
Accelerated AGEing: The Impact of Advanced Glycation End Products on the Prognosis of Chronic Kidney Disease
 , , , and
, , , and
Abstract
:1. Introduction
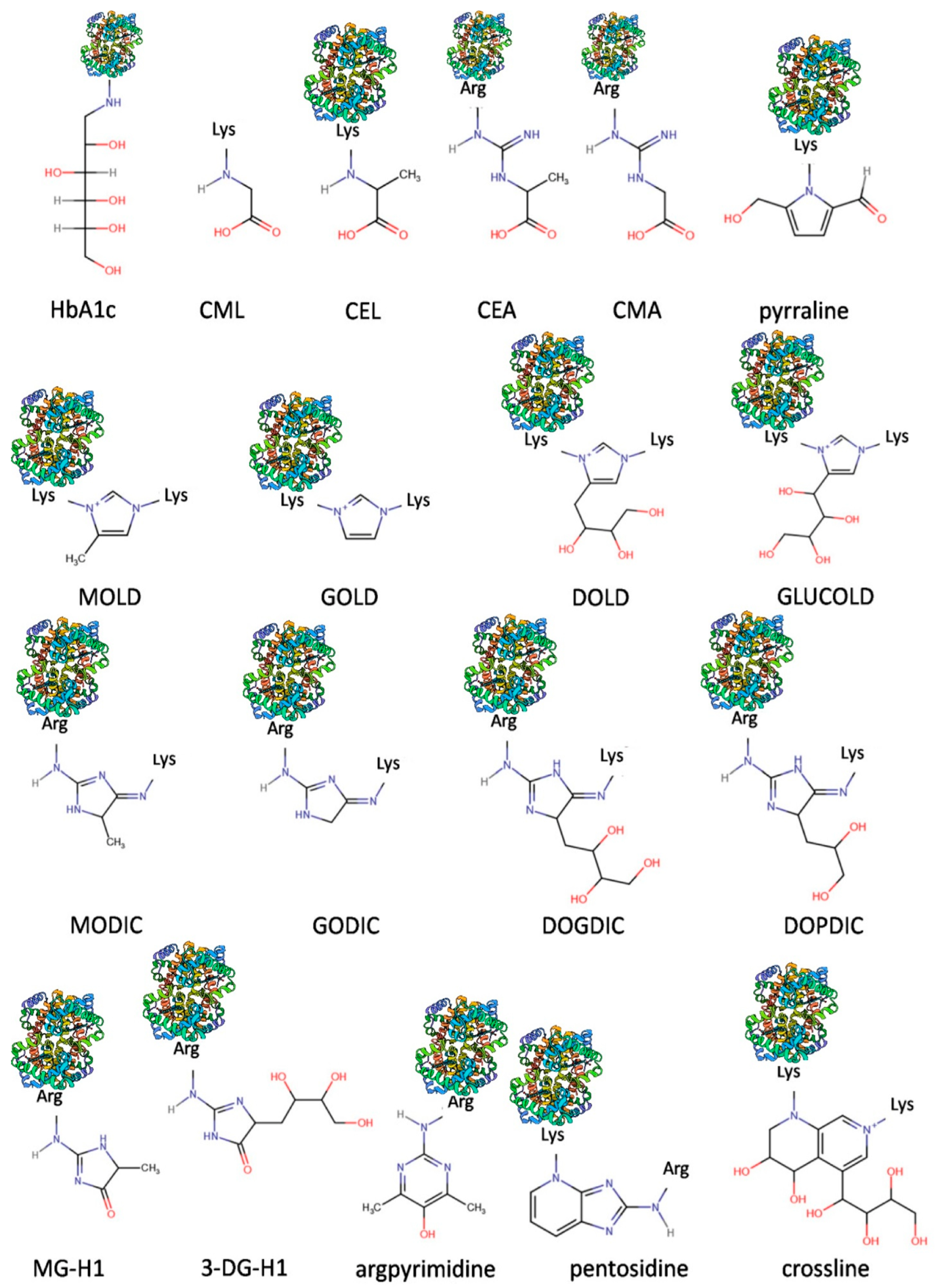
2. Advanced Glycation End Products (AGEs) in Chronic Kidney Disease (CKD): Synthesis and Endogenous Defensive Strategies
3. AGEs in CKD Onset and Progression
4. AGEs as Biomarkers of Mortality in CKD
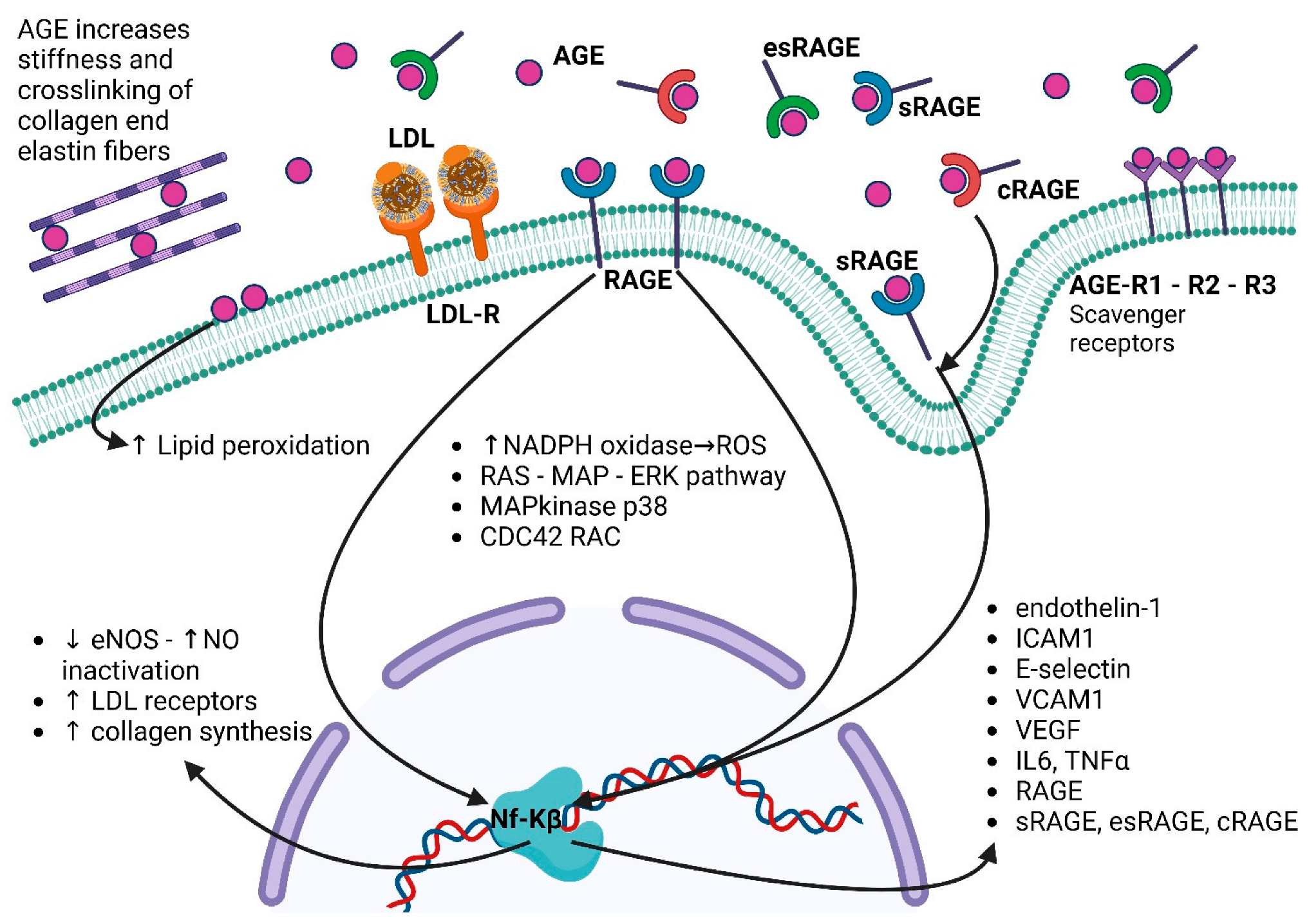
5. AGE–RAGE Pathway in Cardiovascular System: Role in the Etiopathogenesis of Diseases and as Biomarkers
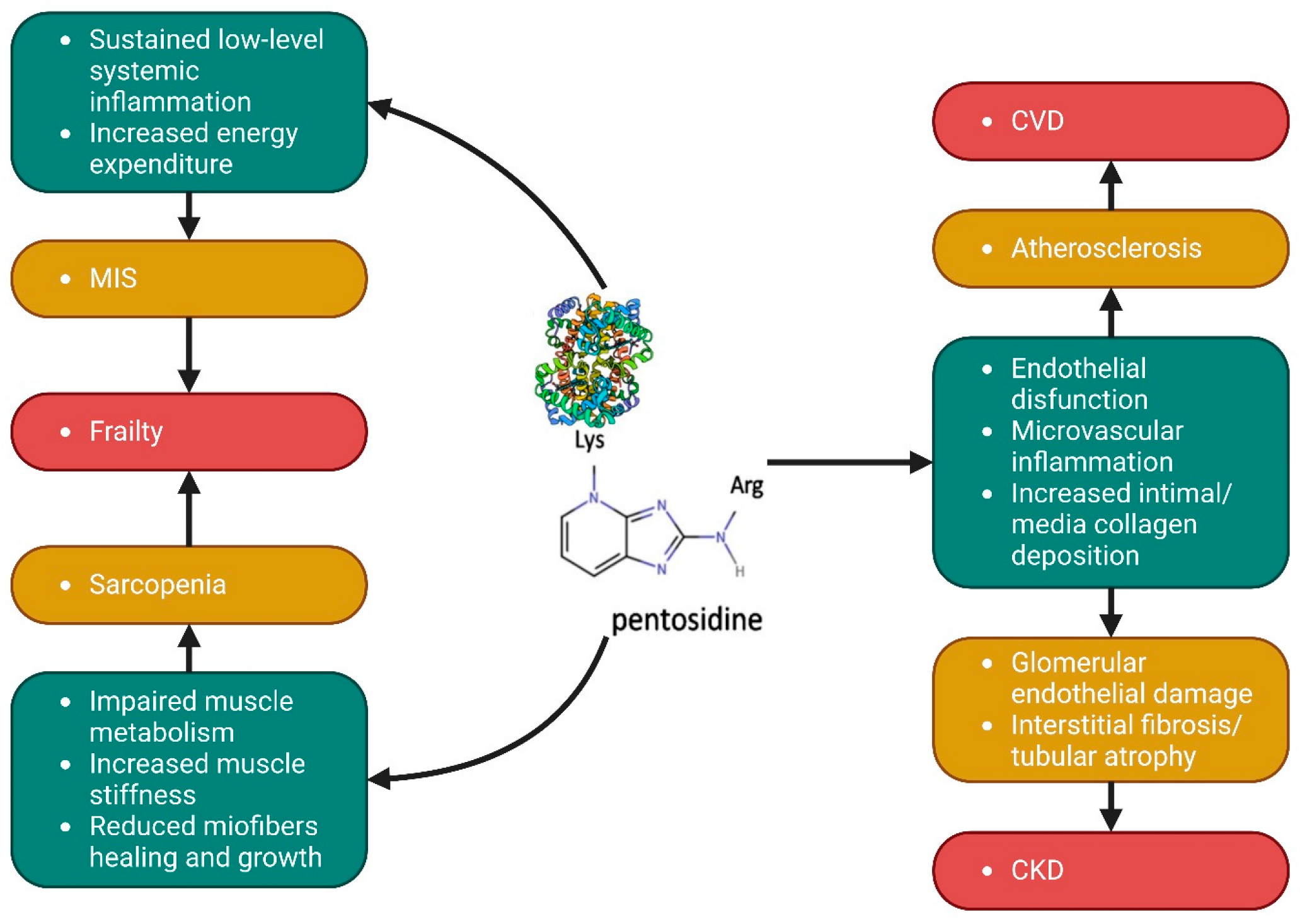
6. AGEs, Sarcopenia, and Nutritional Status: Pathogenic Role and Biomarker of Muscle Wasting
7. Potential Future Therapeutic Solutions
- AGE intake reduction: AGE levels can be lowered by reducing intake of food containing a high number of AGEs and encouraging consumption those containing the lowest amount of AGEs. AGE-rich foods include red meat, animal fat, cheese, sweetened food, and foods cooked at high temperatures in dry heat (frying, broiling, grilling, and roasting) [121]. This nutritional approach could be useful during the early stages of CKD, when patients are suggested which foods to consume and limit. In the advanced stage of CKD, patients are recommended to adhere more strictly to nutritional prescription. The reduced consumption of meat and animal-derived products is an ongoing practice in CKD patients that already limits AGE intake. Other AGE-lowering procedures include stopping cigarette smoking and exercise promotion [122,123].
- 2.
- Reduction of AGE synthesis: Several AGE-lowering compounds and AGE formation inhibitors have been developed and progressed to clinical trials; however, due to side effects or because companies have discontinued activities, these studies had to be retracted. AGE-lowering therapy can be classified into direct and indirect therapies. Names and mechanisms of action of molecules belonging to this group are presented in Table 1. Clinical trials of these anti-AGE drugs have been gradually developed. Despite some studies confirming some beneficial effects on AGE levels, nephropathy progression, endothelial function, inflammation, ventricular mass, and parameters of heart function, other reports produced conflicting results. A more detailed description of these studies and studies on other compounds can be found in the works by Sarmah S. at al. [127,128,129,130];
- 3.
- 4.
