
Membrane Vesicles of Toxigenic Clostridioides difficile Affect the Metabolism of Liver HepG2 Cells
 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients’ Recruitment
2.2. Isolation of EVs from Human Feces
2.3. DNA Extraction, Libraries Preparation and Sequencing
2.4. Bioinformatics, Ordination and Statistical Analysis for the Metagenomic Study
2.5. Cell and Culture Conditions
2.6. C. difficile Culture and MVs Production
2.7. Electron Microscopy of EVs
2.8. Cellular Exposition to C. difficile-Derived MVs
2.9. Fluorescence Microscopy Study of C. difficile-Derived MVs Internalization
2.10. Mitochondrial Membrane Potential Assessment
2.11. Intracellular ROS Level Measurement
2.12. Quantification of Adherent Cells
2.13. RNA Isolation and Quantitative Real-Time PCR
2.14. Statistical Analyses
3. Results
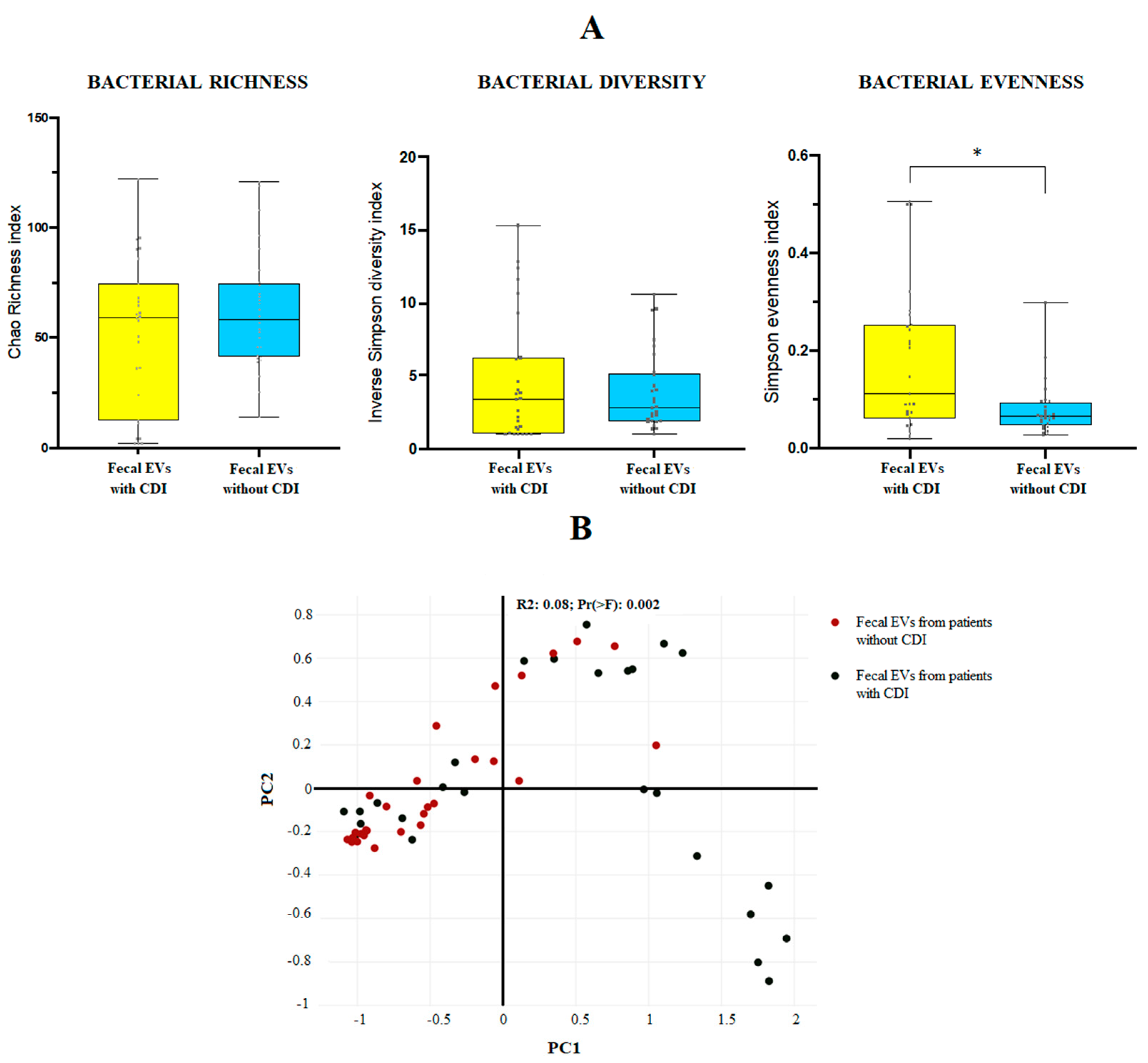
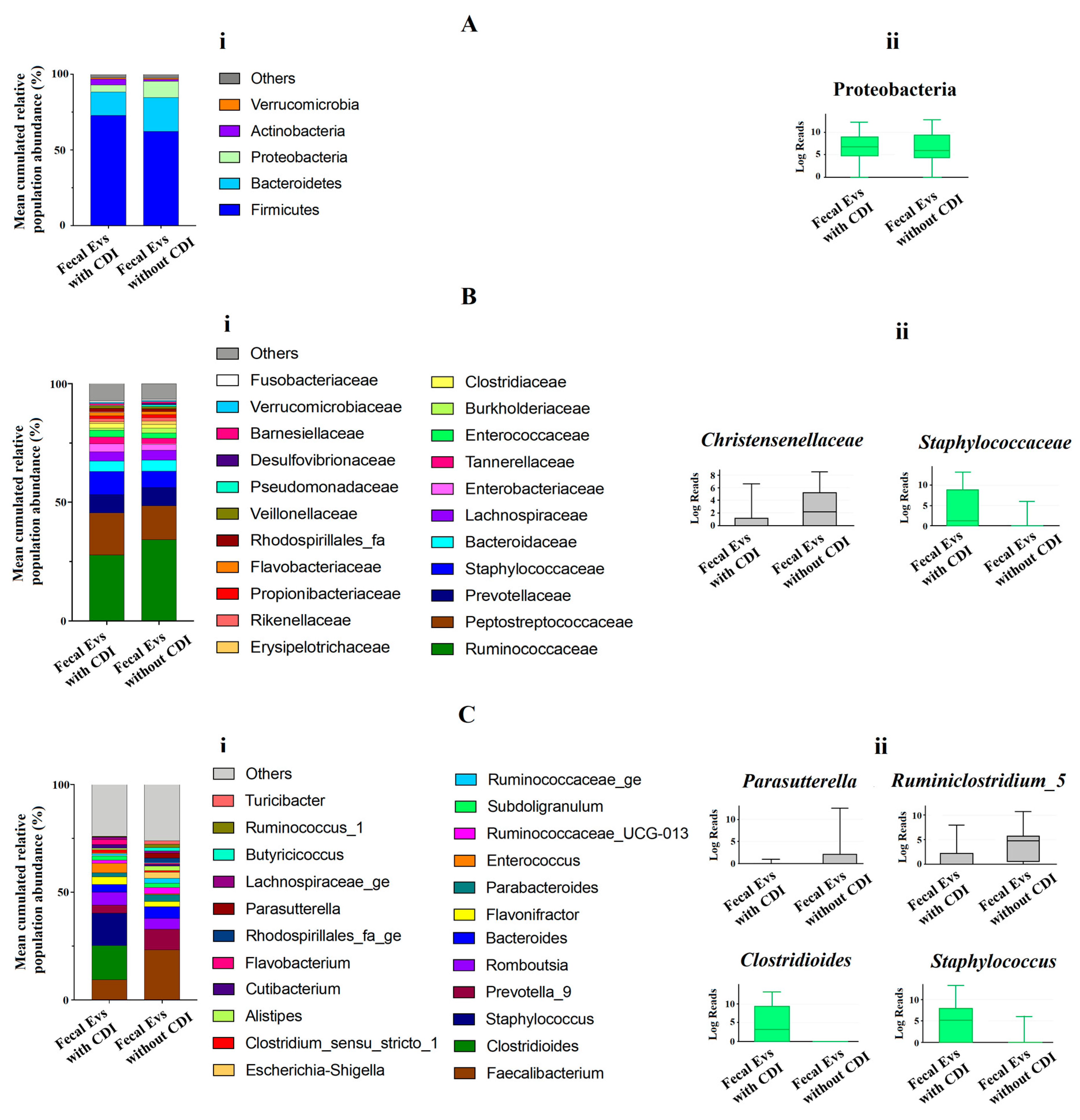
3.1. Microbiome Profile of EVs from Patients with and without CDI
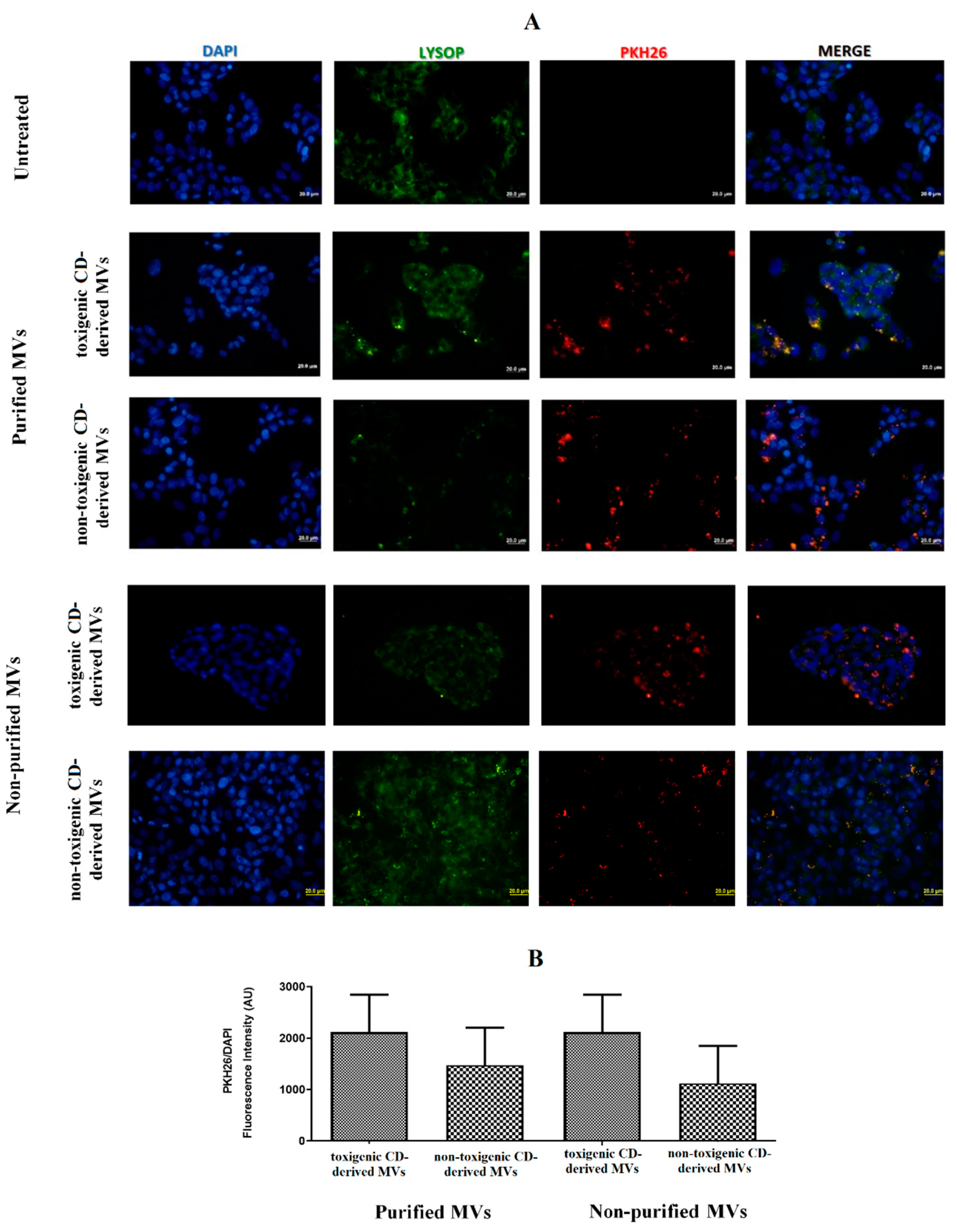
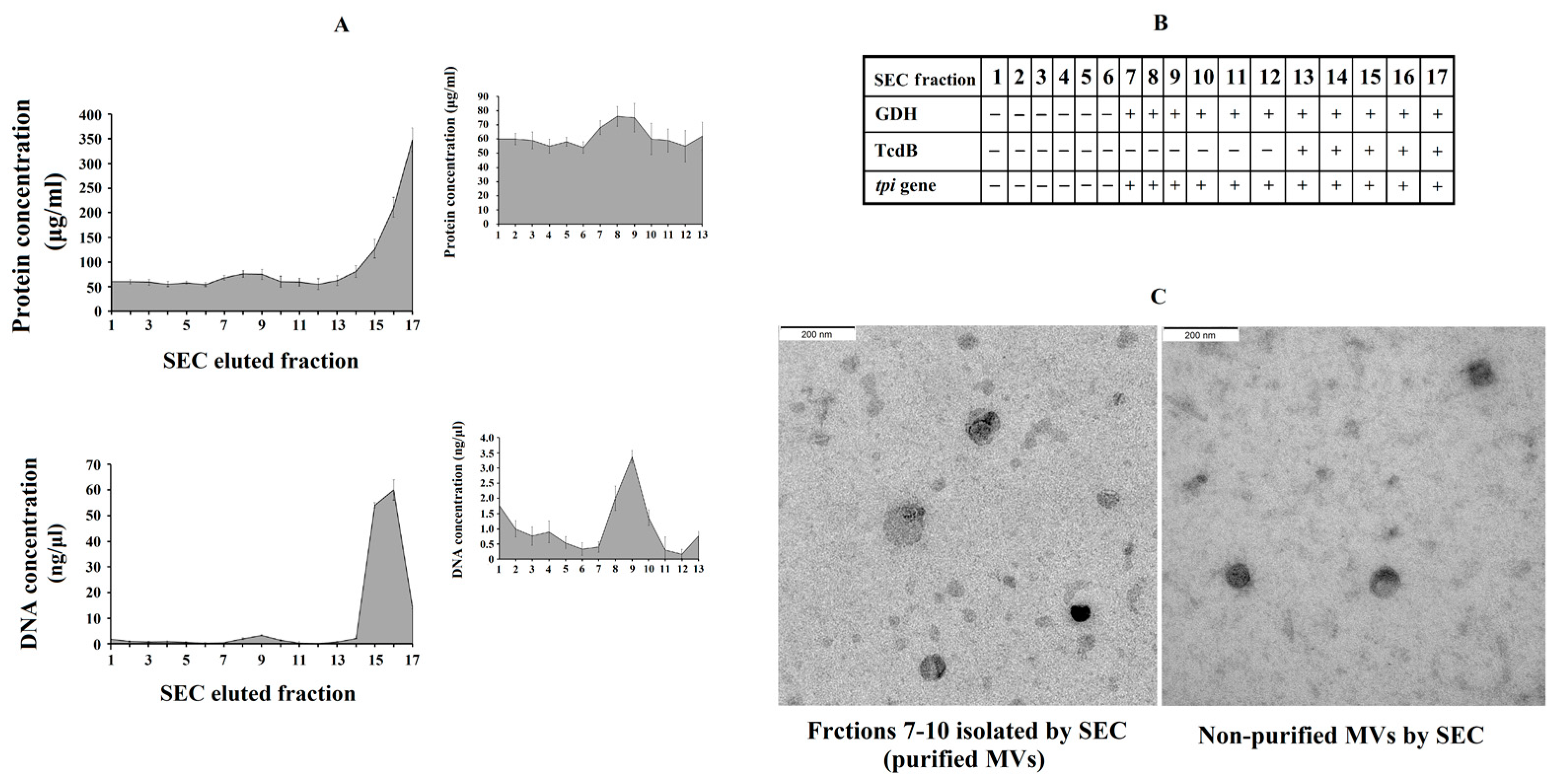
3.2. EVs from C. difficile Are Internalized by HepG2 Cells
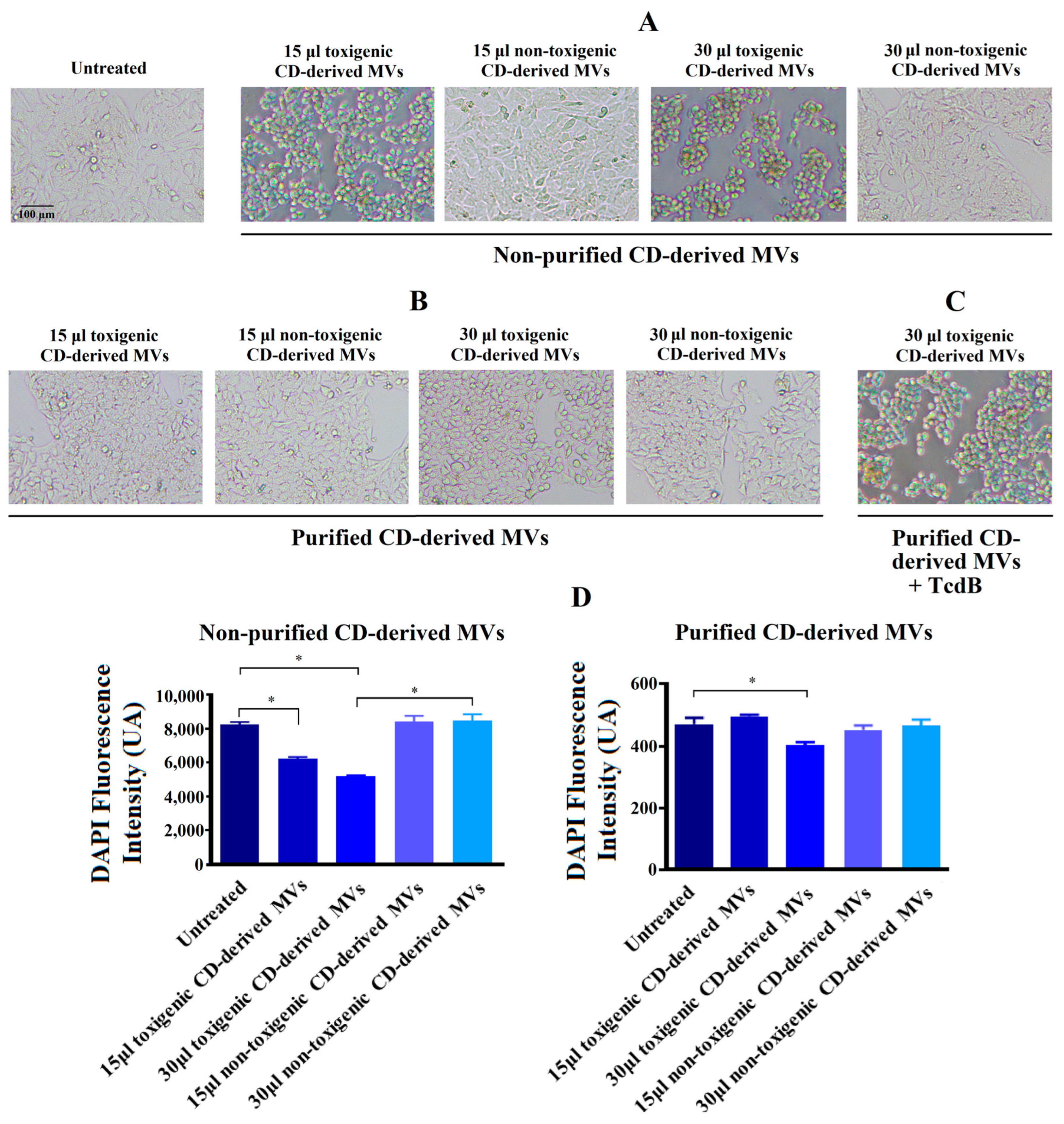
3.3. Effects of Non-Purified C. difficile-Derived MVs on the Morphology of HepG2 Cells
3.4. Effects of Purified C. difficile-Derived MVs on the Morphology of HepG2 Cells
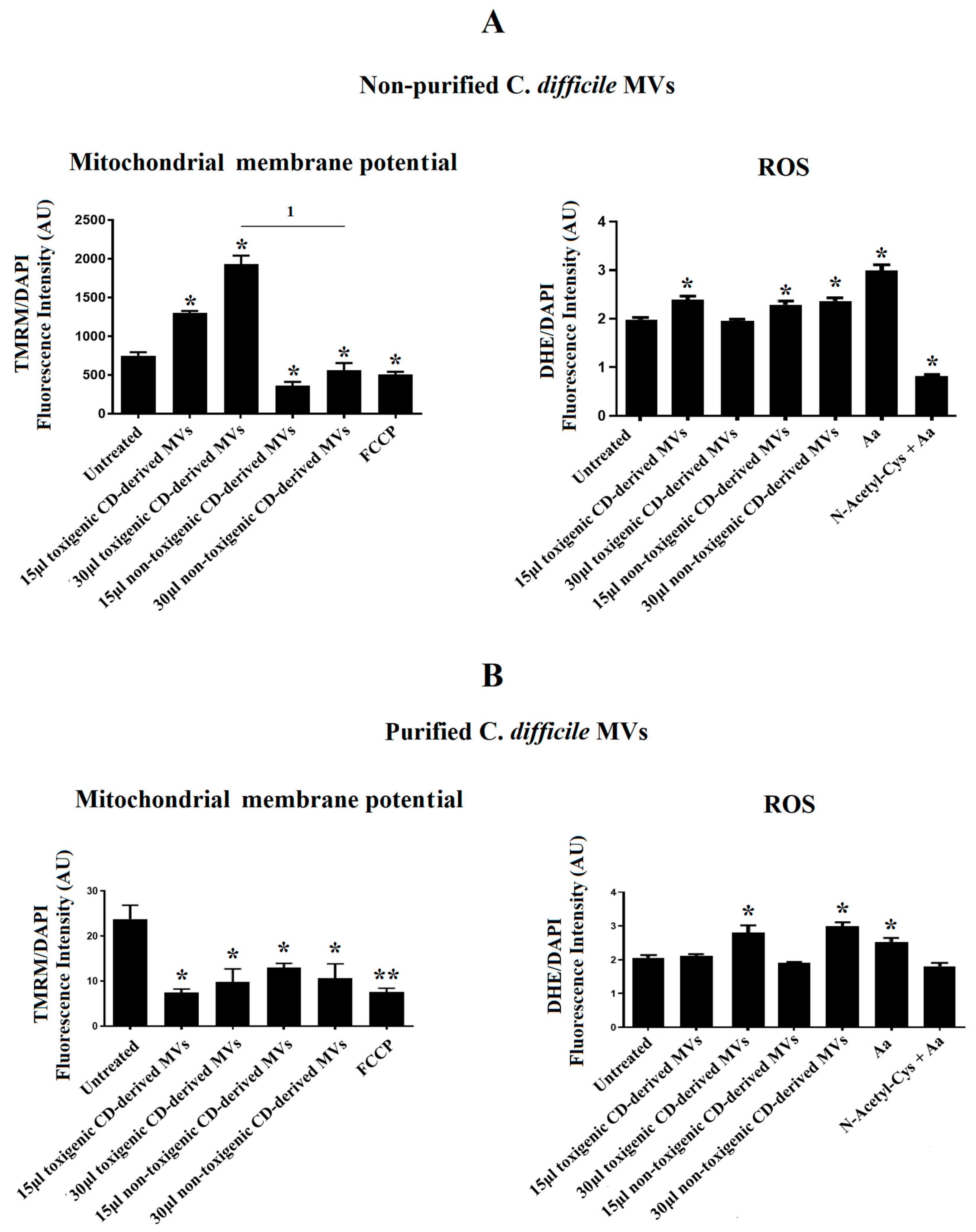
3.5. C. difficile-Derived MVs Induce Mitochondrial Dysfunction and Increase ROS Level in HepG2 Cells
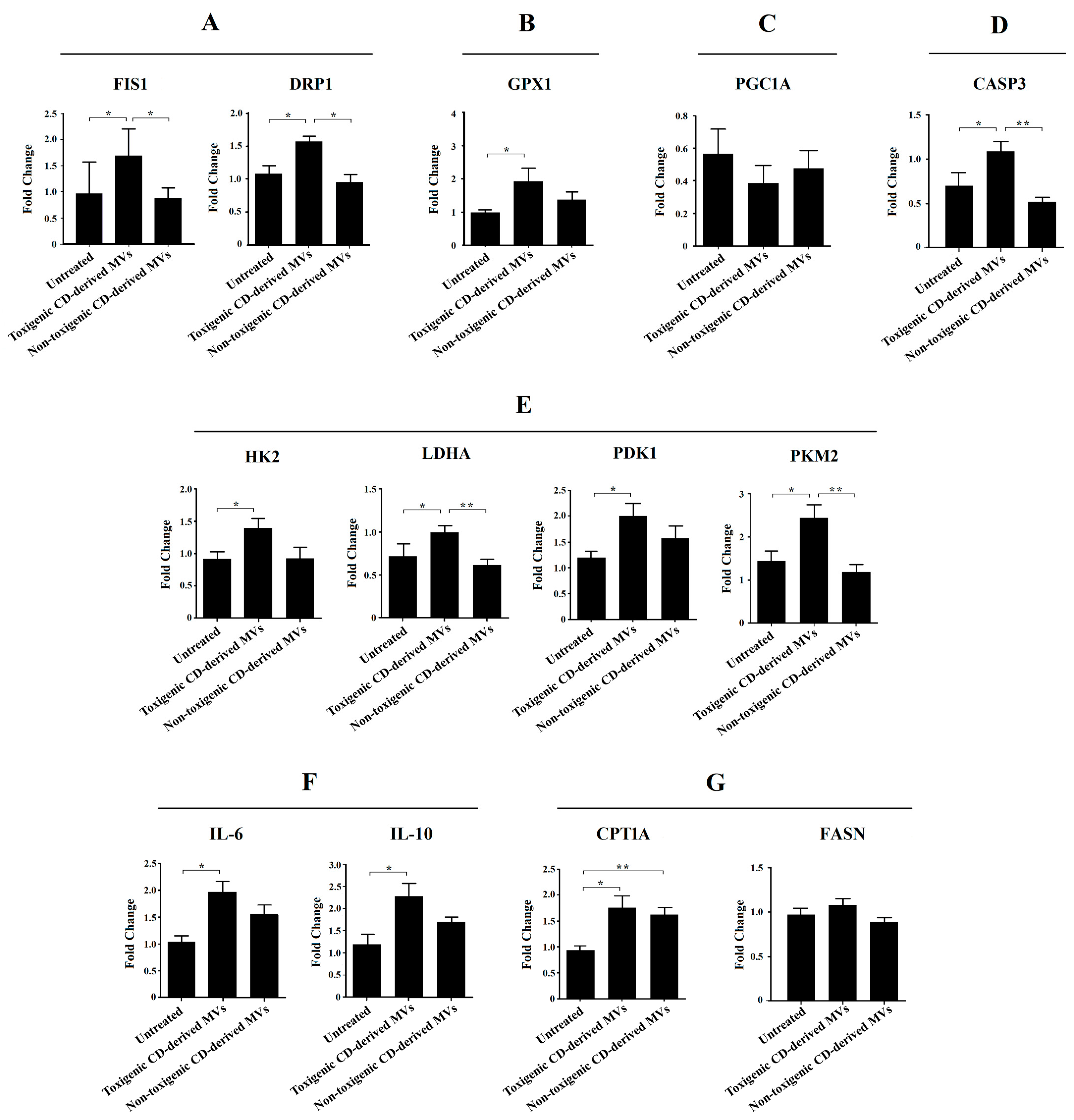
3.6. Mitochondrial Fission Key Genes Are Up-Regulated by Purified Toxigenic C. difficile-Derived MVs in HepG2 Cells
3.7. Purified Toxigenic C. difficile-Derived MVs Do Not Affect Mitochondrial Biogenesis or Trigger Apoptosis in HepG2 Cells
3.8. Glycolysis Key Enzymes Are Up-Regulated by Purified Toxigenic C. difficile-Derived MVs in HepG2 Cells
3.9. Anti- and Pro-Inflammatory Interleukins Are Up-Regulated by Purified Toxigenic C. difficile-Derived MVs in HepG2 Cells
3.10. β-Oxidation, but Not Fatty Acid Synthesis, Is Up-Regulated by Purified C. difficile-Derived MVs in HepG2 Cells
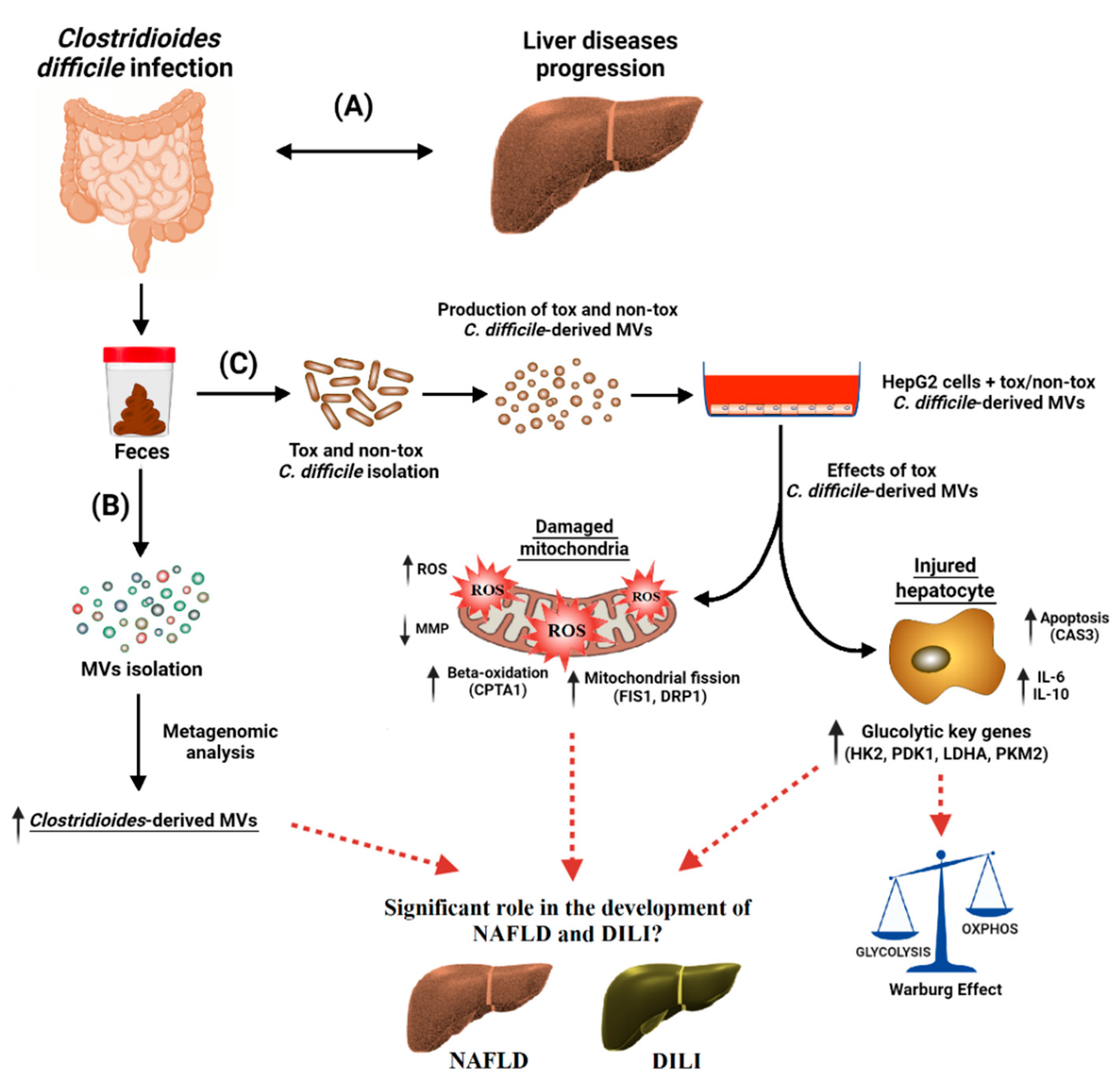
