
Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases

Abstract
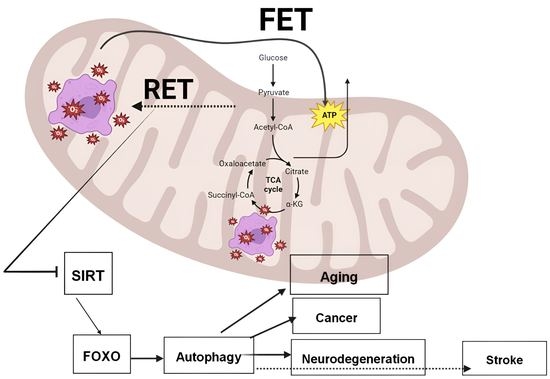
:
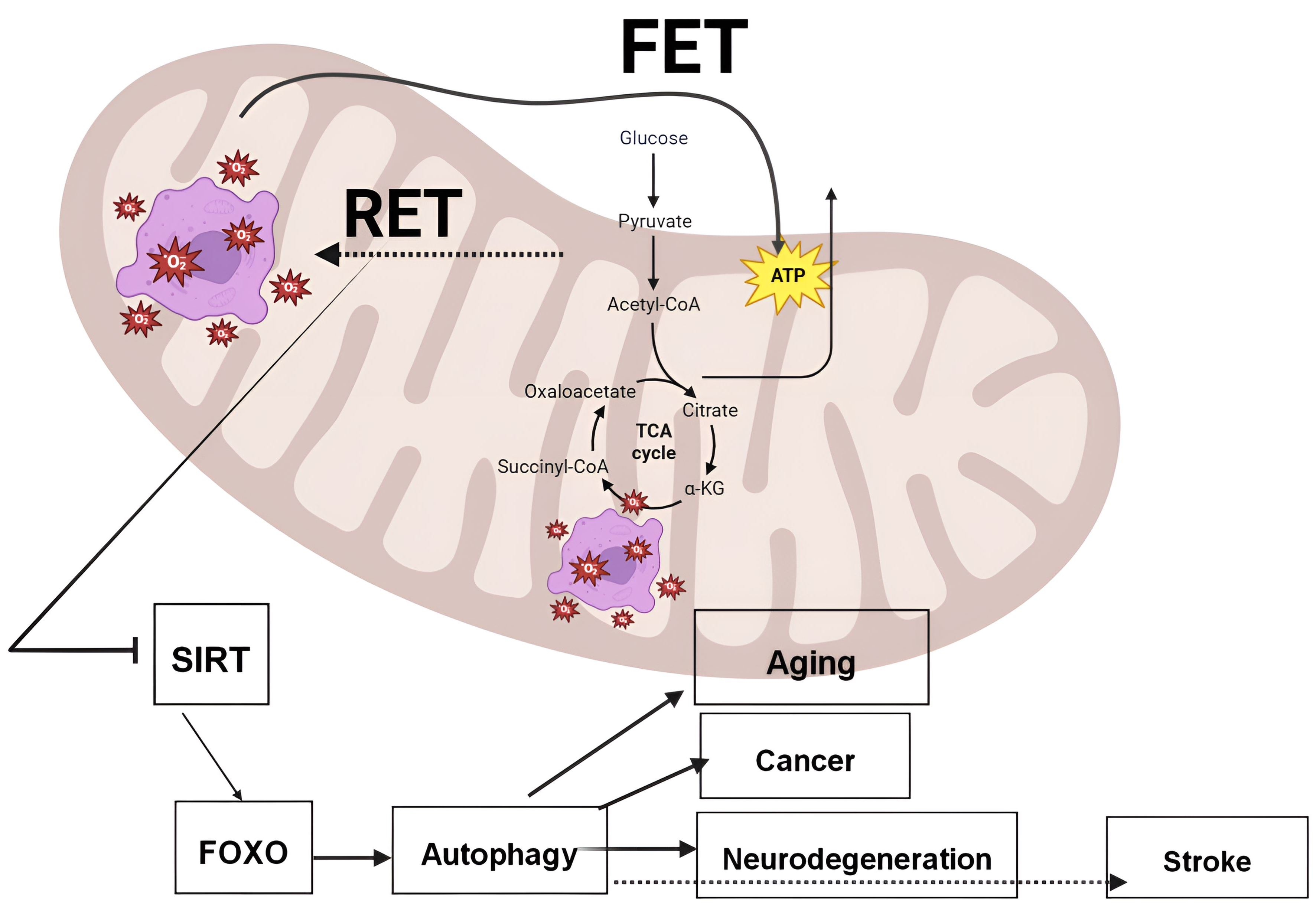{kind=link}
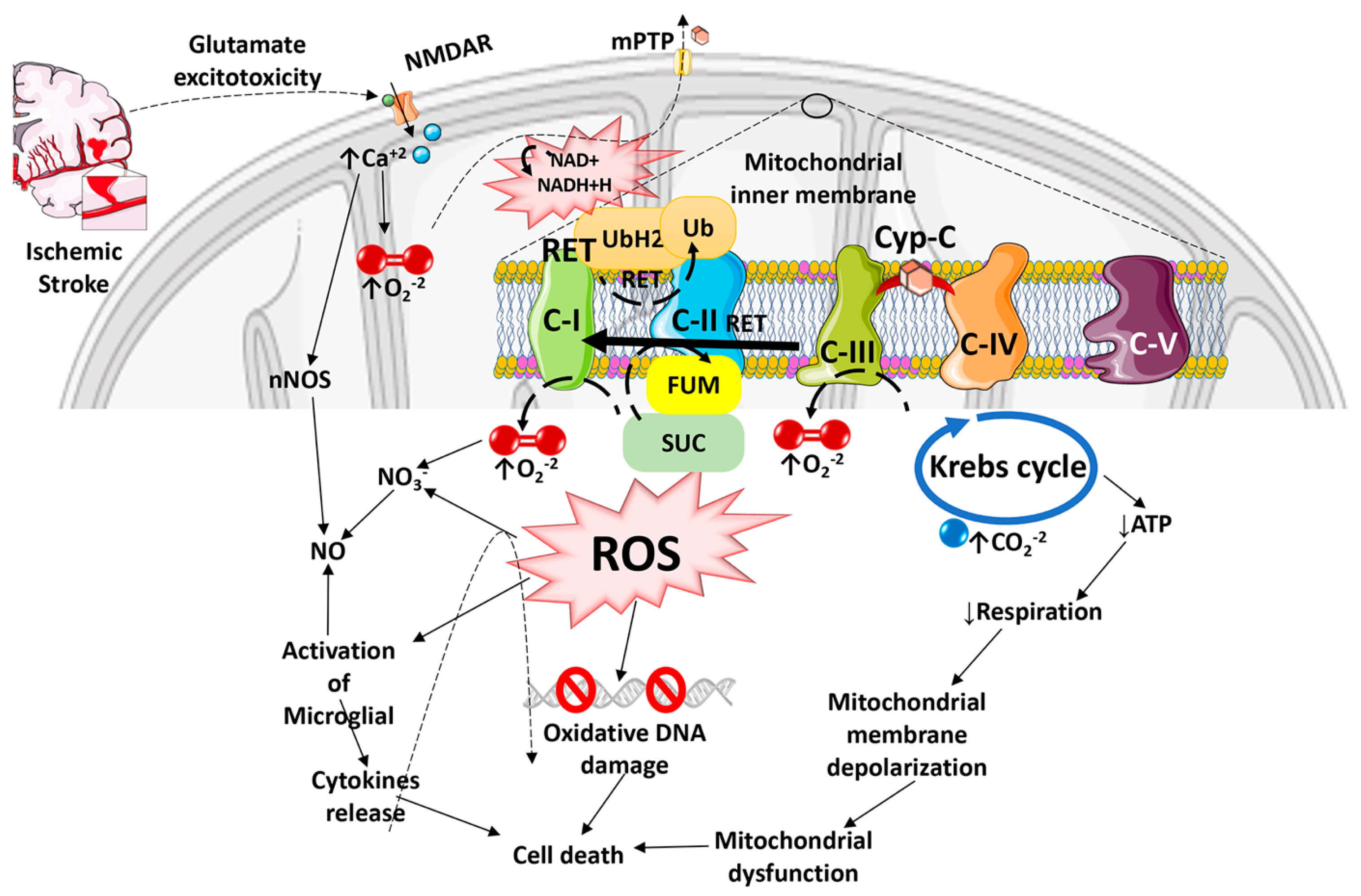{kind=link}
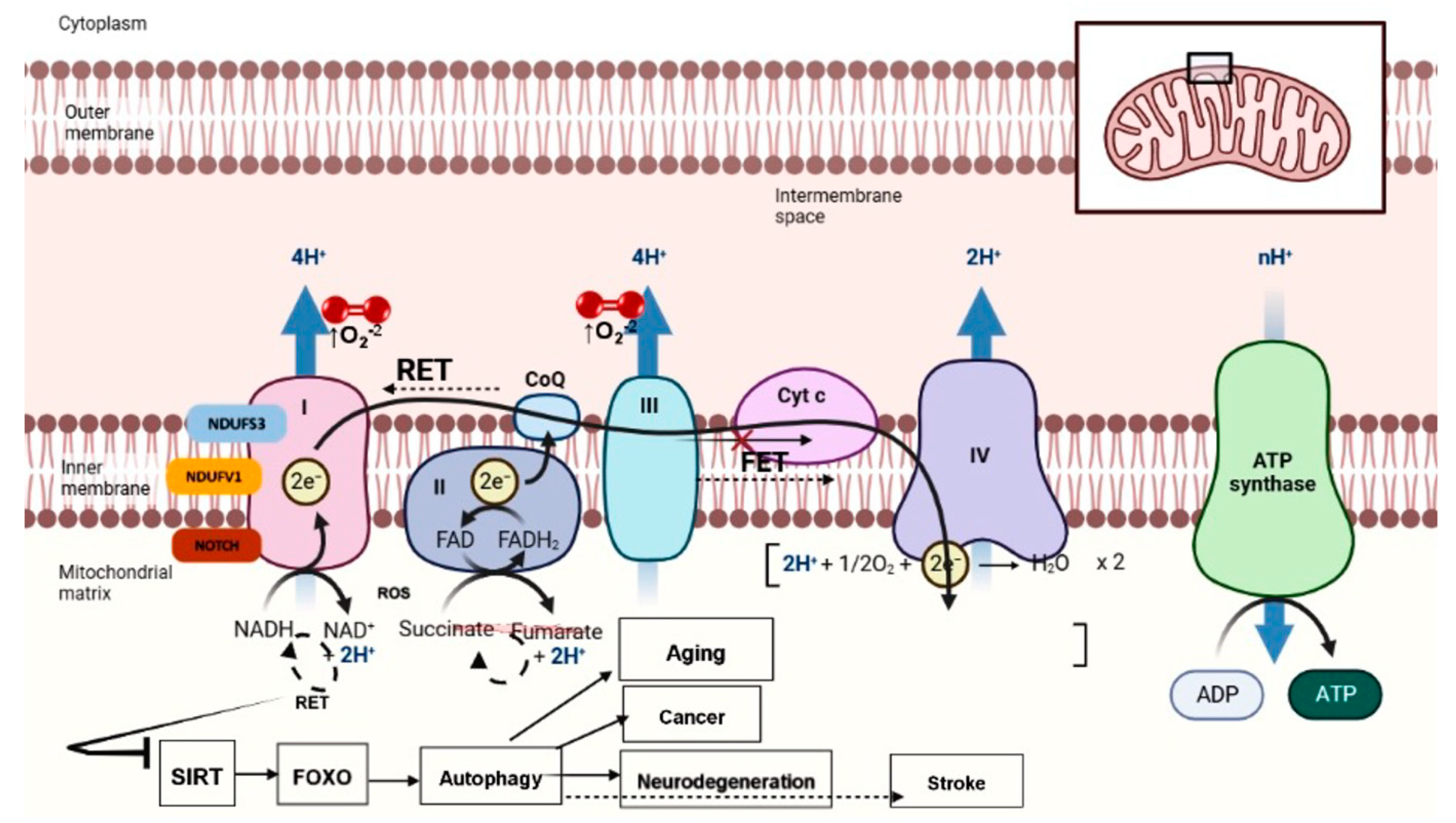{kind=link}
1. Introduction
2. ROS and Oxidative Stress in Stroke
2.1. ROS Generation during Stroke
2.2. ROS-Induced Damages during Stroke
3. Mitochondrial Dysfunction and Ischemic Stroke
4. Mitochondrial Complex I Dysfunction in Ischemia Reperfusion Injury
5. RET in Ischemic Stroke
6. RET in Cancer, Aging, and Age-Related Neurodegenerative Diseases Such as AD
7. RET as a Therapeutic Target for Stroke, Cancer, Aging, and Age-Related Disorders
7.1. RET Inhibition in Stroke
7.2. RET Inhibition in Aging and Age-Related Diseases
7.3. RET Inhibition on the NAD+-Dependent Sirtuin/FOXO/Autophagy Pathway
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owolabi, M.O.; Thrift, A.G.; Mahal, A.; Ishida, M.; Martins, S.; Johnson, W.D.; Pandian, J.; Abd-Allah, F.; Yaria, J.; Phan, H.T.; et al. Primary stroke prevention worldwide: Translating evidence into action. Lancet Public Health 2022, 7, e74–e85. [Google Scholar] [CrossRef]
- Chavda, V.; Chaurasia, B.; Garg, K.; Deora, H.; Umana, G.E.; Palmisciano, P.; Scalia, G.; Lu, B. Molecular mechanisms of oxidative stress in stroke and cancer. Brain Disord. 2022, 5, 100029. [Google Scholar] [CrossRef]
- Monsour, M.; Borlongan, C.V. The central role of peripheral inflammation in ischemic stroke. J. Cereb. Blood Flow Metab. 2023. [Google Scholar] [CrossRef] [PubMed]
- Carinci, M.; Vezzani, B.; Patergnani, S.; Ludewig, P.; Lessmann, K.; Magnus, T.; Casetta, I.; Pugliatti, M.; Pinton, P.; Giorgi, C. Different roles of mitochondriain cell death and inflammation: Focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicines 2021, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, D.; Kaur, H.; Saraf, J.; Vats, K.; Pravalika, K.; Wanve, M.; Kalia, K.; Borah, A.; Kumar, A.; Wang, X.; et al. Mitochondrial dysfunction in stroke: Implications of stem cell therapy. Transl. Stroke Res. 2019, 10, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.; Ustunkaya, T.; Clermont, A.C.; Feener, E.P. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after stroke. Blood J. Am. Soc. Hematol. 2017, 129, 2280–2290. [Google Scholar] [CrossRef]
