
Targeting Mitochondrial Oxidative Stress as a Strategy to Treat Aging and Age-Related Diseases
 , and
, and
Abstract
:1. Introduction
2. Mitochondrial Alterations during the Process of Senescence and Aging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Alteration | Outcome(s) | Experimental Model and References |
|---|---|---|
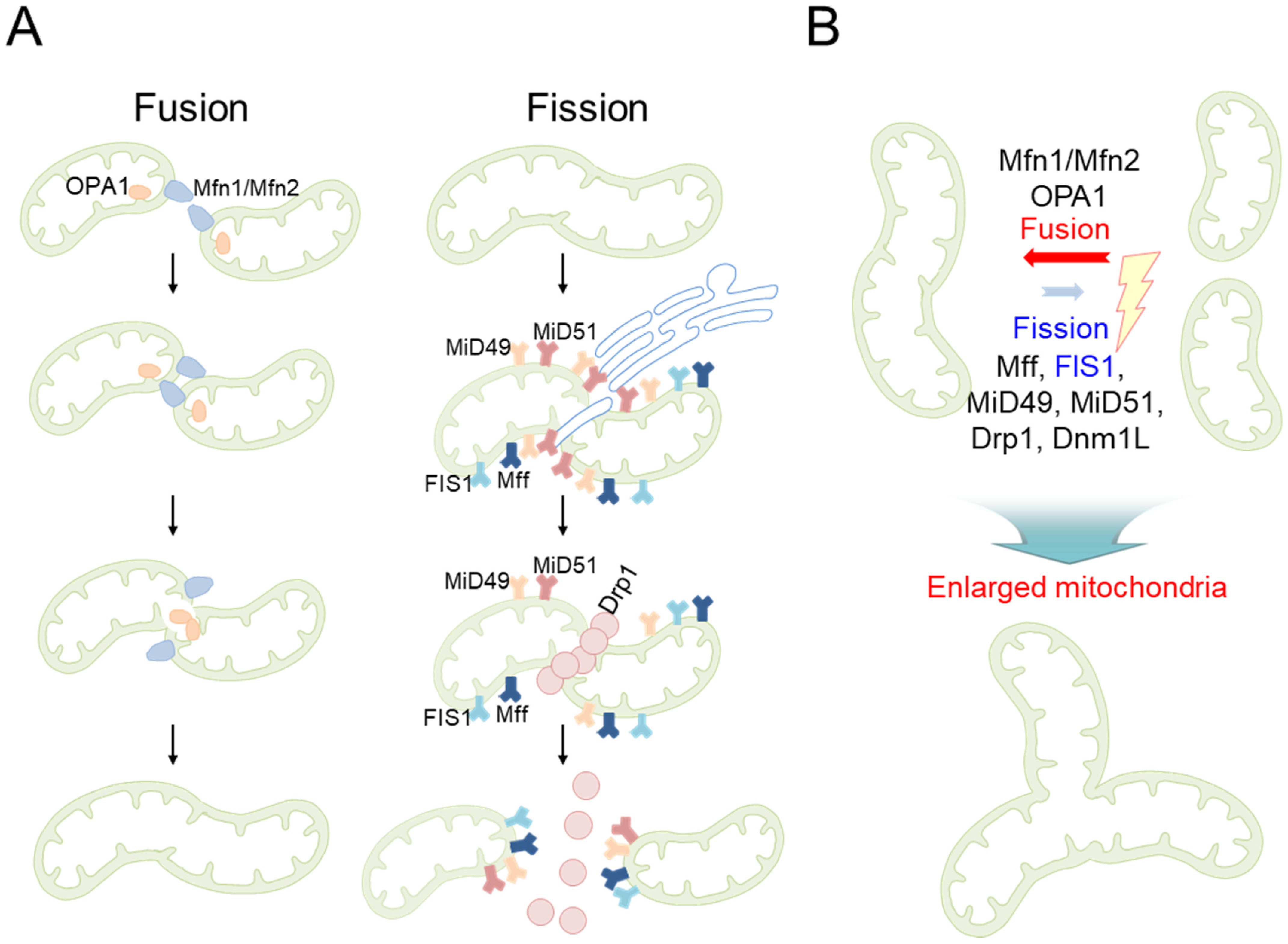
| Mitochondrial morphology | Formation of large mitochondria with highly interconnected network structures | MRC-5 human embryonic lung fibroblasts [19,20] |
| A considerable rise in the percentage of large mitochondria | C57/BL6 mice aged 30 months [26] | |
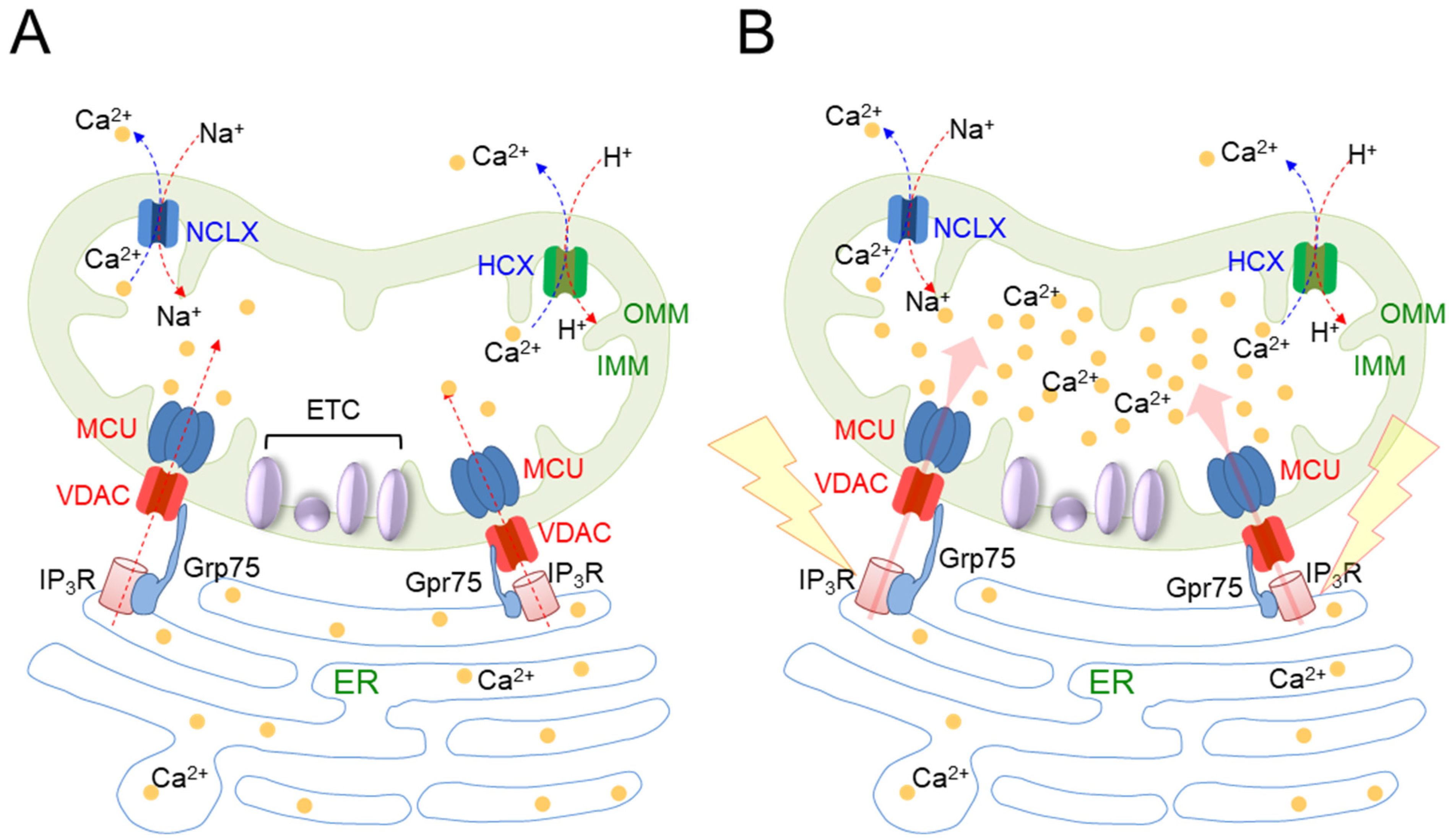
| Mitochondrial Ca2+ homeostasis | Senescent cells show a sustained increase in IP3R-mediated Ca2+ release | Human endometrium-derived stem cells [30] |
| Ca2+ then starts to be transported to the mitochondria through the VDAC/MCU channel | Human endometrial adenocarcinoma cells and WI38 human fibroblasts [31] | |
| Mitochondria overloaded with Ca2+ exhibit increased electron leakage from the ETC and consequently generate mitochondrial ROS | Human endometrial adenocarcinoma cells and WI38 human fibroblasts [31] |
3. Defective Mitochondria Are a Major Cause of Mitochondrial ROS Generation
| Cause of Mitochondrial ROS Generation | Outcome(s) | Experimental Model and References | ROS Related Information |
|---|---|---|---|
| Defective mitochondria | Neurodegenerative diseases such as PD have reduced complex I activity, leading to increased mitochondrial ROS production and complex I oxidation | Fibroblasts from the patient with PINK1 mutation [40] |
|
| Mitochondrial dysfunction manifests as defects in heme and Fe–S cluster biosynthesis. Alterations in iron homeostasis due to these defects lead to mitochondrial iron overload, resulting in the overproduction of free radicals via the Fenton reaction | Chondrocyte C-20/A4 cell lines [47] |
| |
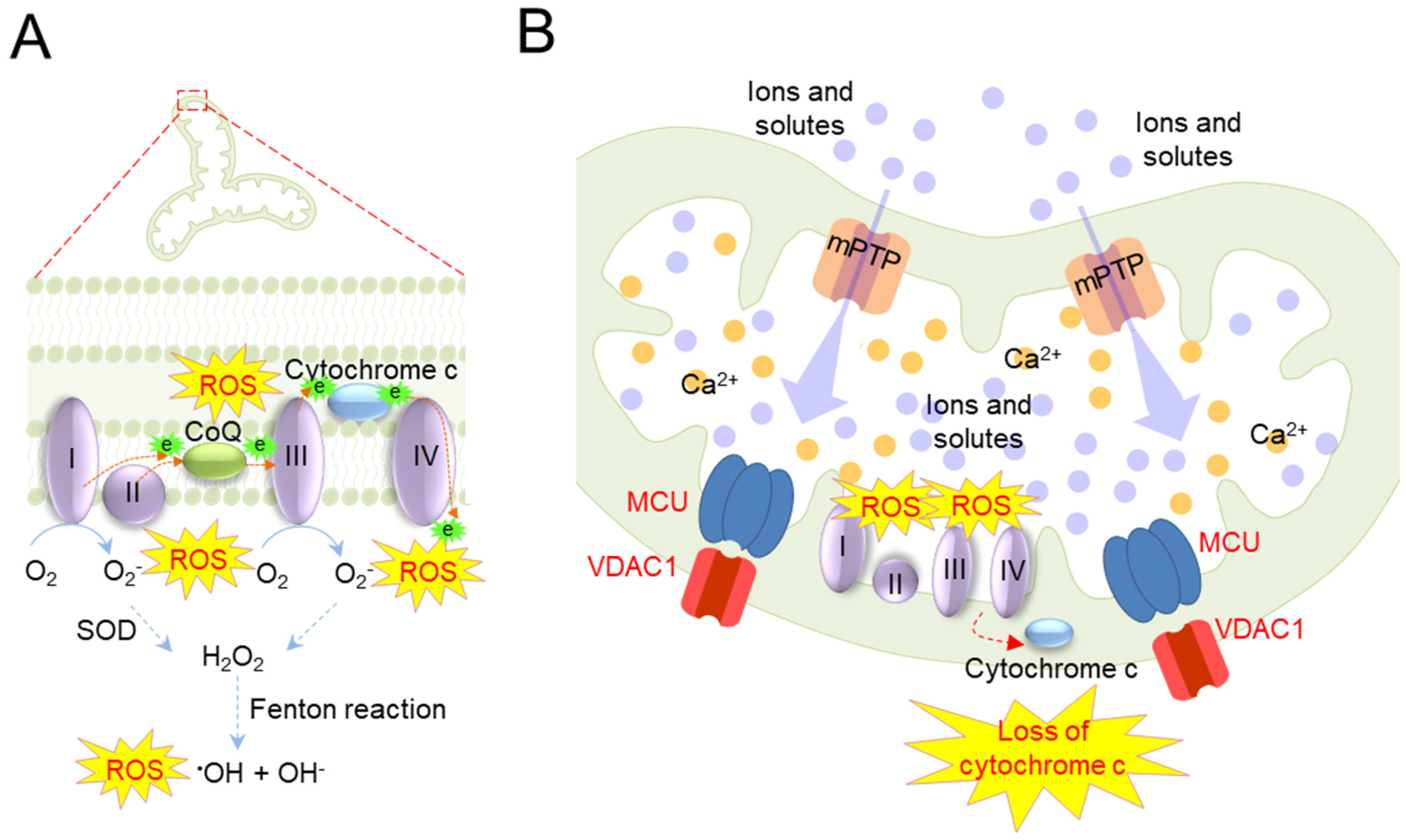
| Mitochondrial Ca2+ overload | Mitochondrial Ca2+ overload stimulates the mitochondrial permeability transition (PT) and opens PT pores (mPTP), allowing ions and other solutes to move freely | Isolated heart mitochondria from the bovine heart [48,49] | |
| The osmotic pressure of the mitochondrial matrix is increased by mPTP opening, which causes swelling of the mitochondria. Enlarged mitochondria readily lose cytochrome c that is loosely bound to the IMM | Astrocytes newborn C57BL/6 mice [53] |
| |
| Loss of cytochrome c impedes electron transport from complex I to IV, resulting in increased electron leakage from the ETC. The leaked electrons react with oxygen to generate large amounts of mitochondrial ROS | Isolated heart mitochondria from male Wistar rats [50] |
|
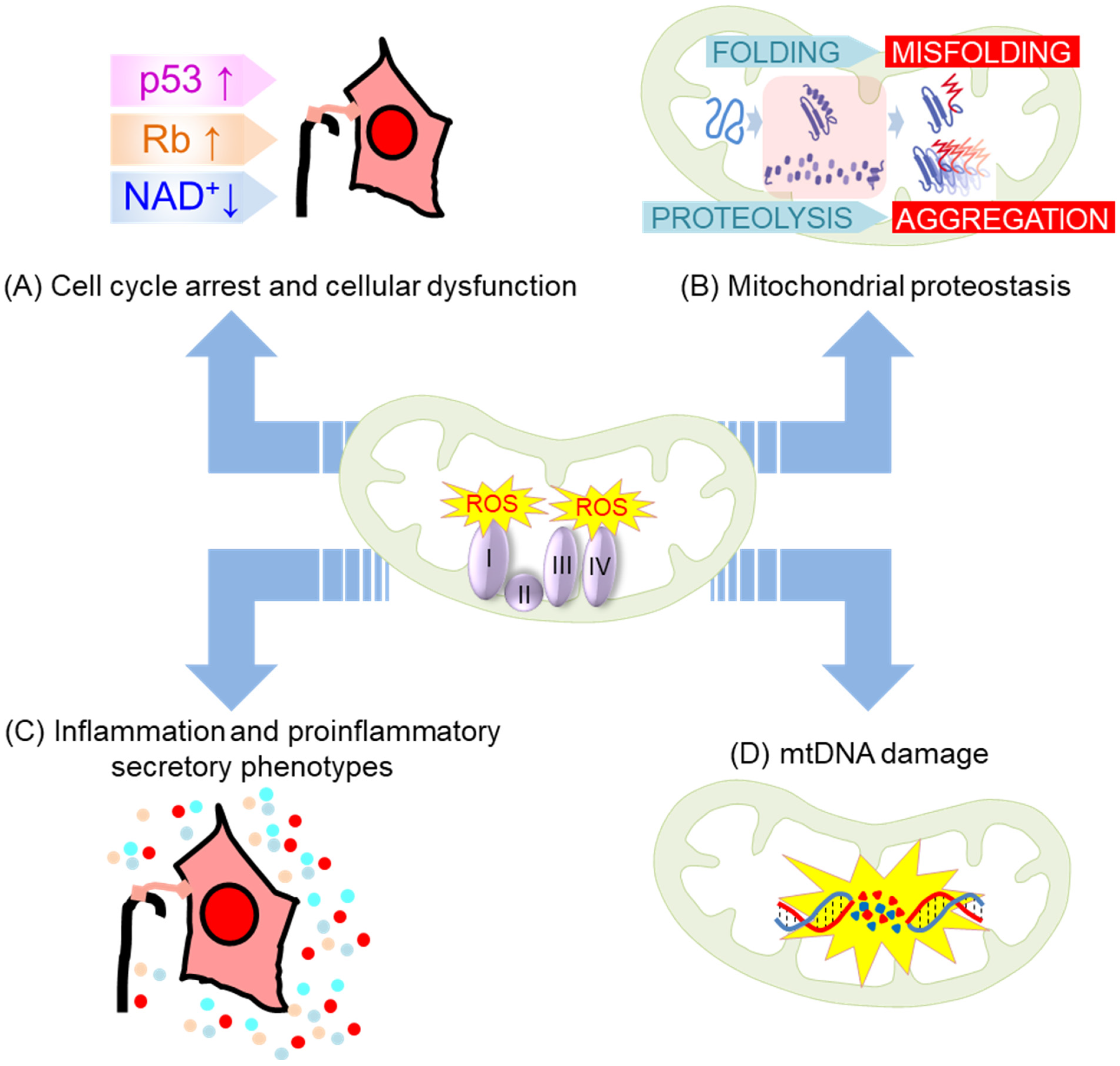
4. Vicious Feedback Loop between Mitochondrial Oxidative Stress and Senescence/Aging
5. Targeting Mitochondrial Oxidative Stress as a Therapeutic Strategy for Aging and Age-Related Diseases
| Therapeutic Strategy | Outcome(s) | Experimental Model and References |
|---|---|---|
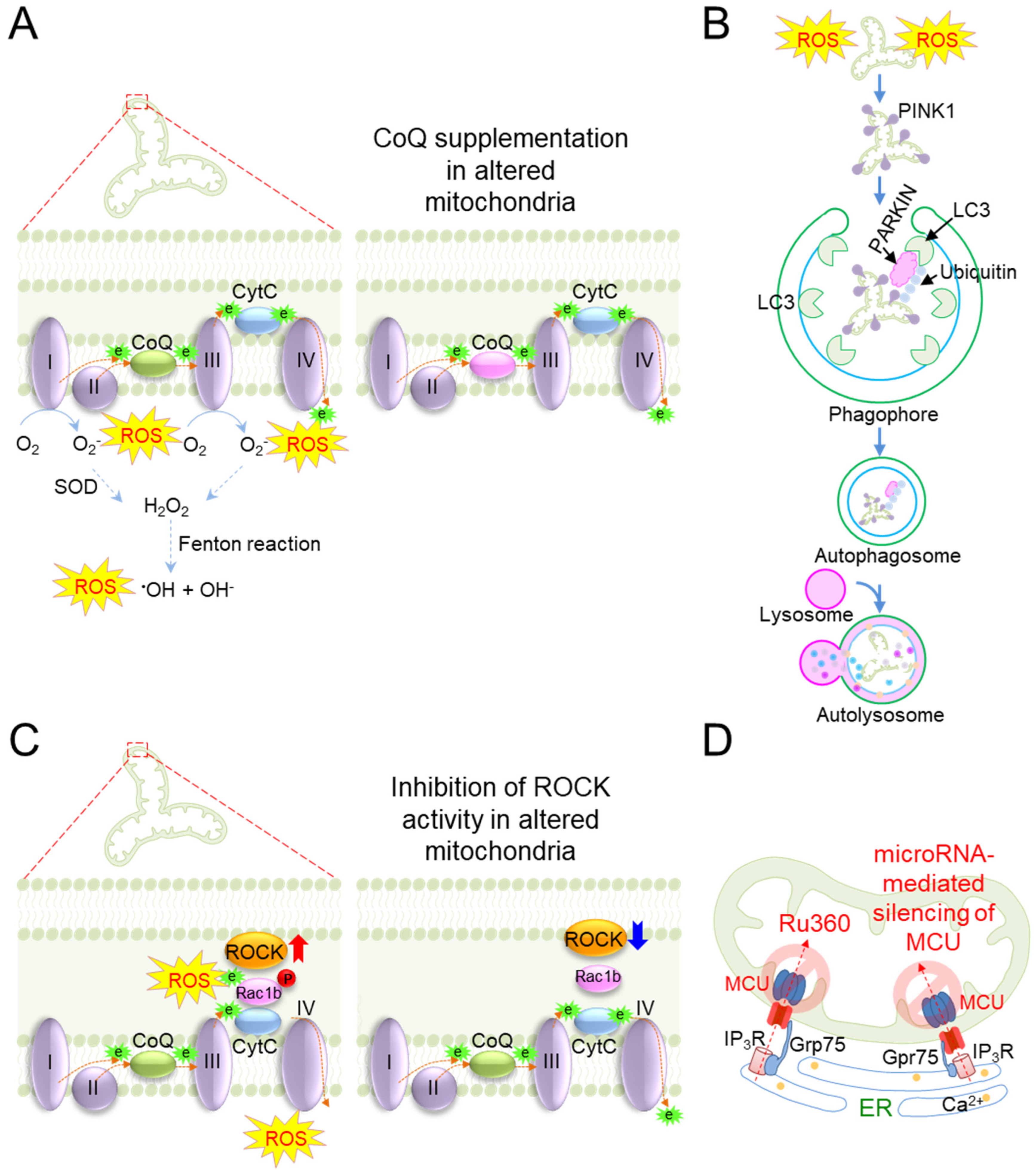
| Antioxidants | Mitochondria-targeted CoQ (MitoQ) treatment lowers mitochondrial ROS production, consequently resulting in reductions in mitochondrial swelling, cristae loss, and oxidative cell death | Male albino rats (Wistar strain) [106] |
