis in hepatocellular carcinoma. Oncotarget 2017, 8, 18872–18884. [Google Scholar] [CrossRef] [Green Version]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarman, E.R.; Khambata, V.S.; Cope, C.; Jones, P.; Roger, J.; Ye, L.Y.; Duggan, N.; Head, D.; Pearce, A.; Press, N.J.; et al. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014, 50, 158–169. [Google Scholar] [CrossRef]
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-beta and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 22, 6107. [Google Scholar] [CrossRef]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the "RAGE" in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.N.; Oury, T.D. The perplexing role of RAGE in pulmonary fibrosis: Causality or casualty? Ther. Adv. Respir. Dis. 2021, 15, 17534666211016071. [Google Scholar] [CrossRef] [PubMed]
- Queisser, M.A.; Kouri, F.M.; Konigshoff, M.; Wygrecka, M.; Schubert, U.; Eickelberg, O.; Preissner, K.T. Loss of RAGE in pulmonary fibrosis: Molecular relations to functional changes in pulmonary cell types. Am. J. Respir. Cell Mol. Biol. 2008, 39, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Sitzia, C.; Pistelli, L.; Cardani, R.; Rigolini, R.; Ranucci, M.; Corsi Romanelli, M.M. Soluble Receptor for Advanced Glycation End Products and Its Forms in COVID-19 Patients with and without Diabetes Mellitus: A Pilot Study on Their Role as Disease Biomarkers. J. Clin. Med. 2020, 9, 3785. [Google Scholar] [CrossRef]
- Buckley, S.T.; Ehrhardt, C. The receptor for advanced glycation end products (RAGE) and the lung. J. Biomed. Biotechnol. 2010, 2010, 917108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhage, T.; Wirth, T.; Schutz, P.; Becker, P.; Lueken, A.; Skryabin, B.V.; Wittkowski, H.; Foell, D. The Receptor for Advanced Glycation Endproducts (RAGE) Contributes to Severe Inflammatory Liver Injury in Mice. Front. Immunol. 2020, 11, 1157. [Google Scholar] [CrossRef]
- Lugade, A.A.; Bogner, P.N.; Thatcher, T.H.; Sime, P.J.; Phipps, R.P.; Thanavala, Y. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J. Immunol. 2014, 192, 5226–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Wang, T.; Shen, Y.; Xu, D.; Li, X.; An, J.; Dong, J.; Li, D.; Wen, F.; Chen, L. Knockout of RAGE ameliorates mainstream cigarette smoke-induced airway inflammation in mice. Int. Immunopharmacol. 2017, 50, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Re, S.L.; Giordano, G.; Yakoub, Y.; Devosse, R.; Uwambayinema, F.; Couillin, I.; Ryffel, B.; Marbaix, E.; Lison, D.; Huaux, F. Uncoupling between inflammatory and fibrotic responses to silica: Evidence from MyD88 knockout mice. PLoS ONE 2014, 9, e99383. [Google Scholar] [CrossRef]
- Kumar, V.; Fleming, T.; Terjung, S.; Gorzelanny, C.; Gebhardt, C.; Agrawal, R.; Mall, M.A.; Ranzinger, J.; Zeier, M.; Madhusudhan, T.; et al. Homeostatic nuclear RAGE-ATM interaction is essential for efficient DNA repair. Nucleic Acids Res. 2017, 45, 10595–10613. [Google Scholar] [CrossRef]