- Blockade of RAGE activation.: Azeliragon has been used as a RAGE blocker in patients with Alzheimer’s disease, but it has not been applied to other clinal diseases [133]. Increasing sRAGE levels have also been proposed as an additional therapy. sRAGE levels can be increased by exogenous administration or by upregulating sRAGE expression with drugs such as statins, ACE inhibitors, and rosiglitazone [134,135,136]. sRAGE administration has been studied in preclinical models of atherosclerosis but not in humans [137]. sRAGE levels are already high in CKD, but since the AGE/sRAGE ratio still favors AGEs, this approach must not be excluded;
- 5.
- AGE degradation in vivo: GLO-1, the endogenous enzyme that blocks AGE synthesis by promoting the degradation of dicarbonyl compounds, is not available for clinical use, but its activity was upregulated in a placebo–control crossover clinical trial by the combined use of trans-resveratrol found in grapes and hesperidin found in orange [138];
- 6.

{kind=link}
{kind=link}
{kind=link}
| Direct AGE-Lowering Compounds | ||
|---|---|---|
| Name | Actions | Clinical Use/Development Stage |
| Algebrium chloride [139,140,141] | A carbonyl scavenger that can also reduce cross links and ROS. | Clinical trials demonstrated beneficial effects in treating cardiovascular disorders, but the company seems to have discontinued operations. |
| Aminoguanidine [142,143] | A scavenger of carbonyl and dicarbonyl compounds and an ROS inhibitor. | Investigated for the treatment of diabetic nephropathy. Clinical trials were started, but commercial efforts to develop aminoguanidine as a drug were stopped by the company. |
| Benfotiamine [144] | A synthetic, fat-soluble S-acyl derivative of thiamine (vitamin B1) that reduces glycolysis and the polyol pathway, ROS generation, and activation of transketolase. | Approved in some countries as a medication/dietary supplement to treat diabetic sensorimotor polyneuropathy. |
| Carnosine [145] | Scavenger of ROS and alpha-beta unsaturated aldehydes created by peroxidation of fatty acid cell membranes during oxidative stress. Carnosine can oppose glycation, and it can chelate divalent metal ions. | Available as a dietary supplement. Clinical trials suggested a renoprotective effect in diabetic nephropathy. |
| OPB-9195 [146] | A carbonyl scavenger that can also reduce ROS levels. | Used in preclinical studies; however, no human data on this agent have been published. |
| Piridoxamine [147] | It reacts with carbonyl groups in Amadori products, thereby preventing AGE synthesis from these intermediates. It can also scavenge and reduce ROS production by chelating transition metals. | In experimental models, reduced AGE accumulation by pyridoxamine improved renal function. Beneficial reduction from baseline in serum creatinine was demonstrated by clinical trials in diabetes. |
| Thiamine [148] | It reduces glycolysis and the polyol pathway, ROS generation, and activation of transketolase. | Beneficial reduction in urinary albumin excretion was demonstrated by clinical trials in diabetes. |
| Direct AGE-Lowering Compounds | ||
| Name | Actions | Clinical Use/Development Stage |
| ACE inhibitors [149] | They inhibit ROS and, by reducing angiotensin II, exert anti-inflammatory effects. | Already used in clinical practice for hypertension. Ramipril reduced fluorescent AGEs, blood pressure, and proteinuria in diabetes. |
| Statins [150,151] | They stimulate RAGE shedding. | Already used in clinical practice for hypercholesterolemia. Simvastin reduced carotid plaque RAGE expression by decreasing AGE generation in diabetes. Atorvastatin reduced proteinuria and AGE levels in CKD. |
| AT1R antagonists [152,153] | They inhibit ROS and exert anti-inflammatory effects by reducing angiotensin II signaling. | Already used in clinical practice for hypertension. In diabetes and hypertension, a significant decrease in AGE levels was demonstrated with valsartan and candesartan, which also reduced albumin excretion. |
| Metformin [154,155] | It controls glucose homeostasis, reduces gluconeogenesis, and traps reactive carbonyl groups. | Already used in clinical practice for diabetes. Research on the anti-AGE effects of metformin mainly focuses on cellular experiments. |
| SGLT-2 inhibitors [156] | They control glucose homeostasis. | Already used in clinical practice for diabetes. Inhibition of oxidative, inflammatory, and fibrotic reactions in the kidney of diabetic rats, partly via suppression of the AGE–RAGE axis, was described. |
| sRAGE [157,158,159] | It blocks AGE from binding to RAGE, thereby working as a decoy receptor. | No human data on this agent have been published. Research on sRAGE effects mainly focuses on preclinical models. |
| Thiazolidinediones [160,161] | They control glucose homeostasis. | An increase in sRAGE levels was observed in clinical trials with pioglitazone and rosiglitazione in diabetes. |
8. Methods for AGE Quantification
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.-J.; Roman, G.; Yeboah, F.; Konishi, Y. The Road to Advanced Glycation End Products: A Mechanistic Perspective. Curr. Med. Chem. 2007, 14, 1653–1671. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Dietary AGEs and ALEs and Risk to Human Health by Their Interaction with the Receptor for Advanced Glycation Endproducts (RAGE)--an Introduction. Mol. Nutr. Food Res. 2007, 51, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard Reaction and AGE Breakers as Therapeutics for Multiple Diseases. Drug. Discov. Today 2006, 11, 646–654. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, Glyoxalase 1 and the Dicarbonyl Proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef]
- Busch, M.; Franke, S.; Rüster, C.; Wolf, G. Advanced Glycation End-Products and the Kidney. Eur. J. Clin. Investig. 2010, 40, 742–755. [Google Scholar] [CrossRef]
- Stitt, A.W.; Burke, G.A.; Chen, F.; McMullen, C.B.T.; Vlassara, H. Advanced Glycation End-Product Receptor Interactions on Microvascular Cells Occur within Caveolin-Rich Membrane Domains. FASEB J. 2000, 14, 2390–2392. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I--Structure, Function and a Critical Role in the Enzymatic Defence against Glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.I.; Zhu, L.; Vlassara, H. Advanced Glycation Endproduct (AGE) Receptor 1 Is a Negative Regulator of the Inflammatory Response to AGE in Mesangial Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [Green Version]
- Ribau, J.C.O.; Hadcock, S.J.; Teoh, K.; DeReske, M.; Richardson, M. Endothelial Adhesion Molecule Expression Is Enhanced in the Aorta and Internal Mammary Artery of Diabetic Patients. J. Res. 1999, 85, 225–233. [Google Scholar] [CrossRef]
- Identification of Galectin-3 as a High-Affinity Binding Protein for Advanced Glycation End Products (AGE): A New Member of the AGE-Receptor Complex. Available online: https://pubmed.ncbi.nlm.nih.gov/8529130/ (accessed on 23 January 2023).