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, L.; Schnabl, B. Gut Microbiota in Liver Disease: What Do We Know and What Do We Not Know? Physiology 2020, 35, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Luo, K.; Li, J.; Li, Y.; Zhang, Y.; Yuan, Z.; Xu, Q.; Wu, X. Role of Intestinal Microbes in Chronic Liver Diseases. Int. J. Mol. Sci. 2022, 23, 12661. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Diaz, C.; Taminiau, B.; García-García, A.; Cueto, A.; Robles-Díaz, M.; Ortega-Alonso, A.; Martín-Reyes, F.; Daube, G.; Sanabria-Cabrera, J.; Jimenez-Perez, M.; et al. Microbiota diversity in nonalcoholic fatty liver disease and in drug-induced liver injury. Pharmacol. Res. 2022, 182, 106348. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.; Min, B.H.; Ganesan, R.; Gebru, Y.A.; Sharma, S.P.; Park, E.; Won, S.M.; Jeong, J.J.; Lee, S.B.; Cha, M.G.; et al. Gut microbiome in non-alcoholic fatty liver disease: From mechanisms to therapeutic role. Biomedicines 2022, 10, 550. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clement, K. Gut microbiota and non-alcoholic fatty liver disease: New insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.; Van Broeck, J.; Taminiau, B.; Delmée, M.; Daube, G. Clostridium difficile infection: Early history, diagnosis and molecular strain typing methods. Microb. Pathog. 2016, 97, 59–78. [Google Scholar] [CrossRef] [Green Version]
- Blanchi, J.; Goret, J.; Mégraud, F. Clostridium difficile Infection: A Model for Disruption of the Gut Microbiota Equilibrium. Dig. Dis. 2016, 34, 217–220. [Google Scholar] [CrossRef]
- Nicholas, A.; Jeon, H.; Selasi, G.N.; Na, S.H.; Kwon, H.I.; Kim, Y.J.; Choi, C.W.; Kim, S.I.; Lee, J.C. Clostridium difficile-derived membrane vesicles induce the expression of pro-inflammatory cytokine genes and cytotoxicity in colonic epithelial cells in vitro. Microb. Pathog. 2017, 107, 6–11. [Google Scholar] [CrossRef]
- Caruana, J.C.; Walper, S.A. Bacterial Membrane Vesicles as Mediators of Microbe—Microbe and Microbe—Host Community Interactions. Front. Microbiol. 2020, 11, 432. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, K.; Hampton, T.H.; Jarek, M.; Scharfe, M.; Gerber, S.A.; Mielcarz, D.W.; Demers, E.G.; Dolber, E.L.; Hammond, J.H.; Hogan, D.A.; et al. A Novel Mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 2016, 12, e1005672. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Garrido, N.; Bonnin, S.; Riera, M.; Gíménez, R.; Badia, J.; Baldomà, L. Transcriptomic microRNA Profiling of Dendritic Cells in Response to Gut Microbiota-Secreted Vesicles. Cells 2020, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, J.P.; Yang, J.; Won, H.K.; Park, C.S.; Song, W.J.; Kwon, H.S.; Kim, T.B.; Kim, Y.K.; Park, H.S.; et al. Metagenome analysis using serum extracellular vesicles identified distinct microbiota in asthmatics. Sci. Rep. 2020, 10, 15125. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Díaz, C.; Martín-Reyes, F.; Taminiau, B.; Ho-Plágaro, A.; Camargo, R.; Fernandez-Garcia, F.; Pinazo-Bandera, J.; Toro-Ortiz, J.P.; Gonzalo, M.; López-Gómez, C.; et al. The Metagenomic Composition and Effects of Fecal-Microbe-Derived Extracellular Vesicles on Intestinal Permeability Depend on the Patient’s Disease. Int. J. Mol. Sci. 2023, 24, 4971. [Google Scholar] [CrossRef]
- Choi, J.W.; Um, J.H.; Cho, J.H.; Lee, H.J. Tiny RNAs and their voyage via extracellular vesicles: Secretion of bacterial small RNA and eukaryotic microRNA. Exp. Biol. Med. 2017, 242, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktories, K.; Papatheodorou, P.; Schwan, C. Binary Clostridium difficile toxin (CDT)—A virulence factor disturbing the cytoskeleton. Anaerobe 2018, 53, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Kong, Q.; Roland, K.L.; Curtiss, R., 3rd. Membrane vesicles of Clostridium perfringens type A strains induce innate and adaptive immunity. Int. J. Med. Microbiol. 2014, 304, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, H.M.; Kuehn, M.J. The extracellular vesicle generation paradox: A bacterial point of view. Embo J. 2021, 40, e108174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, J.; Miao, Y.; Zhang, Q. The effect of extracellular vesicles on the regulation of mitochondria under hypoxia. Cell Death Dis. 2021, 12, 358. [Google Scholar] [CrossRef]
- Trifan, A.; Stoica, O.; Stanciu, C.; Cojocariu, C.; Singeap, A.M.; Girleanu, I.; Miftode, E. Clostridium difficile infection in patients with liver disease: A review. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 2313–2324. [Google Scholar] [CrossRef]
- Šamadan, L.; Jeličić, M.; Vince, A.; Papić, N. Nonalcoholic Fatty Liver Disease-A Novel Risk Factor for Recurrent Clostridioides difficile Infection. Antibiotics 2021, 10, 780. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Kim, S.; Lee, J.; Ha, J.; Choi, Y.; Oh, H.; Kim, Y.; Lee, Y.; Choi, K.H.; et al. Intestinal Clostridioides difficile Can Cause Liver Injury through the Occurrence of Inflammation and Damage to Hepatocytes. Biomed. Res. Int. 2020, 2020, 7929610. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- McKay, D.M.; Mancini, N.L.; Shearer, J.; Shutt, T. Perturbed mitochondrial dynamics, an emerging aspect of epithelial-microbe interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G748–G762. [Google Scholar] [CrossRef] [PubMed]