- Schupp, J.C.; Binder, H.; Jager, B.; Cillis, G.; Zissel, G.; Muller-Quernheim, J.; Prasse, A. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Meyer, A.; Buhl, R.; Magnussen, H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur. Respir. J. 1994, 7, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Comeau, M.R.; Ziegler, S.F. The influence of TSLP on the allergic response. Mucosal Immunol. 2010, 3, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Zhitkovich, A. N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem. Res. Toxicol. 2019, 32, 1318–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9, 22129. [Google Scholar] [CrossRef] [Green Version]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Ask, K.; Bonniaud, P.; Maass, K.; Eickelberg, O.; Margetts, P.J.; Warburton, D.; Groffen, J.; Gauldie, J.; Kolb, M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. Int. J. Biochem. Cell Biol. 2008, 40, 484–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Muller-Quernheim, J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Fortin, M.; D’Anjou, H.; Higgins, M.E.; Gougeon, J.; Aube, P.; Moktefi, K.; Mouissi, S.; Seguin, S.; Seguin, R.; Renzi, P.M.; et al. A multi-target antisense approach against PDE4 and PDE7 reduces smoke-induced lung inflammation in mice. Respir. Res. 2009, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Azuma, A.; Cottin, V.; Hesslinger, C.; Stowasser, S.; Valenzuela, C.; Wijsenbeek, M.S.; Zoz, D.F.; Voss, F.; Maher, T.M.; et al. Trial of a Preferential Phosphodiesterase 4B Inhibitor for Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2022, 386, 2178–2187. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberg, M.; Danovitch, I.; IsHak, W.W. Quality of life and smoking. Am. J. Addict. 2014, 23, 540–562. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27; discussion 18–27. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.D.; Silverman, D.T.; Hollenbeck, A.R.; Schatzkin, A.; Abnet, C.C. Association between smoking and risk of bladder cancer among men and women. JAMA 2011, 306, 737–745. [Google Scholar] [CrossRef]
- Ji, B.T.; Chow, W.H.; Gridley, G.; McLaughlin, J.K.; Dai, Q.; Wacholder, S.; Hatch, M.C.; Gao, Y.T.; Fraumeni, J.F., Jr. Dietary factors and the risk of pancreatic cancer: A case-control study in Shanghai China. Cancer Epidemiol. Biomark. Prev. 1995, 4, 885–893. [Google Scholar]
- Hunt, J.D.; van der Hel, O.L.; McMillan, G.P.; Boffetta, P.; Brennan, P. Renal cell carcinoma in relation to cigarette smoking: Meta-analysis of 24 studies. Int. J. Cancer 2005, 114, 101–108. [Google Scholar] [CrossRef]
- Lagergren, J.; Bergstrom, R.; Lindgren, A.; Nyren, O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N. Engl. J. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef]
- Hashibe, M.; Brennan, P.; Chuang, S.C.; Boccia, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomark. Prev. 2009, 18, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Phillips, D.H. Polycyclic aromatic hydrocarbons in the diet. Mutat. Res. 1999, 443, 139–147. [Google Scholar] [CrossRef]
- Toyokuni, S. Role of iron in carcinogenesis: Cancer as a ferrotoxic disease. Cancer Sci. 2009, 100, 9–16. [Google Scholar] [CrossRef]
- Mengozzi, A.; Pugliese, N.R.; Chiriaco, M.; Masi, S.; Virdis, A.; Taddei, S. Microvascular Ageing Links Metabolic Disease to Age-Related Disorders: The Role of Oxidative Stress and Inflammation in Promoting Microvascular Dysfunction. J. Cardiovasc. Pharmacol. 2021, 78, S78–S87. [Google Scholar] [CrossRef] [PubMed]