- Iacobini, C.; Amadio, L.; Oddi, G.; Ricci, C.; Barsotti, P.; Missori, S.; Sorcini, M.; di Mario, U.; Pricci, F.; Pugliese, G. Role of Galectin-3 in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14, S264–S270. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; du Yan, S.; Yan, S.F.; Stern, D.M. The Multiligand Receptor RAGE as a Progression Factor Amplifying Immune and Inflammatory Responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A Soluble Form of the Receptor for Advanced Glycation Endproducts (RAGE) Is Produced by Proteolytic Cleavage of the Membrane-Bound Form by the Sheddase a Disintegrin and Metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Lee, A.C.H.; Lam, J.K.Y.; Shiu, S.W.M.; Wong, Y.; Betteridge, D.J.; Tan, K.C.B. Serum Level of Soluble Receptor for Advanced Glycation End Products Is Associated with A Disintegrin And Metalloproteinase 10 in Type 1 Diabetes. PLoS ONE 2015, 10, e0137330. [Google Scholar] [CrossRef] [Green Version]
- Jules, J.; Maiguel, D.; Hudson, B.I. Alternative Splicing of the RAGE Cytoplasmic Domain Regulates Cell Signaling and Function. PLoS ONE 2013, 8, e78267. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, H.B.; Luo, M.; Zheng, H. Serum Soluble RAGE Level Inversely Correlates with Left Ventricular Hypertrophy in Essential Hypertension Patients. Genet. Mol. Res. 2016, 15, gmr.15028414. [Google Scholar] [CrossRef]
- Momma, H.; Niu, K.; Kobayashi, Y.; Huang, C.; Chujo, M.; Otomo, A.; Tadaura, H.; Miyata, T.; Nagatomi, R. Lower Serum Endogenous Secretory Receptor for Advanced Glycation End Product Level as a Risk Factor of Metabolic Syndrome among Japanese Adult Men: A 2-Year Longitudinal Study. J. Clin. Endocrinol. Metab. 2014, 99, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Norata, G.D.; Garlaschelli, K.; Grigore, L.; Tibolla, G.; Raselli, S.; Redaelli, L.; Buccianti, G.; Catapano, A.L. Circulating Soluble Receptor for Advanced Glycation End Products Is Inversely Associated with Body Mass Index and Waist/Hip Ratio in the General Population. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 129–134. [Google Scholar] [CrossRef]
- Dozio, E.; Briganti, S.; Delnevo, A.; Vianello, E.; Ermetici, F.; Secchi, F.; Sardanelli, F.; Morricone, L.; Malavazos, A.E.; Corsi Romanelli, M.M. Relationship between Soluble Receptor for Advanced Glycation End Products (SRAGE), Body Composition and Fat Distribution in Healthy Women. Eur. J. Nutr. 2017, 56, 2557–2564. [Google Scholar] [CrossRef] [Green Version]
- Dozio, E.; Corradi, V.; Vianello, E.; Scalzotto, E.; de Cal, M.; Corsi Romanelli, M.M.; Ronco, C. Increased Levels of SRAGE in Diabetic CKD-G5D Patients: A Potential Protective Mechanism against AGE-Related Upregulation of Fibroblast Growth Factor 23 and Inflammation. Mediat. Inflamm. 2017, 2017, 9845175. [Google Scholar] [CrossRef] [Green Version]
- Sabbatinelli, J.; Castiglione, S.; Macrì, F.; Giuliani, A.; Ramini, D.; Vinci, M.C.; Tortato, E.; Bonfigli, A.R.; Olivieri, F.; Raucci, A. Circulating Levels of AGEs and Soluble RAGE Isoforms Are Associated with All-Cause Mortality and Development of Cardiovascular Complications in Type 2 Diabetes: A Retrospective Cohort Study. Cardiovasc. Diabetol. 2022, 21, 95. [Google Scholar] [CrossRef] [PubMed]
- Leurs, P.; Lindholm, B. The AGE-RAGE Pathway and Its Relation to Cardiovascular Disease in Patients with Chronic Kidney Disease. Arch. Med. Res. 2013, 44, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Pietromonaco, S.; Loo, A.K.C.; Farquhar, M.G. Complete Cloning and Sequencing of Rat Gp330/“megalin”, a Distinctive Member of the Low Density Lipoprotein Receptor Gene Family. Proc. Natl. Acad. Sci. USA 1994, 91, 9725–9729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Nagai, R.; Tanuma, A.; Hama, H.; Cho, K.; Takeda, T.; Yoshida, Y.; Toda, T.; Shimizu, F.; Horiuchi, S.; et al. Role of Megalin in Endocytosis of Advanced Glycation End Products: Implications for a Novel Protein Binding to Both Megalin and Advanced Glycation End Products. J. Am. Soc. Nephrol. 2003, 14, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Gugliucci, A.; Bendayan, M. Renal Fate of Circulating Advanced Glycated End Products (AGE): Evidence for Reabsorption and Catabolism of AGE-Peptides by Renal Proximal Tubular Cells. Diabetologia 1996, 39, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Christensen, E.I.; Birn, H. Megalin and Cubilin: Multifunctional Endocytic Receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 258–268. [Google Scholar] [CrossRef]
- Biemesderfer, D.; Nagy, T.; DeGray, B.; Aronson, P.S. Specific Association of Megalin and the Na+/H+ Exchanger Isoform NHE3 in the Proximal Tubule. J. Biol. Chem. 1999, 274, 17518–17524. [Google Scholar] [CrossRef] [Green Version]
- Hryciw, D.H.; Lee, E.M.; Pollock, C.A.; Poronnik, P. Molecular Changes in Proximal Tubule Function in Diabetes Mellitus. Clin. Exp. Pharmacol. Physiol. 2004, 31, 372–379. [Google Scholar] [CrossRef]
- De, S.; Kuwahara, S.; Saito, A. The Endocytic Receptor Megalin and Its Associated Proteins in Proximal Tubule Epithelial Cells. Membranes 2014, 4, 333–335. [Google Scholar] [CrossRef] [Green Version]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased Prevalence of Oxidant Stress and Inflammation in Patients with Moderate to Severe Chronic Kidney Disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating Glycotoxins and Dietary Advanced Glycation Endproducts: Two Links to Inflammatory Response, Oxidative Stress, and Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Genuth, S.; Sun, W.; Cleary, P.; Sell, D.R.; Dahms, W.; Malone, J.; Sivitz, W.; Monnier, V.M. Glycation and Carboxymethyllysine Levels in Skin Collagen Predict the Risk of Future 10-Year Progression of Diabetic Retinopathy and Nephropathy in the Diabetes Control and Complications Trial and Epidemiology of Diabetes Interventions and Complications Participants with Type 1 Diabetes. Diabetes 2005, 54, 3103–3111. [Google Scholar] [CrossRef] [Green Version]
- Miyata, T.; van Ypersele De Strihou, C.; Kurokawa, K.; Baynes, J.W. Alterations in Nonenzymatic Biochemistry in Uremia: Origin and Significance of “Carbonyl Stress” in Long-Term Uremic Complications. Kidney Int. 1999, 55, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Chen, J.; Hua, Y.; Zhang, Y.; Liu, Z. New Insights into the Role of Empagliflozin on Diabetic Renal Tubular Lipid Accumulation. Diabetol. Metab. Syndr. 2022, 14, 121. [Google Scholar] [CrossRef]