- Lagos, L.; Leanti La Rosa, S.; Ø Arntzen, M.; Ånestad, R.; Terrapon, N.; Gaby, J.C.; Westereng, B. Isolation and Characterization of Extracellular Vesicles Secreted In Vitro by Porcine Microbiota. Microorganisms 2020, 8, 983. [Google Scholar] [CrossRef] [PubMed]
- Dauros Singorenko, P.; Chang, V.; Whitcombe, A.; Simonov, D.; Hong, J.; Phillips, A.; Swift, S.; Blenkiron, C. Isolation of membrane vesicles from prokaryotes: A technical and biological comparison reveals heterogeneity. J. Extracell. Vesicles. 2017, 6, 1324731. [Google Scholar] [CrossRef] [Green Version]
- Mehanny, M.; Koch, M.; Lehr, C.M.; Fuhrmann, G. Streptococcal Extracellular Membrane Vesicles Are Rapidly Internalized by Immune Cells and Alter Their Cytokine Release. Front. Immunol. 2020, 11, 80. [Google Scholar] [CrossRef]
- Gérard, A.; El-Hajjaji, S.; Burteau, S.; Fall, P.A.; Pirard, B.; Taminiau, B.; Daube, G.; Sindic, M. Study of the microbial diversity of a panel of Belgian artisanal cheeses associated with challenge studies for Listeria monocytogenes. Food Microbiol. 2021, 100, 103861. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.; Taminiau, B.; Van Broeck, J.; Avesani, V.; Delmée, M.; Daube, G. Clostridium difficile in young farm animals and slaughter animals in Belgium. Anaerobe 2012, 18, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.; Huhulescu, S.; Schneeweis, M.; Hasenberger, P.; Kernbichler, S.; Fiedler, A.; Wewalka, G.; Allerberger, F.; Kuijper, E.J. Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J. Med. Microbiol. 2008, 57 Pt 11, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, C.; Avesani, V.; Van Broeck, J.; Taminiau, B.; Delmée, M.; Daube, G. Presence of Clostridium difficile in pigs and cattle intestinal contents and carcass contamination at the slaughterhouse in Belgium. Int. J. Food Microbiol. 2013, 166, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Hussein, E.; Davies, S.P.; Humphreys, P.N.; Collett, A. Commensal-derived OMVs elicit a mild proinflammatory response in intestinal epithelial cells. Microbiology 2017, 163, 702–711. [Google Scholar] [CrossRef]