- Fleshner, N.E.; Klotz, L.H. Diet, androgens, oxidative stress and prostate cancer susceptibility. Cancer Metastasis Rev. 1998, 17, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxid. Med. Cell Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef] [Green Version]
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal 2020, 33, 435–454. [Google Scholar] [CrossRef]
- Wang, R.; Dashwood, W.M.; Nian, H.; Lohr, C.V.; Fischer, K.A.; Tsuchiya, N.; Nakagama, H.; Ashktorab, H.; Dashwood, R.H. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine (PhIP). Int. J. Cancer 2011, 128, 2581–2590. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Ahn, K.S.; Aggarwal, B.B. Transcription factor NF-kappaB: A sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 2005, 1056, 218–233. [Google Scholar] [CrossRef]
- Hasnis, E.; Bar-Shai, M.; Burbea, Z.; Reznick, A.Z. Mechanisms underlying cigarette smoke-induced NF-kappaB activation in human lymphocytes: The role of reactive nitrogen species. J. Physiol. Pharmacol. 2007, 58 (Suppl. S5), 275–287. [Google Scholar]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of NRF2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [Green Version]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 23. [Google Scholar] [CrossRef]
- Hammad, A.; Namani, A.; Elshaer, M.; Wang, X.J.; Tang, X. “NRF2 addiction” in lung cancer cells and its impact on cancer therapy. Cancer Lett. 2019, 467, 40–49. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control Release 2000, 65, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Heidel, J.D.; Davis, M.E. Clinical developments in nanotechnology for cancer therapy. Pharm. Res. 2011, 28, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.Y.; Zhou, Y.M.; Pinkerton, K.E. NF-kappaB inhibition is involved in tobacco smoke-induced apoptosis in the lungs of rats. Toxicol. Appl. Pharmacol. 2008, 230, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Janowitz, T.; Kleeman, S.; Vonderheide, R.H. Reconsidering Dexamethasone for Antiemesis when Combining Chemotherapy and Immunotherapy. Oncologist 2021, 26, 269–273. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, Q.; Feng, S.; Li, C.; Wang, L.; Zhao, X.; Yang, Z.; Li, Z.; Luo, H.; Liu, R.; et al. Safety and Efficacy of PD-1/PD-L1 inhibitors combined with radiotherapy in patients with non-small-cell lung cancer: A systematic review and meta-analysis. Cancer Med. 2021, 10, 1222–1239. [Google Scholar] [CrossRef]
- Su, S.; Kang, P.M. Recent Advances in Nanocarrier-Assisted Therapeutics Delivery Systems. Pharmaceutics 2020, 12, 837. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly-Battaglini, A.; Hammarstrom, C.; Stankovic, B.; Aamodt, H.; Stjarne, J.; Brustugun, O.T.; Helland, A.; Oynebraten, I.; Corthay, A. Rituximab efficiently depletes B cells in lung tumors and normal lung tissue. F1000Res 2016, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazieres, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qin, C.; An, C.; Zheng, X.; Wen, S.; Chen, W.; Liu, X.; Lv, Z.; Yang, P.; Xu, W.; et al. Application of the CRISPR/Cas9-based gene editing technique in basic research, diagnosis, and therapy of cancer. Mol. Cancer 2021, 20, 126. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Xu, J.; Chen, S.J.; Collins, M.A.; Zhou, J.; Powell, D.J., Jr.; Melenhorst, J.J. Engineering enhanced CAR T-cells for improved cancer therapy. Nat. Cancer 2021, 2, 780–793. [Google Scholar] [CrossRef]