- Kaifu, K.; Ueda, S.; Nakamura, N.; Matsui, T.; Yamada-Obara, N.; Ando, R.; Kaida, Y.; Nakata, M.; Matsukuma-Toyonaga, M.; Higashimoto, Y.; et al. Advanced Glycation End Products Evoke Inflammatory Reactions in Proximal Tubular Cells via Autocrine Production of Dipeptidyl Peptidase-4. Microvasc. Res. 2018, 120, 90–93. [Google Scholar] [CrossRef]
- Haraguchi, R.; Kohara, Y.; Matsubayashi, K.; Kitazawa, R.; Kitazawa, S. New Insights into the Pathogenesis of Diabetic Nephropathy: Proximal Renal Tubules Are Primary Target of Oxidative Stress in Diabetic Kidney. Acta Histochem. Cytochem. 2020, 53, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Sun, T.; Li, H.; Qiu, S.; Li, Y.; Zhang, D. Proximal Tubular RAGE Mediated the Renal Fibrosis in UUO Model Mice via Upregulation of Autophagy. Cell Death Dis. 2022, 13. [Google Scholar] [CrossRef]
- Tan, R.J.; Li, Y.; Rush, B.M.; Cerqueira, D.M.; Zhou, D.; Fu, H.; Ho, J.; Stolz, D.B.; Liu, Y. Tubular Injury Triggers Podocyte Dysfunction by β-Catenin-Driven Release of MMP-7. JCI Insight 2019, 4, e122399. [Google Scholar] [CrossRef] [Green Version]
- Bucala, R.; Vlassara, H. Advanced Glycosylation End Products in Diabetic Renal and Vascular Disease. Am. J. Kidney Dis. 1995, 26, 875–888. [Google Scholar] [CrossRef]
- Shimoike, T.; Inoguchi, T.; Umeda, F.; Nawata, H.; Kawano, K.; Ochi, H. The Meaning of Serum Levels of Advanced Glycosylation End Products in Diabetic Nephropathy. Metabolism 2000, 49, 1030–1035. [Google Scholar] [CrossRef]
- Tezuka, Y.; Nakaya, I.; Nakayama, K.; Nakayama, M.; Yahata, M.; Soma, J. Methylglyoxal as a Prognostic Factor in Patients with Chronic Kidney Disease. Nephrology 2019, 24, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Calviño, J.; Cigarran, S.; Gonzalez-Tabares, L.; Menendez, N.; Latorre, J.; Cillero, S.; Millan, B.; Cobelo, C.; Sanjurjo-Amado, A.; Quispe, J.; et al. Advanced Glycation End Products (AGEs) Estimated by Skin Autofluorescence Are Related with Cardiovascular Risk in Renal Transplant. PLoS ONE 2018, 13, e0201118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced Glycation End Products Induce Glomerular Sclerosis and Albuminuria in Normal Rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.L.; Li, X.C. Proximal Nephron. Compr. Physiol 2013, 3, 1079–1123. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Sato, H.; Iino, N.; Takeda, T. Molecular Mechanisms of Receptor-Mediated Endocytosis in the Renal Proximal Tubular Epithelium. J. Biomed. Biotechnol. 2010, 2010, 403272. [Google Scholar] [CrossRef]
- Kuwahara, S.; Hosojima, M.; Kaneko, R.; Aoki, H.; Nakano, D.; Sasagawa, T.; Kabasawa, H.; Kaseda, R.; Yasukawa, R.; Ishikawa, T.; et al. Megalin-Mediated Tubuloglomerular Alterations in High-Fat Diet-Induced Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1996–2008. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Takeda, T.; Sato, K.; Hama, H.; Tanuma, A.; Kaseda, R.; Suzuki, Y.; Gejyo, F. Significance of Proximal Tubular Metabolism of Advanced Glycation End Products in Kidney Diseases. Ann. N. Y. Acad. Sci 2005, 1043, 637–643. [Google Scholar] [CrossRef]
- Vettoretti, S.; Caldiroli, L.; Armelloni, S.; Ferrari, C.; Cesari, M.; Messa, P. Sarcopenia Is Associated with Malnutrition but Not with Systemic Inflammation in Older Persons with Advanced CKD. Nutrients 2019, 11, 1378. [Google Scholar] [CrossRef] [Green Version]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and Premature Aging in Advanced Chronic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, E.; de Nicola, L.; Gabbai, F.B.; Chiodini, P.; Ravera, M.; Pieracci, L.; Marre, S.; Cassottana, P.; Lucà, S.; Vettoretti, S.; et al. Associations of Left Ventricular Hypertrophy and Geometry with Adverse Outcomes in Patients with CKD and Hypertension. Clin. J. Am. Soc. Nephrol. 2016, 11, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Hörner, D.V.; Taal, M.W. Skin Autofluorescence: An Emerging Biomarker in Persons with Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 507–512. [Google Scholar] [CrossRef]
- Cavero-Redondo, I.; Soriano-Cano, A.; Álvarez-Bueno, C.; Cunha, P.G.; Martínez-Hortelano, J.A.; Garrido-Miguel, M.; Berlanga-Macías, C.; Martínez-Vizcaíno, V. Skin Autofluorescence-Indicated Advanced Glycation End Products as Predictors of Cardiovascular and All-Cause Mortality in High-Risk Subjects: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7, e009833. [Google Scholar] [CrossRef] [Green Version]
- Gerrits, E.G.; Lutgers, H.L.; Smeets, G.H.W.; Groenier, K.H.; Smit, A.J.; Gans, R.O.B.; Bilo, H.J.G. Skin Autofluorescence: A Pronounced Marker of Mortality in Hemodialysis Patients. Nephron Extra 2012, 2, 184–191. [Google Scholar] [CrossRef]
- Nongnuch, A.; Davenport, A. Skin Autofluorescence Advanced Glycosylation End Products as an Independent Predictor of Mortality in High Flux Haemodialysis and Haemodialysis Patients. Nephrology 2015, 20, 862–867. [Google Scholar] [CrossRef]
- Mácsai, E.; Benke, A.; Kiss, I. Skin Autofluorescence and Mortality in Patients on Peritoneal Dialysis. Medicine 2015, 94, e1933. [Google Scholar] [CrossRef]
- Siriopol, D.; Hogas, S.; Veisa, G.; Mititiuc, I.; Volovat, C.; Apetrii, M.; Onofriescu, M.; Busila, I.; Oleniuc, M.; Covic, A. Tissue Advanced Glycation End Products (AGEs), Measured by Skin Autofluorescence, Predict Mortality in Peritoneal Dialysis. Int. Urol. Nephrol. 2015, 47, 563–569. [Google Scholar] [CrossRef]
- McIntyre, N.J.; Chesterton, L.J.; John, S.G.; Jefferies, H.J.; Burton, J.O.; Taal, M.W.; Fluck, R.J.; McIntyre, C.W. Tissue-Advanced Glycation End Product Concentration in Dialysis Patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Oleniuc, M.; Schiller, A.; Secara, I.; Onofriescu, M.; Hogas, S.; Apetrii, M.; Siriopol, D.; Covic, A. Evaluation of Advanced Glycation End Products Accumulation, Using Skin Autofluorescence, in CKD and Dialysis Patients. Int. Urol. Nephrol. 2012, 44, 1441–1449. [Google Scholar] [CrossRef]