| Treatment of senescent fibroblasts with N-acetylcysteine (NAC), a ROS scavenger, ameliorates senescence-associated phenotypes | Laminopathy progeria fibroblasts [11] | |
| Treatment with SS-31 boosts electron transport in the ETC, reducing the generation of free radicals in mitochondria | Primary neurons from C57BL/6 mice [109] | |
| Mitophagy | Overexpression of PARKIN reduces mitochondrial oxidative stress with the protection of age-related loss of skeletal muscle in aged mice | Skeletal muscle of aged mice [117] |
| Knocking out PARKIN accumulates markers of oxidative stress and deteriorates the contractile function of skeletal muscles | PARKIN knockout mice [118] | |
| Mice given the mitophagy inducer trehalose showed reduced superoxide production and improved age-related atherosclerosis | C57/BL6 mice aged 27–28 months [122] | |
| Therapy with the autophagy inducer lithium in C. elegans activates mitochondrial turnover with a decrease in mitochondrial oxidative stress | C. elegans [123] | |
| Inhibition of ATM activity activates mitophagy and consequently lowers mitochondrial ROS levels | Human fibroblasts [127] | |
| Genes that regulate the activity of ETC components | Inhibition of the ROCK activity facilitates complex IV activity, thereby reducing mitochondrial ROS production | Progeria skin fibroblasts [129] |
| Long-term caloric restriction (CR) upregulates the activity of complex IV, which partially offsets electron leak and reduces mitochondrial ROS production | Female Swiss Albino balb/c mice [131] | |
| Mitochondrial Ca2+ homeostasis | Inhibition of MCU by Ru360 reduces the percentage of mitochondria exhibiting Ca2+ overload and subsequently reduces the production of mitochondrial ROS | Isolated brain mitochondria from male Wistar rats [146] |
| MicroRNA-mediated silencing of MCU shields cardiomyocytes from mitochondrial oxidative stress | Rat cardiac myoblast H9c2 cell line [147] | |
| Sirtuin | Reduction in mitochondrial ROS levels by sirtuin 1 ameliorates symptoms of age-related neurodegeneration by protecting cultured neurons from oxidative stress-mediated death | PGC-1α null cell [148] |
| Overexpression of sirtuin 2 extends the shortened lifespan caused by hydrogen peroxide (H2O2) treatment | Yeast cells [150] |
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; An, J.; Zou, M.H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolden, J.E.; Lowe, S.W. 15—Cellular Senescence. In The Molecular Basis of Cancer, 4th ed.; Mendelsohn, J., Gray, J.W., Howley, P.M., Israel, M.A., Thompson, C.B., Eds.; Content Repository Only: Philadelphia, PA, USA, 2015; pp. 229–238.e222. [Google Scholar]
- Bouska, M.; Huang, K.; Kang, P.; Bai, H. Organelle aging: Lessons from model organisms. J. Genet. Genom. Yi Chuan Xue Bao 2019, 46, 171–185. [Google Scholar] [CrossRef]
- Boffoli, D.; Scacco, S.C.; Vergari, R.; Solarino, G.; Santacroce, G.; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1994, 1226, 73–82. [Google Scholar] [CrossRef]