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W.; et al. Solitaire™ with the Intention for Thrombectomy as Primary Endovascular Treatment for Acute Ischemic Stroke (SWIFTPRIME) trial: Protocol for a randomized, controlled, multicenter study comparing the Solitaire revascularization device with IV tPA with IV tPA alone in acute ischemic stroke. Int. J. Stroke 2015, 10, 439–448. [Google Scholar]
- Grossberg, J.A.; Rebello, L.C.; Haussen, D.C.; Bouslama, M.; Bowen, M.; Barreira, C.M.; Belagaje, S.R.; Frankel, M.R.; Nogueira, R.G. Beyond large vessel occlusion strokes: Distal occlusion thrombectomy. Stroke 2018, 49, 1662–1668. [Google Scholar] [CrossRef]
- Albers, G.W.; Clark, W.M.; Madden, K.P.; Hamilton, S.A. ATLANTIS trial: Results for patients treated within 3 hours of stroke onset. Stroke 2002, 33, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, A.P.; Desai, S.M.; Kenmuir, C.L.; Rocha, M.; Starr, M.T.; Molyneaux, B.J.; Gross, B.A.; Jankowitz, B.T.; Jovin, T.G. Eligibility for endovascular trial enrollment in the 6-to-24 hour time window: Analysis of a single comprehensive stroke center. Stroke 2018, 49, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Martynov, M.Y.; Zhuravleva, M.V.; Vasyukova, N.S.; Kuznetsova, E.V.; Kameneva, T.R. Oxidative stress in the pathogenesis of stroke and its correction. Zhurnal Nevrol. Psikhiatrii Im. Korsakova 2023, 123, 16–27. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Gerke Duncan, M.B.; Holsinger, R.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants 2023, 12, 517. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.; Nalapko, Y. Ageing brain and neurodegeneration: Preventive and regenerative medicine. In Handbook of Anti-Aging Medicine; European Wellness Academy: London, UK, 2023. [Google Scholar]
- Shehjar, F.; Maktabi, B.; Rahman, Z.A.; Bahader, G.A.; James, A.W.; Naqvi, A.; Mahajan, R.; Shah, Z.A. Stroke: Molecular mechanisms and therapies: Update on recent developments. Neurochem. Int. 2023, 162, 105458. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Danieli, M.G.; Antonelli, E.; Piga, M.A.; Cozzi, M.F.; Allegra, A.; Gangemi, S. Oxidative stress, mitochondrial dysfunction, and respiratory chain enzyme defects in inflammatory myopathies. Autoimmun. Rev. 2023, 21, 103308. [Google Scholar] [CrossRef]
- García-Sánchez, A.; Miranda-Díaz, A.G.; Cardona-Muñoz, E.G. The role of oxidative stress in physiopathology and pharmacological treatment with pro-and antioxidant properties in chronic diseases. Oxidative Med. Cell. Longev. 2020, 2020, 2082145. [Google Scholar] [CrossRef]