- Mac-Way, F.; Couture, V.; Utescu, M.S.; Ignace, S.; de Serres, S.A.; Loignon, R.C.; Marquis, K.; Larivière, R.; Agharazii, M. Advanced Glycation End Products, Aortic Stiffness, and Wave Reflection in Peritoneal Dialysis as Compared to Hemodialysis. Int. Urol. Nephrol. 2014, 46, 817–824. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, P.; Chen, J.; Yu, X.; Xie, D.; Mei, C.; Xiong, F.; Shi, W.; Zhou, W.; Liu, X.; et al. Accumulation of Tissue Advanced Glycation End Products Correlated with Glucose Exposure Dose and Associated with Cardiovascular Morbidity in Patients on Peritoneal Dialysis. Atherosclerosis 2012, 224, 187–194. [Google Scholar] [CrossRef]
- Uflacker, R.; Paolini, R.M.; Nobrega, M. Ablation of Tumor and Inflammatory Tissue with Absolute Ethanol. Acta Radiol. Diagn. 1986, 27, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Svedberg, O.; Lindholm, B.; Dai, L.; Heimbürger, O.; Barany, P.; Anderstam, B.; Stenvinkel, P.; Qureshi, A.R. Skin Autofluorescence, Arterial Stiffness and Framingham Risk Score as Predictors of Clinical Outcome in Chronic Kidney Disease Patients: A Cohort Study. Nephrol. Dial. Transplant. 2019, 34, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.D.S.; Roderick, P.J.; McIntyre, N.J.; Harris, S.; McIntyre, C.W.; Fluck, R.J.; Taal, M.W. Skin Autofluorescence and All-Cause Mortality in Stage 3 CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 1361–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenbeke, M.; Speeckaert, R.; Desmedt, S.; Glorieux, G.; Delanghe, J.R.; Speeckaert, M.M. The Role of Advanced Glycation End Products and Its Soluble Receptor in Kidney Diseases. Int. J. Mol. Sci. 2022, 23, 3439. [Google Scholar] [CrossRef]
- Dozio, E.; Ambrogi, F.; de Cal, M.; Vianello, E.; Ronco, C.; Corsi Romanelli, M.M. Role of the Soluble Receptor for Advanced Glycation End Products (SRAGE) as a Prognostic Factor for Mortality in Hemodialysis and Peritoneal Dialysis Patients. Mediat. Inflamm. 2018, 2018, 1347432. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.S.; Chung, W.; Kim, A.J.; Ro, H.; Chang, J.H.; Lee, H.H.; Jung, J.Y. Associations between Soluble Receptor for Advanced Glycation End Products (SRAGE) and S100A12 (EN-RAGE) with Mortality in Long-Term Hemodialysis Patients. J. Korean Med. Sci. 2017, 32, 54–59. [Google Scholar] [CrossRef]
- Dozio, E.; Vettoretti, S.; Caldiroli, L.; Nerini-Molteni, S.; Tacchini, L.; Ambrogi, F.; Messa, P.; Corsi Romanelli, M.M. Advanced Glycation End Products (AGE) and Soluble Forms of AGE Receptor: Emerging Role as Mortality Risk Factors in CKD. Biomedicines 2020, 8, 638. [Google Scholar] [CrossRef]
- Dozio, E.; Corradi, V.; Proglio, M.; Vianello, E.; Menicanti, L.; Rigolini, R.; Caprara, C.; de Cal, M.; Corsi Romanelli, M.M.; Ronco, C. Usefulness of Glycated Albumin as a Biomarker for Glucose Control and Prognostic Factor in Chronic Kidney Disease Patients on Dialysis (CKD-G5D). Diabetes Res. Clin. Pract. 2018, 140, 9–17. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Johansen, K.L.; Chertow, G.M.; Foley, R.N.; Gilbertson, D.T.; Herzog, C.A.; Ishani, A.; Israni, A.K.; Ku, E.; Kurella Tamura, M.; Li, S.; et al. US Renal Data System 2020 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2021, 77, A7–A8. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney Disease as a Risk Factor for Development of Cardiovascular Disease: A Statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef]
- Yamagishi, S.; Sotokawauchi, A.; Matsui, T. Pathological Role of Advanced Glycation End Products (AGEs) and Their Receptor Axis in Atrial Fibrillation. Mini Rev. Med. Chem. 2019, 19, 1040–1048. [Google Scholar] [CrossRef]
- Baumann, M. Role of Advanced Glycation End Products in Hypertension and Cardiovascular Risk: Human Studies. J. Am. Soc. Hypertens. 2012, 6, 427–435. [Google Scholar] [CrossRef]
- Matsushita, K.; Kwak, L.; Sang, Y.; Ballew, S.H.; Skali, H.; Shah, A.M.; Coresh, J.; Solomon, S. Kidney Disease Measures and Left Ventricular Structure and Function: The Atherosclerosis Risk in Communities Study. J. Am. Heart Assoc. 2017, 6, e006259. [Google Scholar] [CrossRef]
- Park, M.; Hsu, C.Y.; Li, Y.; Mishra, R.K.; Keane, M.; Rosas, S.E.; Dries, D.; Xie, D.; Chen, J.; He, J.; et al. Associations between Kidney Function and Subclinical Cardiac Abnormalities in CKD. J. Am. Soc. Nephrol. 2012, 23, 1725–1734. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, S.; Hartog Jasper, W.L.; Heiner-Fokkema, M.R.; van Veldhuisen, D.J.; Voors, A.A. Advanced Glycation End-Products, a Pathophysiological Pathway in the Cardiorenal Syndrome. Heart Fail. Rev. 2012, 17, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 Induces Left Ventricular Hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, S.; Huber, J.; Wihler, C.; Rütten, H.; Busch, A.E.; Linz, W. Impaired Left Ventricular Relaxation in Type 2 Diabetic Rats Is Related to Myocardial Accumulation of N(Epsilon)-(Carboxymethyl) Lysine. Eur. J. Heart Fail. 2006, 8, 2–6. [Google Scholar] [CrossRef]
- Petrova, R.; Yamamoto, Y.; Muraki, K.; Yonekura, H.; Sakurai, S.; Watanabe, T.; Li, H.; Takeuchi, M.; Makita, Z.; Kato, I.; et al. Advanced Glycation Endproduct-Induced Calcium Handling Impairment in Mouse Cardiac Myocytes. J. Mol. Cell. Cardiol. 2002, 34, 1425–1431. [Google Scholar] [CrossRef]
- Koyama, Y.; Takeishi, Y.; Arimoto, T.; Niizeki, T.; Shishido, T.; Takahashi, H.; Nozaki, N.; Hirono, O.; Tsunoda, Y.; Nitobe, J.; et al. High Serum Level of Pentosidine, an Advanced Glycation End Product (AGE), Is a Risk Factor of Patients with Heart Failure. J. Card. Fail. 2007, 13, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartog, J.W.L.; Voors, A.A.; Schalkwijk, C.G.; Scheijen, J.; Smilde, T.D.J.; Damman, K.; Bakker, S.J.L.; Smit, A.J.; van Veldhuisen, D.J. Clinical and Prognostic Value of Advanced Glycation End-Products in Chronic Heart Failure. Eur. Heart J. 2007, 28, 2879–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshi, B.; Chen, J.; Ikram, M.A.; Zillikens, M.C.; Kavousi, M. Advanced Glycation End-Products, Cardiac Function and Heart Failure in the General Population: The Rotterdam Study. Diabetologia 2022, 66, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Pentosidine, Carotid Atherosclerosis and Alterations in Left Ventricular Geometry in Hemodialysis. Available online: https://pubmed.ncbi.nlm.nih.gov/11506253/ (accessed on 23 January 2023).