- Brandes, V.; Schelle, I.; Brinkmann, S.; Schulz, F.; Schwarz, J.; Gerhard, R.; Genth, H. Protection from Clostridium difficile toxin B-catalysed Rac1/Cdc42 glucosylation by tauroursodeox-ycholic acid-induced Rac1/Cdc42 phosphorylation. Biol. Chem. 2012, 393, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Ligasová, A.; Koberna, K. Quantification of fixed adherent cells using a strong enhancer of the fluorescence of DNA dyes. Sci. Rep. 2019, 9, 8701. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.S.; Schnellmann, R.G. Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. 2004, 25, 12.8.1–12.8.22. [Google Scholar] [CrossRef] [Green Version]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Ni, Q.; Wang, C.; Zhang, L.; Li, Z.; Jiang, C.; Mao, E.; Peng, Y. Effects of intestinal colonization by Clostridium difficile and Staphylococcus aureus on microbiota diversity in healthy individuals in China. BMC Infect. Dis. 2018, 18, 207. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Mikkelsen, L.S.; Brandsborg, E.; Marcotte, H. Clostridium difficile, the Difficult “Kloster” Fuelled by Antibiotics. Curr. Microbiol. 2019, 76, 774–782. [Google Scholar] [CrossRef]
- Azimirad, M.; Krutova, M.; Balaii, H.; Kodori, M.; Shahrokh, S.; Azizi, O.; Yadegar, A.; Aghdaei, H.A.; Zali, M.R. Coexistence of Clostridioides difficile and Staphylococcus aureus in gut of Iranian outpatients with underlying inflammatory bowel disease. Anaerobe 2020, 61, 102113. [Google Scholar] [CrossRef] [PubMed]
- Vincent, C.; Manges, A.R. Antimicrobial Use, Human Gut Microbiota and Clostridium difficile Colonization and Infection. Antibiotics 2015, 4, 230–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.; Kong, J.Y.; Stothard, P.; Willing, B.P. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019, 13, 1520–1534. [Google Scholar] [CrossRef]
- Schadt, H.S.; Wolf, A.; Pognan, F.; Chibout, S.D.; Merz, M.; Kullak-Ublick, G.A. Bile acids in drug induced liver injury: Key players and surrogate markers. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Paz, M.; Morán, L.; López-Alcántara, N.; Freixo, C.; Andrade, R.J.; Lucena, M.I.; Cubero, F.J. Oxidative Stress in Drug-Induced Liver Injury (DILI): From Mechanisms to Biomarkers for Use in Clinical Practice. Antioxidants 2021, 10, 390. [Google Scholar] [CrossRef]