- Roleira, F.M.; Tavares-da-Silva, E.J.; Garrido, J.; Borges, F. Antioxidants and stroke: Success and pitfalls. Transl. Stroke Res. Target Sel. Clin. Trials 2012, 3, 117–143. [Google Scholar]
- Shirley, R.; Ord, E.N.; Work, L.M. Oxidative stress and the use of antioxidants in stroke. Antioxidants 2014, 3, 472–501. [Google Scholar] [CrossRef] [Green Version]
- Ascherio, A. Antioxidants and stroke. Am. J. Clin. Nutr. 2000, 72, 337–338. [Google Scholar]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system:A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative stress-mediated blood-brain barrier (BBB) disruption in neurological diseases. Oxidative Med. Cell. Longev. 2020, 4356386. [Google Scholar] [CrossRef]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 6. [Google Scholar]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef]
- Brouns, R.; Verkerk, R.; Aerts, T.; DeSurgeloose, D.; Wauters, A.; Scharpé, S.; DeDeyn, P.P. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef]
- Hazell, A.S. Excitotoxic mechanisms in stroke: An update of concepts and treatment strategies. Neurochem. Int. 2007, 50, 941–953. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP-1and PARthanatos in ischemic stroke. J. Neurochem. 2022, 160, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Endo, H.; Nito, C.; Myer, D.J.; Chan, P.H. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke 2009, 40, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Saeed, S.A.; Shad, K.F.; Saleem, T.; Javed, F.; Khan, M.U. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Exp. Brain Res. 2007, 182, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zuo, W.; Guo, X.F.; He, W.B.; Chen, N.H. Cerebral glucose transporter: The possible therapeutic target for ischemic stroke. Neurochem. Int. 2014, 70, 22–29. [Google Scholar] [CrossRef]
- Shao, Z.; Dou, S.; Zhu, J.; Wang, H.; Xu, D.; Wang, C.; Cheng, B.; Bai, B. The role of mitophagy in ischemic stroke. Front. Neurol. 2020, 11, 608610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, Y.; Liu, C. Autophagy and ischemic stroke. In Autophagy: Biology and Diseases: Clinical Science; Springer Nature: Singapore, 2020; pp. 111–134. [Google Scholar]