- Lazo, M.; Halushka, M.K.; Shen, L.; Maruthur, N.; Rebholz, C.M.; Rawlings, A.M.; Hoogeveen, R.C.; Brinkley, T.E.; Ballantyne, C.M.; Astor, B.C.; et al. Soluble Receptor for Advanced Glycation End Products and the Risk for Incident Heart Failure: The Atherosclerosis Risk in Communities Study. Am. Heart J. 2015, 170, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Takeishi, Y.; Niizeki, T.; Suzuki, S.; Kitahara, T.; Sasaki, T.; Kubota, I. Soluble Receptor for Advanced Glycation End Products (RAGE) Is a Prognostic Factor for Heart Failure. J. Card. Fail. 2008, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Raposeiras-Roubín, S.; Rodiño-Janeiro, B.K.; Grigorian-Shamagian, L.; Moure-González, M.; Seoane-Blanco, A.; Varela-Román, A.; Almenar-Bonet, L.; Álvarez, E.; González-Juanatey, J.R. Relation of Soluble Receptor for Advanced Glycation End Products to Predict Mortality in Patients with Chronic Heart Failure Independently of Seattle Heart Failure Score. Am. J. Cardiol. 2011, 107, 938–944. [Google Scholar] [CrossRef]
- Willemsen, S.; Hartog, J.W.L.; van Veldhuisen, D.J.; van der Meer, P.; Roze, J.F.; Jaarsma, T.; Schalkwijk, C.; van der Horst, I.C.C.; Hillege, H.L.; Voors, A.A. The Role of Advanced Glycation End-Products and Their Receptor on Outcome in Heart Failure Patients with Preserved and Reduced Ejection Fraction. Am. Heart J. 2012, 164, 742–749.e3. [Google Scholar] [CrossRef]
- Leonardis, D.; Basta, G.; Mallamaci, F.; Cutrupi, S.; Pizzini, P.; Tripepi, R.; Tripepi, G.; de Caterina, R.; Zoccali, C. Circulating Soluble Receptor for Advanced Glycation End Product (SRAGE) and Left Ventricular Hypertrophy in Patients with Chronic Kidney Disease (CKD). Nutr. Metab. Cardiovasc. Dis. 2012, 22, 748–755. [Google Scholar] [CrossRef]
- Zanoli, L.; Empana, J.P.; Perier, M.C.; Alivon, M.; Ketthab, H.; Castellino, P.; Laude, D.; Thomas, F.; Pannier, B.; Laurent, S.; et al. Increased Carotid Stiffness and Remodelling at Early Stages of Chronic Kidney Disease. J. Hypertens. 2019, 37, 1176–1182. [Google Scholar] [CrossRef]
- Briet, M.; Bozec, E.; Laurent, S.; Fassot, C.; London, G.M.; Jacquot, C.; Froissart, M.; Houillier, P.; Boutouyrie, P. Arterial Stiffness and Enlargement in Mild-to-Moderate Chronic Kidney Disease. Kidney Int. 2006, 69, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Townsend, R.R. Arterial Stiffness in CKD: A Review. Am. J. Kidney Dis. 2019, 73, 240–247. [Google Scholar] [CrossRef]
- de Faria Fonseca, L.; Araújo, A.B.; da Silva Quadros, K.R.; Carbonara, C.E.M.; Dertkigil, S.S.J.; Sposito, A.C.; de Oliveira, R.B. AGEs Accumulation Is Related to Muscle Degeneration and Vascular Calcification in Peritoneal Dialysis Patients. J. Bras. Nefrol. 2021, 43, 191–199. [Google Scholar] [CrossRef]
- Wang, A.Y.M.; Wong, C.K.; Yau, Y.Y.; Wong, S.; Chan, I.H.S.; Lam, C.W.K. Skin Autofluorescence Associates with Vascular Calcification in Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1784–1790. [Google Scholar] [CrossRef] [Green Version]
- Bucala, R.; Makita, Z.; Vega, G.; Grundy, S.; Koschinsky, T.; Cerami, A.; Vlassara, H. Modification of Low Density Lipoprotein by Advanced Glycation End Products Contributes to the Dyslipidemia of Diabetes and Renal Insufficiency. Proc. Natl. Acad. Sci. USA 1994, 91, 9441–9445. [Google Scholar] [CrossRef] [Green Version]
- Makita, Z.; Yanagisawa, K.; Kuwajima, S.; Bucala, R.; Vlassara, H.; Koike, T. The Role of Advanced Glycosylation End-Products in the Pathogenesis of Atherosclerosis. Nephrol. Dial. Transplant. 1996, 11 (Suppl. S5), 31–33. [Google Scholar] [CrossRef] [Green Version]
- Immunohistochemical Localization of Advanced Glycosylation End Products in Coronary Atheroma and Cardiac Tissue in Diabetes Mellitus. Available online: https://pubmed.ncbi.nlm.nih.gov/8256853/ (accessed on 23 January 2023).