- Park, J.W.; Kim, S.E.; Lee, N.Y.; Kim, J.H.; Jung, J.H.; Jang, M.K.; Park, S.H.; Lee, M.S.; Kim, D.J.; Kim, H.S.; et al. Role of Microbiota-Derived Metabolites in Alcoholic and Non-Alcoholic Fatty Liver Diseases. Int. J. Mol. Sci. 2021, 23, 426. [Google Scholar] [CrossRef]
- Richard, J.F.; Petit, L.; Gibert, M.; Marvaud, J.C.; Bouchaud, C.; Popoff, M.R. Bacterial toxins modifying the actin cytoskeleton. Int. Microbiol. 1999, 2, 185–194. [Google Scholar]
- Buddle, J.E.; Fagan, R.P. Pathogenicity and virulence of Clostridioides difficile. Virulence 2023, 14, 2150452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chowdhury, S.; Xu, B.H.; Meybodi, M.A.; Damiris, K.; Devalaraju, S.; Pyrsopoulos, N. Nonalcoholic fatty liver disease is associated with worse intestinal complications in patients hospitalized for Clostridioides difficile infection. World J. Hepatol. 2021, 13, 1777–1790. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, M. Clostridioides difficile Infection in Liver Cirrhosis: A Concise Review. Can. J. Gastroenterol. Hepatol. 2022, 2022, 4209442. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.H.; Chen, B.H.; Liu, Q.Y.; Le, X.Y.; He, L. Synthesis, structural studies, interaction with DNA/HSA and antitumor evaluation of new Cu(II) complexes containing 2-(1H-imidazol-2-yl)pyridine and amino acids. Dalton Trans. 2022, 51, 16574–16586. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell. Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Deo, P.; Chow, S.H.; Hay, I.D.; Kleifeld, O.; Costin, A.; Elgass, K.D.; Jiang, J.H.; Ramm, G.; Gabriel, K.; Dougan, G.; et al. Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLoS Pathog. 2018, 14, e1006945. [Google Scholar] [CrossRef] [Green Version]
- To, E.E.; O’Leary, J.J.; O’Neill, L.A.J.; Vlahos, R.; Bozinovski, S.; Porter, C.J.H.; Brooks, R.D.; Brooks, D.A.; Selemidis, S. Spatial Properties of Reactive Oxygen Species Govern Pathogen-Specific Immune System Responses. Antioxid. Redox Signal. 2020, 32, 982–992. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Fondevila, M.F.; Fernandez, U.; Heras, V.; Parracho, T.; Gonzalez-Rellan, M.J.; Novoa, E.; Porteiro, B.; Alonso, C.; Mayo, R.; da Silva Lima, N.; et al. Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J. Hepatol. 2022, 77, 15–28. [Google Scholar] [CrossRef]