- Hwang, E.; Yoon, G.; Kang, H. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 2009, 66, 2503–2524. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A.; Smit-McBride, Z.; Guinan, K.J.; Litovchenko, M.; Moskalev, A. Potential therapeutic approaches for modulating expression and accumulation of defective lamin A in laminopathies and age-related diseases. J. Mol. Med. 2012, 90, 1361–1389. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.A.; Muter, J.; Ritchie, P.; Lattanzi, G.; Hutchison, C.J. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum. Mol. Genet. 2011, 20, 3997–4004. [Google Scholar] [CrossRef] [Green Version]
- Henze, K.; Martin, W. Evolutionary biology: Essence of mitochondria. Nature 2003, 426, 127–128. [Google Scholar] [CrossRef]
- Bernhardt, D.; Müller, M.; Reichert, A.S.; Osiewacz, H.D. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci. Rep. 2015, 5, 7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Al Ojaimi, M.; Salah, A.; El-Hattab, A.W. Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders. Membranes 2022, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef] [Green Version]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Chi, C.W.; Wei, Y.H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Navarro, L.; Jansen-Dürr, P.; Cavinato, M. Age-Related Lysosomal Dysfunctions. Cells 2022, 11, 1977. [Google Scholar] [CrossRef]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2020, 14, 612757. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Jeong, S.Y.; Lim, W.C.; Kim, S.; Park, Y.Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.D.; Franks, L.M. The effect of age on mitochondrial ultrastructure. Gerontologia 1975, 21, 81–94. [Google Scholar] [CrossRef]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca2+ transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP(3)R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Griukova, A.A.; Abushik, P.A.; Antonov, S.M.; Nikolsky, N.N.; Burova, E.B. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging 2016, 8, 3400–3418. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calvé, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef] [Green Version]
- Tretter, L.; Adam-Vizi, V. High Ca2+ load promotes hydrogen peroxide generation via activation of α-glycerophosphate dehydrogenase in brain mitochondria. Free. Radic. Biol. Med. 2012, 53, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Takacs, K.; Kövér, K.; Adam-Vizi, V. Stimulation of H2O2 generation by calcium in brain mitochondria respiring on alpha-glycerophosphate. J. Neurosci. Res. 2007, 85, 3471–3479. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Choksi, K.B.; Nuss, J.E.; Deford, J.H.; Papaconstantinou, J. Age-related alterations in oxidatively damaged proteins of mouse skeletal muscle mitochondrial electron transport chain complexes. Free. Radic. Biol. Med. 2008, 45, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Choksi, K.B.; Boylston, W.H.; Rabek, J.P.; Widger, W.R.; Papaconstantinou, J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim. Biophys. Acta 2004, 1688, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; De Rasmo, D. Complex I deficiencies in neurological disorders. Trends Mol. Med. 2013, 19, 61–69. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, C.; Sardanelli, A.; Scrima, R.; Ripoli, M.; Quarato, G.; D’Aprile, A.; Bellomo, F.; Scacco, S.; De Michele, G.; Filla, A.; et al. Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem. Res. 2008, 33, 2565–2574. [Google Scholar] [CrossRef]