- Vosler, P.S.; Graham, S.H.; Wechsler, L.R.; Chen, J. Mitochondrial targets for stroke: Focusing basic science research toward development of clinically translatable therapeutics. Stroke 2009, 40, 3149–3155. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Trojel Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target forcommon pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and celldeath in stroke. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2010, 1802, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Ding, Z.H.; Zhan, F.X.; Cai, L.; Yin, X.; Ling, J.L.; Ye, J.J.; Hou, S.Y.; Lu, Z.; Wang, Z.H.; et al. The NLRP3 inflammasome and stroke. Int. J. Clin. Exp. Med. 2015, 8, 4787. [Google Scholar] [PubMed]
- Gao, L.; Dong, Q.; Song, Z.; Shen, F.; Shi, J.; Li, Y. NLRP3 inflammasome: A promising target in ischemic stroke. Inflamm. Res. 2017, 66, 17–24. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP 3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Rimal, S.; Tantray, I.; Li, Y.; PalKhaket, T.; Li, Y.; Bhurtel, S.; Li, W.; Zeng, C.; Lu, B. Reverse electron transfer is activated during aging and contributes to aging and age-related disease. EMBO Rep. 2023, e55548. [Google Scholar] [CrossRef]
- Roy, T.; Chatterjee, A.; Swarnakar, S. Rotenone induced neurodegeneration is mediated via cytoskeleton degradation and necroptosis. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2023, 1870, 119417. [Google Scholar] [CrossRef]
- Mathur, S.; Gawas, C.; Ahmad, I.Z.; Wani, M.; Tabassum, H. Neurodegenerative disorders: Assessing the impact of natural vs drug-induced treatment options. Aging Med. 2023, 6, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Bahire, K.L.; Maļuhins, R.; Bello, F.; Freitag, S.V.; Jeļisejevs, I.; Gile, R.; Upīte, J.; Plesnila, N.; Jansone, B. He mispheric analysis of mitochondrial Complex I and II activity in the mouse model of ischemia-reperfusion-induced injury. Mitochondrion 2023, 69, 147–158. [Google Scholar] [CrossRef]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Seta, K.; Araki, H.; Handa, H. The effect of ischemia on mitochondrial metabolism. J. Biochem. 1967, 61, 512–514. [Google Scholar] [CrossRef]
- Ozawa, K.; Seta, K.; Handa, H. Biochemical studies on brain swelling, I. Influence of brain swelling and ischemia on the formation of anendogenous inhibitor in mitochondria. Psychiatry Clin. Neurosci. 1966, 20, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Seta, K.; Takeda, H.; Ando, K.; Handa, H.; Araki, C. On the isolation of mitochondria with high respiratory control from rat brain. J. Biochem. 1966, 59, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.D.; Mela, L.; Wrobel-Kuhl, K.; Reivich, M. Mitochondrial metabolism following bilateral cerebral ischemia in the gerbil. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1977, 1, 519–527. [Google Scholar] [CrossRef]