- Qu, J.; Mei, Q.; Chen, L.; Zhou, J. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell lung cancer (NSCLC): Current status and future perspectives. Cancer Immunol. Immunother. 2021, 70, 619–631. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Sadeghi, S.; Tehrani, F.R.; Tahmasebi, S.; Shafiee, A.; Hashemi, S.M. Exosome engineering in cell therapy and drug delivery. Inflammopharmacology 2023, 31, 145–169. [Google Scholar] [CrossRef]
- Raghav, A.; Khan, Z.A.; Upadhayay, V.K.; Tripathi, P.; Gautam, K.A.; Mishra, B.K.; Ahmad, J.; Jeong, G.B. Mesenchymal Stem Cell-Derived Exosomes Exhibit Promising Potential for Treating SARS-CoV-2-Infected Patients. Cells 2021, 10, 587. [Google Scholar] [CrossRef]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Q.; Liu, X.; Yang, T.; Cui, K.; Kong, L.; Yang, C.; Zhang, Z. Nanomedicine for acute respiratory distress syndrome: The latest application, targeting strategy, and rational design. Acta Pharm. Sin. B 2021, 11, 3060–3091. [Google Scholar] [CrossRef] [PubMed]
- Moazed, F.; Hendrickson, C.; Jauregui, A.; Gotts, J.; Conroy, A.; Delucchi, K.; Zhuo, H.; Arambulo, M.; Vessel, K.; Ke, S.; et al. Cigarette Smoke Exposure and Acute Respiratory Distress Syndrome in Sepsis: Epidemiology, Clinical Features, and Biologic Markers. Am. J. Respir. Crit. Care Med. 2022, 205, 927–935. [Google Scholar] [CrossRef]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Mantelli, F.; Allegretti, M.; Balk, R.A. The Role of Interleukin-8 in Lung Inflammation and Injury: Implications for the Management of COVID-19 and Hyperinflammatory Acute Respiratory Distress Syndrome. Front. Pharmacol. 2021, 12, 808797. [Google Scholar] [CrossRef]
- Moazed, F.; Burnham, E.L.; Vandivier, R.W.; O’Kane, C.M.; Shyamsundar, M.; Hamid, U.; Abbott, J.; Thickett, D.R.; Matthay, M.A.; McAuley, D.F.; et al. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax 2016, 71, 1130–1136. [Google Scholar] [CrossRef] [Green Version]
- Aslan, A.; Aslan, C.; Zolbanin, N.M.; Jafari, R. Acute respiratory distress syndrome in COVID-19: Possible mechanisms and therapeutic management. Pneumonia 2021, 13, 14. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2021, 253, 17–30. [Google Scholar] [CrossRef]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco Smoking Increases the Lung Gene Expression of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Voinsky, I.; Gurwitz, D. Smoking and COVID-19: Similar bronchial ACE2 and TMPRSS2 expression and higher TMPRSS4 expression in current versus never smokers. Drug Dev. Res. 2020, 81, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Nawijn, M.C.; Timens, W. Can ACE2 expression explain SARS-CoV-2 infection of the respiratory epithelia in COVID-19? Mol. Syst. Biol. 2020, 16, e9841. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Kashiouris, M.G.; L’Heureux, M.; Fisher, B.J.; Leichtle, S.W.; Truwit, J.D.; Nanchal, R.; Hite, R.D.; Morris, P.E.; Martin, G.S.; et al. Biological Effects of Intravenous Vitamin C on Neutrophil Extracellular Traps and the Endothelial Glycocalyx in Patients with Sepsis-Induced ARDS. Nutrients 2022, 14, 4415. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Panel on Dietary Antioxidants and Related Compounds. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids; Institute of Medicine (US) Panel on Dietary Antioxidants and Related Compounds: Washington, DC, USA, 2000.