- Prousek, J. Fenton chemistry in biology and medicine. Pure Appl. Chem. 2007, 79, 2325–2338. [Google Scholar] [CrossRef]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A.; Tong, W.-H. Iron–sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxidants 2021, 10, 1864. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Kung, G.P.; Gnana-Prakasam, J.P. Role of Iron in Aging Related Diseases. Antioxidants 2022, 11, 865. [Google Scholar] [CrossRef]
- Karim, A.; Bajbouj, K.; Shafarin, J.; Qaisar, R.; Hall, A.C.; Hamad, M. Iron Overload Induces Oxidative Stress, Cell Cycle Arrest and Apoptosis in Chondrocytes. Front. Cell Dev. Biol. 2022, 10, 821014. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A.; Southard, J.H. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 1976, 251, 5069–5077. [Google Scholar] [CrossRef]
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem. J. 2011, 436, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Markevich, N.I.; Hoek, J.B. Computational modeling analysis of mitochondrial superoxide production under varying substrate conditions and upon inhibition of different segments of the electron transport chain. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1847, 656–679. [Google Scholar] [CrossRef] [Green Version]
- Tam, Z.Y.; Cai, Y.H.; Gunawan, R. Elucidating cytochrome C release from mitochondria: Insights from an in silico three-dimensional model. Biophys. J. 2010, 99, 3155–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.S.; Li, R.H.; Stadlin, A. Oxidative stress induced by MPTP and MPP(+): Selective vulnerability of cultured mouse astrocytes. Brain Res. 1999, 836, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Goodell, S.; Cortopassi, G. Analysis of oxygen consumption and mitochondrial permeability with age in mice. Mech. Ageing Dev. 1998, 101, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Scialò, F.; Sriram, A.; Fernández-Ayala, D.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Zhang, F.; Xie, R.; Munoz, F.M.; Lau, S.S.; Monks, T.J. PARP-1 Hyperactivation and Reciprocal Elevations in Intracellular Ca2+ During ROS-Induced Nonapoptotic Cell Death. Toxicol. Sci. 2014, 140, 118–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Kang, J.; Wang, H.; Huang, T. Mitochondrial ROS contribute to oridonin-induced HepG2 apoptosis through PARP activation. Oncol. Lett. 2018, 15, 2881–2888. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senoo-Matsuda, N.; Yasuda, K.; Tsuda, M.; Ohkubo, T.; Yoshimura, S.; Nakazawa, H.; Hartman, P.S.; Ishii, N. A Defect in the Cytochrome b Large Subunit in Complex II Causes Both Superoxide Anion Overproduction and Abnormal Energy Metabolism in Caenorhabditis elegans *. J. Biol. Chem. 2001, 276, 41553–41558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, H.; Ishii, N.; Ishida, H.; Ichimori, K.; Nakazawa, H.; Suzuki, K. Rapid accumulation of fluorescent material with aging in an oxygen-sensitive mutant mev-1 of Caenorhabditis elegans. Mech. Ageing Dev. 1994, 74, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Fujiwara, Y.; Ishii, N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1998, 53, B240–B244. [Google Scholar] [CrossRef] [Green Version]