- Nordström, C.H.; Rehncrona, S.; Siesjö, B.K. Effects of phenobarbital in cerebral ischemia. Part II: Restitution of cerebral energystate, as well as of glycolytic metabolites, citric acid cycle intermediates and associated amino acids after pronounced in complete ischemia. Stroke 1978, 9, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.L.; Almeida, A.; Bates, T.E.; Clark, J.B. Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischaemia. J. Neurochem. 1995, 64, 2222–2229. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Kristián, T.; Hu, B.; Ouyang, Y.B.; Siesjö, B.K. Effect of NXY-059 on secondary mitochondrial dysfunction after transient focal ischemia: Comparison with cyclosporin A. Brain Res. 2002, 932, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Niatsetskaya, Z.V.; Sosunov, S.A.; Matsiukevich, D.; Utkina Sosunova, I.V.; Ratner, V.I.; Starkov, A.A.; Ten, V.S. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia–ischemia in neonatal mice. J. Neurosci. 2012, 32, 3235–3244. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, H.; Ohba, H.; Nishiyama, S.; Kanazawa, M.; Kakiuchi, T.; Harada, N. PET imaging of ischemia-induced impairment of mitochondrial complex I function in monkey brain. J. Cereb. Blood Flow Metab. 2014, 34, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. Aunifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Ten, V.; Galkin, A. Mechanism of mitochondrial complex I damage in brain ischemia/reperfusion injury—A hypothesis. Mol. Cell. Neurosci. 2019, 100, 103408. [Google Scholar] [CrossRef]
- Moro, M.A.; Almeida, A.; Bolaños, J.P.; Lizasoain, I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med. 2005, 39, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; Schapira, A.H. Mitochondrial respiratory chain disorders I: Mitochondrial DNA defects. Lancet 2000, 355, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; Schapira, A.H. Mitochondrial respiratory chain disorders II: Neurodegenerative disorders and nuclear gene defects. Lancet 2000, 355, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Becker, N.H. The cytochemistry of anoxic and anoxio ischemic encephalopathy in rats. II. Alterations in neuronal mitochondria indentified by diphosphopyridine and triphosphopyridine nucleotide diaphorases. Am. J. Pathol. 1961, 38, 587–597. [Google Scholar] [PubMed]