- Azzouz, D.; Khan, M.A.; Palaniyar, N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. 2021, 7, 113. [Google Scholar] [CrossRef]
- Jiang, S.; Li, S.; Hu, J.; Xu, X.; Wang, X.; Kang, X.; Qi, J.; Lu, X.; Wu, J.; Du, Y.; et al. Combined delivery of angiopoietin-1 gene and simvastatin mediated by anti-intercellular adhesion molecule-1 antibody-conjugated ternary nanoparticles for acute lung injury therapy. Nanomedicine 2019, 15, 25–36. [Google Scholar] [CrossRef]
- MacIntyre, N.; Huang, Y.C. Acute exacerbations and respiratory failure in chronic obstructive pulmonary disease. Proc. Am. Thorac Soc. 2008, 5, 530–535. [Google Scholar] [CrossRef]
- Bach, P.B.; Brown, C.; Gelfand, S.E.; McCrory, D.C.; American College of Physicians-American Society of Internal Medicine. Management of acute exacerbations of chronic obstructive pulmonary disease: A summary and appraisal of published evidence. Ann. Intern Med. 2001, 134, 600–620. [Google Scholar] [CrossRef]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef] [Green Version]
- Fagon, J.Y.; Chastre, J.; Trouillet, J.L.; Domart, Y.; Dombret, M.C.; Bornet, M.; Gibert, C. Characterization of distal bronchial microflora during acute exacerbation of chronic bronchitis. Use of the protected specimen brush technique in 54 mechanically ventilated patients. Am. Rev. Respir. Dis. 1990, 142, 1004–1008. [Google Scholar] [CrossRef]
- Monso, E.; Ruiz, J.; Rosell, A.; Manterola, J.; Fiz, J.; Morera, J.; Ausina, V. Bacterial infection in chronic obstructive pulmonary disease. A study of stable and exacerbated outpatients using the protected specimen brush. Am. J. Respir. Crit. Care Med. 1995, 152, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Evans, N.; Grant, B.J.; Murphy, T.F. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N. Engl. J. Med. 2002, 347, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Ma, A.; Zhang, L.; Guo, J.; Liu, Q.; Petersen, F.; Wang, Z.; Yu, X. Acute exacerbations of chronic obstructive pulmonary disease in a cohort of Chinese never smokers goes along with decreased risks of recurrent acute exacerbation, emphysema and comorbidity of lung cancer as well as decreased levels of circulating eosinophils and basophils. Front. Med. 2022, 9, 907893. [Google Scholar] [CrossRef]
- Li, X.; Wu, Z.; Xue, M.; Du, W. Smoking status affects clinical characteristics and disease course of acute exacerbation of chronic obstructive pulmonary disease: A prospectively observational study. Chron. Respir. Dis. 2020, 17, 1479973120916184. [Google Scholar] [CrossRef] [Green Version]
- Mortality, G.B.D.; Causes of Death, C. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef] [Green Version]
- Messner, B.; Bernhard, D. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, J.A.; Barua, R.S. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J. Am. Coll. Cardiol. 2004, 43, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Nakano, Y.; Adachi, S.; Murohara, T. Effects of Tobacco Smoking on Cardiovascular Disease. Circ. J. 2019, 83, 1980–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahdah, A.; Jaggers, R.M.; Sreejit, G.; Johnson, J.; Kanuri, B.; Murphy, A.J.; Nagareddy, P.R. Immunological Insights into Cigarette Smoking-Induced Cardiovascular Disease Risk. Cells 2022, 11, 3190. [Google Scholar] [CrossRef]