- Yoon, G.; Kim, H.J.; Yoon, Y.S.; Cho, H.; Lim, I.K.; Lee, J.H. Iron chelation-induced senescence-like growth arrest in hepatocyte cell lines: Association of transforming growth factor beta1 (TGF-beta1)-mediated p27Kip1 expression. Biochem. J. 2002, 366, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.S.; Byun, H.O.; Cho, H.; Kim, B.K.; Yoon, G. Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced senescence-associated growth arrest. J. Biol. Chem. 2003, 278, 51577–51586. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.O.; Jung, H.J.; Kim, M.J.; Yoon, G. PKCδ phosphorylation is an upstream event of GSK3 inactivation-mediated ROS generation in TGF-β1-induced senescence. Free. Radic. Res. 2014, 48, 1100–1108. [Google Scholar] [CrossRef]
- Byun, H.O.; Jung, H.J.; Seo, Y.H.; Lee, Y.K.; Hwang, S.C.; Hwang, E.S.; Yoon, G. GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) β1-induced senescence. Exp. Cell Res. 2012, 318, 1808–1819. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou, G.K.; Pye, D.; Vasilaki, A.; Zibrik, L.; Palomero, J.; Kabayo, T.; McArdle, F.; Van Remmen, H.; Richardson, A.; Tidball, J.G.; et al. Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu, Zn superoxide dismutase. Aging Cell 2011, 10, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanos-González, E.; Henares-Chavarino, Á.A.; Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Sancho-Bielsa, F.J.; Alcain, F.J.; Peinado, J.R.; Rabanal-Ruíz, Y.; Durán-Prado, M. Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1444. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.B.; Jasper, H. Mitochondrial Proteostasis in the Control of Aging and Longevity. Cell Metab. 2014, 20, 214–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.L.; Mena, N.P.; Auchère, F.; Lee, I.; Prigent, A.; Lobsiger, C.S.; Camadro, J.M.; Hirsch, E.C. Dysfunction of mitochondrial Lon protease and identification of oxidized protein in mouse brain following exposure to MPTP: Implications for Parkinson disease. Free. Radic. Biol. Med. 2017, 108, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased proteolytic activity of the mitochondrial amyloid-β degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J. Alzheimer’s Dis. JAD 2011, 27, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, Y.; Inoue, I.; Nakano, T.; Ikeda, M.; Kakinuma, Y. Prolonged disturbance of proteostasis induces cellular senescence via temporal mitochondrial dysfunction and subsequent mitochondrial accumulation in human fibroblasts. FEBS J. 2022, 289, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Paez, H.G.; Pitzer, C.R.; Alway, S.E. Age-Related Dysfunction in Proteostasis and Cellular Quality Control in the Development of Sarcopenia. Cells 2023, 12, 249. [Google Scholar] [CrossRef]
- Ichimura, H.; Parthasarathi, K.; Quadri, S.; Issekutz, A.C.; Bhattacharya, J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J. Clin. Investig. 2003, 111, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [Green Version]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, R.A.; Suponitsky-Kroyter, I.; Selak, M.A. Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to beta-cell failure. J. Biol. Chem. 2005, 280, 28785–28791. [Google Scholar] [CrossRef] [Green Version]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Aliev, G.; Seyidova, D.; Lamb, B.T.; Obrenovich, M.E.; Siedlak, S.L.; Vinters, H.V.; Friedland, R.P.; Siedlak, S.L.; Vinters, H.V.; Friedland, R.P.; et al. Mitochondria and vascular lesions as a central target for the development of Alzheimer’s disease and Alzheimer disease-like pathology in transgenic mice. Neurol. Res. 2003, 25, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, L.; Kujoth, G.C.; Prolla, T.A.; Van Leuven, F.; Vassar, R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onur, S.; Niklowitz, P.; Fischer, A.; Metges, C.C.; Grune, T.; Menke, T.; Rimbach, G.; Döring, F. A comparative study into alterations of coenzyme Q redox status in ageing pigs, mice, and worms. BioFactors 2014, 40, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Kwong, L.; Sohal, R.S. Mitochondrial coenzyme Q content and aging. BioFactors 1999, 9, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Kamzalov, S.; Sohal, R.S. Effect of age and caloric restriction on coenzyme Q and alpha-tocopherol levels in the rat. Exp. Gerontol. 2004, 39, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- De la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavón, A.; Alcocer-Gómez, E.; de Lavera, I.; Ybot-González, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [Green Version]
- Wani, W.Y.; Gudup, S.; Sunkaria, A.; Bal, A.; Singh, P.P.; Kandimalla, R.J.; Sharma, D.R.; Gill, K.D. Protective efficacy of mitochondrial targeted antioxidant MitoQ against dichlorvos induced oxidative stress and cell death in rat brain. Neuropharmacology 2011, 61, 1193–1201. [Google Scholar] [CrossRef]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Xie, J.; Lin, J.; Wei, M.; Teng, Y.; He, Q.; Yang, G.; Yang, X. Sustained Akt signaling in articular chondrocytes causes osteoarthritis via oxidative stress-induced senescence in mice. Bone Res. 2019, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Reddy, T.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Reddy, A.P.; Shirendeb, U.; Park, B.; Reddy, P.H. Toxicity of neurons treated with herbicides and neuroprotection by mitochondria-targeted antioxidant SS31. Int. J. Environ. Res. Public Health 2011, 8, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Manczak, M.; Reddy, P.H. Mitochondria-Targeted Antioxidant SS31 Prevents Amyloid Beta-Induced Mitochondrial Abnormalities and Synaptic Degeneration in Alzheimer’s Disease. Pharmaceuticals 2012, 5, 1103–1119. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Szeto, H.H.; Kim, E.; Kim, H.; Tolhurst, A.T.; Pinto, J.T. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J. Biol. Chem. 2007, 282, 4634–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Schriner, S.E.; Linford, N.J. Extension of mouse lifespan by overexpression of catalase. Age 2006, 28, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Zhang, X.; Zheng, L.; Li, Z.; Zhao, X.; Lai, W.; Shen, H.; Lv, J.; Yang, G.; Wang, Q.; et al. Peroxiredoxin 6 Is a Crucial Factor in the Initial Step of Mitochondrial Clearance and Is Upstream of the PINK1-Parkin Pathway. Antioxid. Redox Signal. 2016, 24, 486–501. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Liu, H.; Zang, C.; Yuan, F.; Ju, C.; Shang, M.; Ning, J.; Yang, Y.; Ma, J.; Li, G.; Bao, X.; et al. The role of FUNDC1 in mitophagy, mitochondrial dynamics and human diseases. Biochem. Pharmacol. 2022, 197, 114891. [Google Scholar] [CrossRef]