- Sun, W.; Nie, T.; Li, K.; Wu, W.; Long, Q.; Feng, T.; Mao, L.; Gao, Y.; Liu, Q.; Gao, X.; et al. Hepatic CPT1A Facilitates Liver-Adipose Cross-Talk via Induction of FGF21 in Mice. Diabetes 2021, 71, 31–42. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free. Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.; Wu, B.; Nguyen, H.B.; Thai, T.Q.; Yamasaki, R.; Lu, H.; Rietsch, A.M.; Zorlu, M.M.; Shinozaki, Y.; Saitoh, Y.; et al. Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci. Rep. 2017, 7, 4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramond, E.; Jamet, A.; Coureuil, M.; Charbit, A. Pivotal Role of Mitochondria in Macrophage Response to Bacterial Pathogens. Front. Immunol. 2019, 10, 2461. [Google Scholar] [CrossRef] [PubMed]
- Legaki, A.I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Umbaugh, D.S.; Jaeschke, H. Mitochondrial Dynamics in Drug-Induced Liver Injury. Livers 2021, 1, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell. Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell. Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell. Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, S.; Yu, D. Protein kinase function of pyruvate kinase M2 and cancer. Cancer Cell. Int. 2020, 20, 523. [Google Scholar] [CrossRef]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef] [Green Version]
- Inomata, Y.; Oh, J.W.; Taniguchi, K.; Sugito, N.; Kawaguchi, N.; Hirokawa, F.; Lee, S.W.; Akao, Y.; Takai, S.; Kim, K.P.; et al. Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2022, 23, 5230. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, S.; Zhao, Y.; Sun, Q.; Zhang, J.; Shen, D.; Wu, J.; Shen, N.; Fu, X.; Sun, X.; et al. Geranylgeranyl diphosphate synthase (GGPPS) regulates non-alcoholic fatty liver disease (NAFLD)-fibrosis progression by determining hepatic glucose/fatty acid preference under high-fat diet conditions. J. Pathol. 2018, 246, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, K.; Yao, W.; Zheng, R.; He, Q.; Xia, J.; Li, J.; Shao, Y.; Zhang, L.; Huang, L.; et al. Acetylation of lactate dehydrogenase B drives NAFLD progression by impairing lactate clearance. J. Hepatol. 2021, 74, 1038–1052. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene ID (ENSEMBL) | Sequence (5′-3′) | TM (°C) |
| DRP1 | ENSG00000087470 | forward: GTGAACCCGTGGATGATAAA reverse: GAAACCTCAGGCACAAATAAAG | 62 |