- Morris, P.B.; Ference, B.A.; Jahangir, E.; Feldman, D.N.; Ryan, J.J.; Bahrami, H.; El-Chami, M.F.; Bhakta, S.; Winchester, D.E.; Al-Mallah, M.H.; et al. Cardiovascular Effects of Exposure to Cigarette Smoke and Electronic Cigarettes: Clinical Perspectives from the Prevention of Cardiovascular Disease Section Leadership Council and Early Career Councils of the American College of Cardiology. J. Am. Coll. Cardiol. 2015, 66, 1378–1391. [Google Scholar] [CrossRef] [Green Version]
- Celermajer, D.S.; Sorensen, K.E.; Georgakopoulos, D.; Bull, C.; Thomas, O.; Robinson, J.; Deanfield, J.E. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation 1993, 88, 2149–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, R.S.; Ambrose, J.A.; Eales-Reynolds, L.J.; DeVoe, M.C.; Zervas, J.G.; Saha, D.C. Dysfunctional endothelial nitric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation 2001, 104, 1905–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Han, W.; Giraldo, C.; De Li, Y.; Block, E.R. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, N.; Toda, H. Nitric oxide-mediated blood flow regulation as affected by smoking and nicotine. Eur. J. Pharmacol. 2010, 649, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.A.; Weisbrod, R.M.; Gericke, M.; Yaghoubi, M.; Bierl, C.; Bolotina, V.M. Mechanism of nitric oxide-induced vasodilatation: Refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ. Res. 1999, 84, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Barua, R.S.; Ambrose, J.A.; Srivastava, S.; DeVoe, M.C.; Eales-Reynolds, L.J. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: An in vitro demonstration in human coronary artery endothelial cells. Circulation 2003, 107, 2342–2347. [Google Scholar] [CrossRef] [Green Version]
- Ismaeel, A.; Brumberg, R.S.; Kirk, J.S.; Papoutsi, E.; Farmer, P.J.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Sawicki, I.; Koutakis, P. Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease. Antioxidants 2018, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bian, Y.; Zhang, R.; Liu, X.; Ni, L.; Ma, B.; Zeng, R.; Zhao, Z.; Song, X.; Liu, C. Melatonin alleviates cigarette smoke-induced endothelial cell pyroptosis through inhibiting ROS/NLRP3 axis. Biochem. Biophys. Res. Commun. 2019, 519, 402–408. [Google Scholar] [CrossRef]
- Mazzone, A.; Cusa, C.; Mazzucchelli, I.; Vezzoli, M.; Ottini, E.; Ghio, S.; Tossini, G.; Pacifici, R.; Zuccaro, P. Cigarette smoking and hypertension influence nitric oxide release and plasma levels of adhesion molecules. Clin. Chem. Lab. Med. 2001, 39, 822–826. [Google Scholar] [CrossRef]