- Schmid, E.T.; Pyo, J.-H.; Walker, D.W. Neuronal induction of BNIP3-mediated mitophagy slows systemic aging in Drosophila. Nat. Aging 2022, 2, 494–507. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choo, O.-S.; Lee, J.-S.; Jang, J.H.; Woo, H.G.; Choung, Y.-H. BCL2 Interacting Protein 3-like/NIX-mediated Mitophagy Plays an Important Role in the Process of Age-related Hearing Loss. Neuroscience 2021, 455, 39–51. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Hearon, C.M., Jr.; Henson, G.D.; Seals, D.R. Mitochondrial quality control and age-associated arterial stiffening. Exp. Gerontol. 2014, 58, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Tam, Z.Y.; Gruber, J.; Ng, L.F.; Halliwell, B.; Gunawan, R. Effects of lithium on age-related decline in mitochondrial turnover and function in Caenorhabditis elegans. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfi, P.; Tse, D.Y.; Di Ronza, A.; Seymour, M.L.; Martano, G.; Cooper, J.D.; Pereira, F.A.; Passafaro, M.; Wu, S.M.; Sardiello, M. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 2018, 14, 1419–1434. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.-H.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD+ supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 2021, 20, e13329. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Kuk, M.U.; Kim, J.W.; Lee, Y.S.; Cho, K.A.; Park, J.T.; Park, S.C. Alleviation of Senescence via ATM Inhibition in Accelerated Aging Models. Mol. Cells 2019, 42, 210–217. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Choi, H.J.C.; Jung, C.W.; Kim, G.R.; Lee, Y.S.; Park, S.C. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell 2017, 16, 541–550. [Google Scholar] [CrossRef]
- Park, J.T.; Kang, H.T.; Park, C.H.; Lee, Y.S.; Cho, K.A.; Park, S.C. A crucial role of ROCK for alleviation of senescence-associated phenotype. Exp. Gerontol. 2018, 106, 8–15. [Google Scholar] [CrossRef]
- Olgun, A.; Akman, S.; Serdar, M.A.; Kutluay, T. Oxidative phosphorylation enzyme complexes in caloric restriction. Exp. Gerontol. 2002, 37, 639–645. [Google Scholar] [CrossRef]
- Masoro, E.J. Retardation of aging processes by food restriction: An experimental tool. Am. J. Clin. Nutr. 1992, 55, 1250S–1252S. [Google Scholar] [CrossRef]
- Sayer, A.A.; Cooper, C. Undernutrition and Aging. Gerontology 1997, 43, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Sharma, A.; Kumari, A.; Kulurkar, P.M.; Raj, R.; Gulati, A.; Padwad, Y.S. Consumption of green tea epigallocatechin-3-gallate enhances systemic immune response, antioxidative capacity and HPA axis functions in aged male swiss albino mice. Biogerontology 2017, 18, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Na, L.; Feng, R.; Gong, L.; Zhao, Y.; Li, Q.; Li, Y.; Sun, C. The phytochemical, EGCG, extends lifespan by reducing liver and kidney function damage and improving age-associated inflammation and oxidative stress in healthy rats. Aging Cell 2013, 12, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.K.; Evans, J.L.; Luo, Y. Beneficial effects of natural antioxidants EGCG and alpha-lipoic acid on life span and age-dependent behavioral declines in Caenorhabditis elegans. Pharmacol. Biochem. Behav. 2006, 85, 620–628. [Google Scholar] [CrossRef]
- Zhang, L.; Jie, G.; Zhang, J.; Zhao, B. Significant longevity-extending effects of EGCG on Caenorhabditis elegans under stress. Free. Radic. Biol. Med. 2009, 46, 414–421. [Google Scholar] [CrossRef]