| FIS1 | ENSG00000214253 | forward: AGCGGGATTACGTCTTCTA reverse: CCACGAGTCCATCTTTCTTC | 60 |
| GPX1 | ENSG00000233276 | forward: CCCTGCGGGGCAAGGTACTA reverse: GGGCATCAGGAGAACGCCAA | 60 |
| LDHA | ENSG00000134333 | forward: TATGGAGTGGAATGAATGTTG reverse: CCCTTAATCATGGTGGAAACT | 60 |
| HK2 | ENSG00000159399 | forward: TAGCCTTCTTTGTGCGCCGT reverse: GGTCAACCTTCTGCACTTGGTCA | 69 |
| PDK1 | ENSG00000152256 | forward: TGCCTCTGGCTGGTTTTGGTTAT reverse: TGTCTAGGCACTGCGGAACG | 68 |
| PKM2 | ENSG00000067225 | forward: ACTCACTCTGGGCTGTAA reverse: CCTCCTTCTTCCCTTGATTG | 60 |
| PGC1A | ENSG00000109819 | forward: CACCAGCCAACACTCAGCTA reverse: GTGTGAGGAGGGTCATCGTT | 62 |
| CASP3 | ENSG00000164305 | forward: TGCCCTGACTTCTCTGTAGC reverse: TTGGAGCCACACAGACCTAG | 60 |
| CPT1A | ENSG00000110090 | forward: CAGCATATGTATCGCCTCGC reverse: CTGGACACGTACTCTGGGTT | 60 |
| FASN | ENSG00000169710 | forward: CCCTCATCTCCCCACTCATC reverse: CAGCGTCTTCCACACTATGC | 60 |
| IL-6 | ENSG00000136244 | forward: CACACAGACAGCCACTCACC reverse: TTTTCTGCCAGTGCCTCTTT | 62 |
| IL-10 | ENSG00000136634 | forward: GCTCTGTTGCCTGGTCCTC reverse: TGTCTGGGTCTTGGTTCTCA | 63 |
| β-actin | ENSG00000075624 | forward: TACAGCTTCACCACCACGGC reverse: AAGGAAGGCTGGAAGAGTGC | 64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballano-Infantes, E.; Ho-Plágaro, A.; López-Gómez, C.; Martín-Reyes, F.; Rodríguez-Pacheco, F.; Taminiau, B.; Daube, G.; Garrido-Sánchez, L.; Alcaín-Martínez, G.; Andrade, R.J.; et al. Membrane Vesicles of Toxigenic Clostridioides difficile Affect the Metabolism of Liver HepG2 Cells. Antioxidants 2023, 12, 818. https://doi.org/10.3390/antiox12040818
Caballano-Infantes E, Ho-Plágaro A, López-Gómez C, Martín-Reyes F, Rodríguez-Pacheco F, Taminiau B, Daube G, Garrido-Sánchez L, Alcaín-Martínez G, Andrade RJ, et al. Membrane Vesicles of Toxigenic Clostridioides difficile Affect the Metabolism of Liver HepG2 Cells. Antioxidants. 2023; 12(4):818. https://doi.org/10.3390/antiox12040818
Chicago/Turabian StyleCaballano-Infantes, Estefanía, Ailec Ho-Plágaro, Carlos López-Gómez, Flores Martín-Reyes, Francisca Rodríguez-Pacheco, Bernard Taminiau, Georges Daube, Lourdes Garrido-Sánchez, Guillermo Alcaín-Martínez, Raúl J. Andrade, and et al. 2023. "Membrane Vesicles of Toxigenic Clostridioides difficile Affect the Metabolism of Liver HepG2 Cells" Antioxidants 12, no. 4: 818. https://doi.org/10.3390/antiox12040818