- Cryer, A.; Bartley, W. Changes in enzyme activities in tissues of rats exposed to hypoxia. Biochem. J. 1973, 134, 1119. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.S.; Parvez, S.; Tabassum, H. Ischemic stroke and mitochondria: Mechanisms and targets. Protoplasma 2020, 257, 335–343. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.J.; Brand, M.D. Superoxide production by NADH: Ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004, 382, 511–517. [Google Scholar] [CrossRef]
- Murphy, M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016, 44, 1219–1226. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Vlkolinsky, R.; Cairns, N.; Lubec, G. Decreased levels of complex-III core protein1 and complex V β chain in brains from patients with Alzheimer’s disease and Down syndrome. Cell. Mol. Life Sci. 2000, 57, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Hollunger, G. The interaction of energy and electron transfer reactions in mitochondria: I. General properties and nature of the products of succinate-linked reduction of pyridinenucleotide. J. Biol. Chem. 1961, 236, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactiveoxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, K.S.; Ashraf, S.; Lomax, T.M.; Wiseman, J.M.; Hall, M.E.; Gava, F.N.; Hall, J.E.; Hosler, J.P.; Harmancey, R. Uncoupling protein-3 deficiency impairs myocardial fatty acid oxidation and contractile recovery following ischemia/reperfusion. Basic Res. Cardiol. 2018, 113, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Clearing the brain’s amyloid cobwebs. Neuron 2001, 32, 177–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toan, S.; Zhou, H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 2020, 23, 299–314. [Google Scholar] [CrossRef] [PubMed]
- McAvoy, K.; Kawamata, H. Glial mitochondrial function and dysfunction in health and neurodegeneration. Mol. Cell. Neurosci. 2019, 101, 103417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.; Yamazaki, Y.; et al. Modulation of mitochondrial complex-I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. EBioMedicine 2015, 2, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; McKeehan, N.; Fillit, H.M. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 2019, 92, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Lu, B. Regulation of cell growth by Notch signaling and its differential requirement in normal vs.tumor-forming stemcells in Drosophila. Genes Dev. 2011, 25, 2644–2658. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Wu, Z.; Song, Y.; Mitra, S.S.; Feroze, A.H.; Cheshier, S.H.; Lu, B. Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev. 2013, 27, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- Ojha, R.; Tantray, I.; Rimal, S.; Mitra, S.; Cheshier, S.; Lu, B. Regulation of reverse electron transfer at mitochondrial complexI by unconventional Notch actionin cancer stem cells. Dev. Cell 2022, 57, 260–276. [Google Scholar] [CrossRef]
- Ho, D.M.; Artavanis-Tsakonas, S.; Louvi, A. The Notch pathway in CNS homeostasis and neurodegeneration. Wiley Interdis. Rev. Dev. Biol. 2020, 9, e358. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Chan, S.L.; Jo, D.G.; Yilmaz, G.; Tang, S.C.; Cheng, A.; Gleichmann, M.; Okun, E.; Dixit, V.D.; Chigurupati, S.; et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat. Med. 2006, 12, 621–623. [Google Scholar] [CrossRef]
- Kim, M.; Stepanova, A.; Niatsetskaya, Z.; Sosunov, S.; Arndt, S.; Murphy, M.P.; Galkin, A.; Ten, V.S. Attenuation of oxidative damage by targeting mitochondrial complex-I in neonatal hypoxic-ischemic brain injury. Free Radic. Biol. Med. 2018, 124, 517–524. [Google Scholar] [CrossRef]
- Drose, S.; Stepanova, A.; Galkin, A. Ischemic A/D transition of mitochondrial complex I and its role in ROS generation. Biochim. Biophys. Acta 2016, 1857, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardio protection by S-nitrosation of acysteine switch on mitochondrial complex-I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, C.C.; Zeidler, J.D.; Kashyap, S.; Warner, G.; Chini, E.N. Evolving concepts in NAD+ metabolism. Cell Metab. 2021, 33, 1076–1087. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and health span. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.I. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet-and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavda, V.; Lu, B. Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases. Antioxidants 2023, 12, 895. https://doi.org/10.3390/antiox12040895
Chavda V, Lu B. Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases. Antioxidants. 2023; 12(4):895. https://doi.org/10.3390/antiox12040895
Chicago/Turabian StyleChavda, Vishal, and Bingwei Lu. 2023. "Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases" Antioxidants 12, no. 4: 895. https://doi.org/10.3390/antiox12040895