- Marcu, R.; Wiczer, B.M.; Neeley, C.K.; Hawkins, B.J. Mitochondrial matrix Ca2+ accumulation regulates cytosolic NAD;+/NADH metabolism, protein acetylation, and sirtuin expression. Mol. Cell. Biol. 2014, 34, 2890–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated with Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca2+ Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef]
- Jahangir, A.; Sagar, S.; Terzic, A. Aging and cardioprotection. J. Appl. Physiol. 2007, 103, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jesús García-Rivas, G.; Guerrero-Hernández, A.; Guerrero-Serna, G.; Rodríguez-Zavala, J.S.; Zazueta, C. Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J. 2005, 272, 3477–3488. [Google Scholar] [CrossRef] [PubMed]
- Sripetchwandee, J.; Sanit, J.; Chattipakorn, N.; Chattipakorn, S.C. Mitochondrial calcium uniporter blocker effectively prevents brain mitochondrial dysfunction caused by iron overload. Life Sci. 2013, 92, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.M.; Liang, D.D.; et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Wang, W.J.; Song, X.; Chu, W.M.; et al. SIRT1 confers protection against UVB- and H2O2-induced cell death via modulation of p53 and JNK in cultured skin keratinocytes. J. Cell. Mol. Med. 2009, 13, 3632–3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merksamer, P.I.; Liu, Y.; He, W.; Hirschey, M.D.; Chen, D.; Verdin, E. The sirtuins, oxidative stress and aging: An emerging link. Aging 2013, 5, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Lozada-Delgado, J.G.; Torres-Ramos, C.A.; Ayala-Peña, S. Chapter 4—Aging, oxidative stress, mitochondrial dysfunction, and the liver. In Aging, 2nd ed.; Preedy, V.R., Patel, V.B., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 37–46. [Google Scholar]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, C.; Lymp, J.; Thompson, V.; Dunn, C.; Davies, Z.; Chatfield, B.; Nichols, D.; Clancy, J.; Vender, R.; Egan, M.E.; et al. Long-term treatment with oral N-acetylcysteine: Affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2015, 14, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Tao, A.; Bai, Y.; Deng, Y.; Chen, G. Effectiveness of N-Acetylcysteine for the Prevention of Contrast-Induced Nephropathy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2016, 5, e003968. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.H.; Kuk, M.U.; So, M.K.; Song, E.S.; Lee, H.; Ahn, S.K.; Kwon, H.W.; Park, J.T.; Park, S.C. Targeting Mitochondrial Oxidative Stress as a Strategy to Treat Aging and Age-Related Diseases. Antioxidants 2023, 12, 934. https://doi.org/10.3390/antiox12040934
Lee YH, Kuk MU, So MK, Song ES, Lee H, Ahn SK, Kwon HW, Park JT, Park SC. Targeting Mitochondrial Oxidative Stress as a Strategy to Treat Aging and Age-Related Diseases. Antioxidants. 2023; 12(4):934. https://doi.org/10.3390/antiox12040934
Chicago/Turabian StyleLee, Yun Haeng, Myeong Uk Kuk, Moon Kyoung So, Eun Seon Song, Haneur Lee, Soon Kil Ahn, Hyung Wook Kwon, Joon Tae Park, and Sang Chul Park. 2023. "Targeting Mitochondrial Oxidative Stress as a Strategy to Treat Aging and Age-Related Diseases" Antioxidants 12, no. 4: 934. https://doi.org/10.3390/antiox12040934