- Craig, W.Y.; Palomaki, G.E.; Haddow, J.E. Cigarette smoking and serum lipid and lipoprotein concentrations: An analysis of published data. BMJ 1989, 298, 784–788. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, T.; Yla-Herttuala, S.; Luoma, J.; Kurz, S.; Munzel, T.; Just, H.; Olschewski, M.; Drexler, H. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia. Role of oxidized LDL. Circulation 1996, 93, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calo, L.A. The role of oxidized low-density lipoproteins in atherosclerosis: The myths and the facts. Mediat. Inflamm. 2013, 2013, 714653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, M.; Wada, H.; Ono, K.; Yamakage, H.; Satoh-Asahara, N.; Shimada, S.; Akao, M.; Morimoto, T.; Shimatsu, A.; Takahashi, Y.; et al. Smoking cessation reduces the lectin-like low-density lipoprotein receptor index, an independent cardiovascular risk marker of vascular inflammation. Heart Vessels 2018, 33, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]


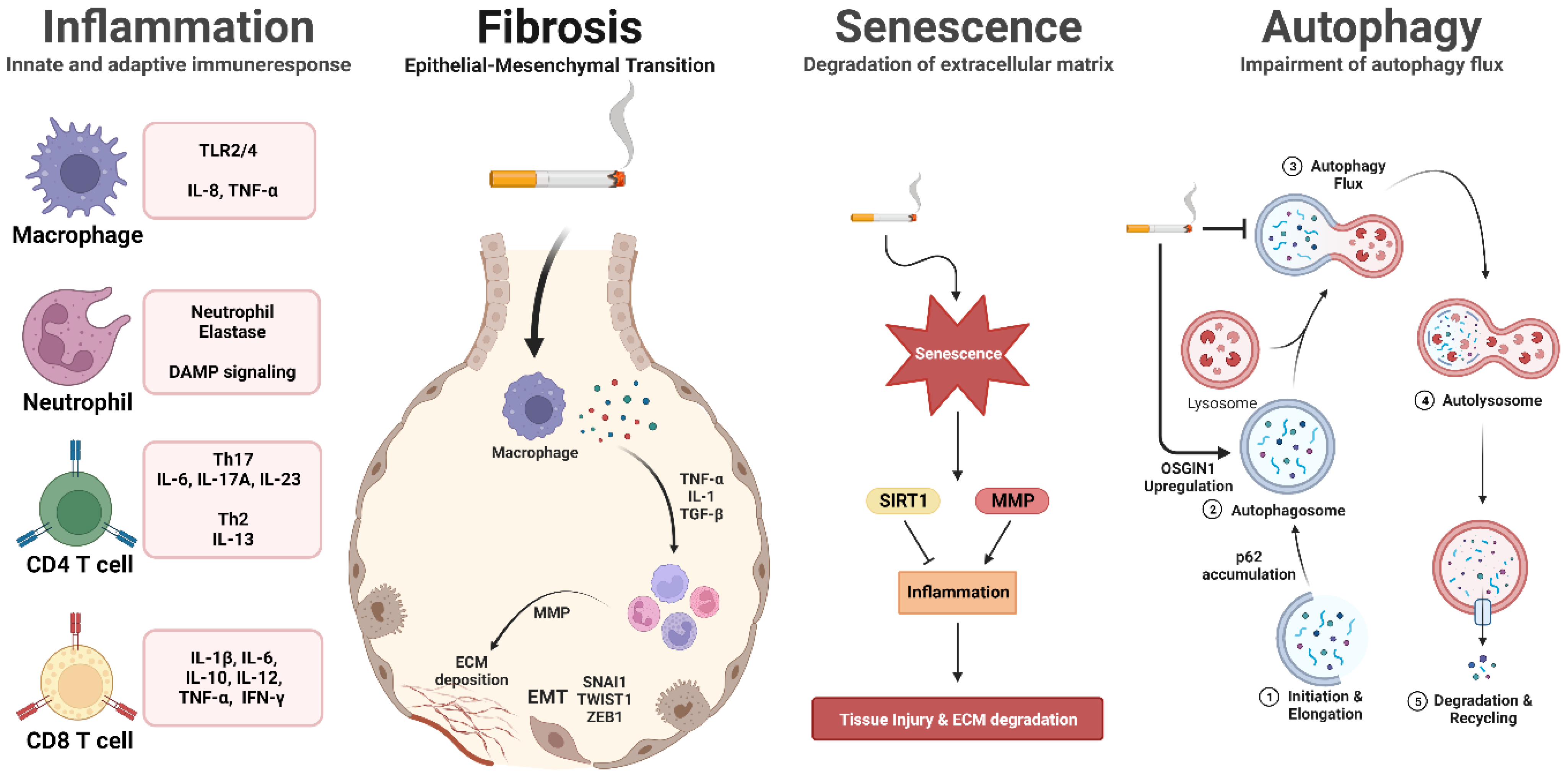{kind=link}
| Respiratory Disease | Therapy | CS-Induced Mechanism | Reference |
|---|---|---|---|
| COPD | Metformin | Inhibition of apoptosis through regulation of AMP kinase. | [226] |
| Astaxanthin | Inhibition of Nrf2-modulated oxidative stress and regulation of NF-κB-related inflammatory responses through binding with SIRT1. | [231] | |
| HUC-MSC- derived EVs | Alleviation of airway inflammation in the CS-induced rat model. | [232] | |
| LPHNPs | Upregulation of cytocompatibility toward bronchial epithelial cells and macrophages. | [233] | |
| Pulmonary Fibrosis | N-acetylcysteine | Replenish the levels of the antioxidant glutathione and reduce reactive oxygen species by targeting pro-inflammatory cytokines such as TNF-α, IL-1, and TGF-β. | [262,263] |
| PDE4B inhibitors | Reducing inflammation and fibrotic processes by inhibiting the degradation of cAMP. | [269] | |
| Cancer | PD-1/PD-L1 inhibitors | Enhance the immune system’s ability to target cancer cells and restore immune cell functionality by combining PD-L1 from cancer and PD-1 from T cells. | [309] |
| Trastuzumab | Durable anticancer activity in patients with previously treated HER2-mutant of NSCLC. | [291] | |
| Rituximab | Depletes CD20-positive B cells in lung tumors. | [292] | |
| CRISPR-Cas9 | Modify or correct disease-related genes causing mutations in cancer cells. | [316] | |
| CAR-T-cell | improve the survival and functionality of engineered T-cells by reducing ROS and eliminating cancer cells. | [318] | |
| MSCs-exosome | Cell-to-cell communication within the tumor microenvironment and suppression of angiogenesis. | [321] | |
| ARDS and AE-COPD | Vitamin C | Antioxidant vitamin C inhibits ROS-mediated NET by inactivating NADPH oxidase. | [335] |
| ICAM/NLC | Decrease pro-inflammatory cytokines in the ARDS mouse model. | [338] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cha, S.-R.; Jang, J.; Park, S.-M.; Ryu, S.M.; Cho, S.-J.; Yang, S.-R. Cigarette Smoke-Induced Respiratory Response: Insights into Cellular Processes and Biomarkers. Antioxidants 2023, 12, 1210. https://doi.org/10.3390/antiox12061210
Cha S-R, Jang J, Park S-M, Ryu SM, Cho S-J, Yang S-R. Cigarette Smoke-Induced Respiratory Response: Insights into Cellular Processes and Biomarkers. Antioxidants. 2023; 12(6):1210. https://doi.org/10.3390/antiox12061210
Chicago/Turabian StyleCha, Sang-Ryul, Jimin Jang, Sung-Min Park, Se Min Ryu, Seong-Joon Cho, and Se-Ran Yang. 2023. "Cigarette Smoke-Induced Respiratory Response: Insights into Cellular Processes and Biomarkers" Antioxidants 12, no. 6: 1210. https://doi.org/10.3390/antiox12061210