- Sánchez, E.; Betriu, À.; Arroyo, D.; López, C.; Hernández, M.; Rius, F.; Fernández, E.; Lecube, A. Skin Autofluorescence and Subclinical Atherosclerosis in Mild to Moderate Chronic Kidney Disease: A Case-Control Study. PLoS ONE 2017, 12, e0170778. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Toyoguchi, T.; Inage, K.; Fujimoto, K.; Orita, S.; Suzuki, M.; Kanamoto, H.; Abe, K.; Norimoto, M.; Umimura, T.; et al. Advanced Glycation End Products Are Associated with Sarcopenia in Older Women: Aging Marker Dynamics. J. Women Aging 2021, 33, 328–340. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE Prevents Muscle Wasting and Prolongs Survival in Cancer Cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef] [Green Version]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.-i.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of Advanced Glycation End Products with Sarcopenia and Frailty in Chronic Kidney Disease. Sci. Rep. 2020, 10, 17647. [Google Scholar] [CrossRef]
- Diesel, W.; Knight, B.K.; Noakes, T.D.; Swanepoel, C.R.; van Zyl Smit, R.; Kaschula, R.O.C.; Sinclair-Smith, C.C. Morphologic Features of the Myopathy Associated with Chronic Renal Failure. Am. J. Kidney Dis. 1993, 22, 677–684. [Google Scholar] [CrossRef]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Dal Bello, F.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-Derived Advanced Glycation End-Products Drive Lipogenesis and Skeletal Muscle Reprogramming via SREBP-1c Dysregulation in Mice. Free Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Yang, R.S.; Sheu, M.L.; Chan, D.C.; Yang, T.H.; Tsai, K.S.; Chiang, C.K.; Liu, S.H. Advanced Glycation End-Products Induce Skeletal Muscle Atrophy and Dysfunction in Diabetic Mice via a RAGE-Mediated, AMPK-down-Regulated, Akt Pathway. J. Pathol. 2016, 238, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.M.; Essex, A.L.; Valdez, S.; Deosthale, P.J.; Aref, M.W.; Allen, M.R.; Bonetto, A.; Plotkin, L.I. Short-Term Pharmacologic RAGE Inhibition Differentially Affects Bone and Skeletal Muscle in Middle-Aged Mice. Bone 2019, 124, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Vettoretti, S.; Lungarella, G.; Messa, P.; Romanelli, M.M.C. Sarcopenia in Chronic Kidney Disease: Focus on Advanced Glycation End Products as Mediators and Markers of Oxidative Stress. Biomedicines 2021, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kubo, A.; Sugioka, Y.; Mitsui, R.; Fukuhara, N.; Nihei, F.; Takeda, Y. Relationship between Advanced Glycation End-Product Accumulation and Low Skeletal Muscle Mass in Japanese Men and Women. Geriatr. Gerontol. Int. 2017, 17, 785–790. [Google Scholar] [CrossRef]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Relationship of an Advanced Glycation End Product, Plasma Carboxymethyl-Lysine, with Slow Walking Speed in Older Adults: The InCHIANTI Study. Eur. J. Appl. Physiol. 2010, 108, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Proud, C.G. Regulation of Protein Synthesis by Insulin. Biochem. Soc. Trans. 2006, 34, 213–216. [Google Scholar] [CrossRef]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin Resistance and Type 2 Diabetes in High-Fat-Fed Mice Are Linked to High Glycotoxin Intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Heimbürger, O.; Lindholm, B.; Kaysen, G.A.; Bergström, J. Are There Two Types of Malnutrition in Chronic Renal Failure? Evidence for Relationships between Malnutrition, Inflammation and Atherosclerosis (MIA Syndrome). Nephrol. Dial. Transplant. 2000, 15, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Carrero, J.J.; Stenvinkel, P.; Cuppari, L.; Ikizler, T.A.; Kalantar-Zadeh, K.; Kaysen, G.; Mitch, W.E.; Price, S.R.; Wanner, C.; Wang, A.Y.M.; et al. Etiology of the Protein-Energy Wasting Syndrome in Chronic Kidney Disease: A Consensus Statement from the International Society of Renal Nutrition and Metabolism (ISRNM). J. Ren. Nutr. 2013, 23, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Semba, R.D.; Arab, L.; Sun, K.; Nicklett, E.J.; Ferrucci, L. Fat Mass Is Inversely Associated with Serum Carboxymethyl-Lysine, an Advanced Glycation End Product, in Adults. J. Nutr. 2011, 141, 1726–1730. [Google Scholar] [CrossRef] [Green Version]
- Suliman, M.E.; Heimbürger, O.; Bárány, P.; Anderstam, B.; Pecoits-Filho, R.; Ayala, E.R.; Qureshi, A.R.; Fehrman-Ekholm, I.; Lindholm, B.; Stenvinkel, P. Plasma Pentosidine Is Associated with Inflammation and Malnutrition in End-Stage Renal Disease Patients Starting on Dialysis Therapy. J. Am. Soc. Nephrol. 2003, 14, 1614–1622. [Google Scholar] [CrossRef] [Green Version]
- Nediani, C.; Dinu, M. Oxidative Stress and Inflammation as Targets for Novel Preventive and Therapeutic Approaches in Non-Communicable Diseases II. Antioxidants 2022, 11, 824. [Google Scholar] [CrossRef]
- Flores, E.A.; Bistrian, B.R.; Pomposelli, J.J.; Dinarello, C.A.; Blackburn, G.L.; Istfan, N.W. Infusion of Tumor Necrosis Factor/Cachectin Promotes Muscle Catabolism in the Rat. A Synergistic Effect with Interleukin 1. J. Clin. Investig. 1989, 83, 1614–1622. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Ikizler, T.A.; Block, G.; Avram, M.M.; Kopple, J.D. Malnutrition-Inflammation Complex Syndrome in Dialysis Patients: Causes and Consequences. Am. J. Kidney Dis. 2003, 42, 864–881. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Kushner, I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Caldiroli, L.; Molinari, P.; Dozio, E.; Rigolini, R.; Giubbilini, P.; Romanelli, M.M.C.; Castellano, G.; Vettoretti, S. In Patients with Chronic Kidney Disease Advanced Glycation End-Products Receptors Isoforms (SRAGE and EsRAGE) Are Associated with Malnutrition. Antioxidants 2022, 11, 1253. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [Green Version]
- Dual Responses to Increase Validity in Case-Control Studies. Available online: https://pubmed.ncbi.nlm.nih.gov/2181566/ (accessed on 24 January 2023).
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of Advanced Glycation End Products and Its Receptors in the Pathogenesis of Cigarette Smoke-Induced Cardiovascular Disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Fotheringham, A.K.; Gallo, L.A.; Borg, D.J.; Forbes, J.M. Advanced Glycation End Products (AGEs) and Chronic Kidney Disease: Does the Modern Diet AGE the Kidney? Nutrients 2022, 14, 2675. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced Glycation End Products Dietary Restriction Effects on Bacterial Gut Microbiota in Peritoneal Dialysis Patients; a Randomized Open Label Controlled Trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margiotta, E.; Caldiroli, L.; Callegari, M.L.; Miragoli, F.; Zanoni, F.; Armelloni, S.; Rizzo, V.; Messa, P.; Vettoretti, S. Association of Sarcopenia and Gut Microbiota Composition in Older Patients with Advanced Chronic Kidney Disease, Investigation of the Interactions with Uremic Toxins, Inflammation and Oxidative Stress. Toxins 2021, 13, 472. [Google Scholar] [CrossRef] [PubMed]
- Nenna, A.; Nappi, F.; Avtaar Singh, S.; Sutherland, F.; di Domenico, F.; Chello, M.; Spadaccio, C. Pharmacologic Approaches Against Advanced Glycation End Products (AGEs) in Diabetic Cardiovascular Disease. Res. Cardiovasc. Med. 2015, 4, 5. [Google Scholar] [CrossRef]
- Sarmah, S.; Roy, A.S. A Review on Prevention of Glycation of Proteins: Potential Therapeutic Substances to Mitigate the Severity of Diabetes Complications. Int. J. Biol. Macromol. 2022, 195, 565–588. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.C.; Watson, A.; Jandeleit-Dahm, K. Inhibitors of Advanced Glycation End Product (AGE) Formation and Accumulation. Handb. Exp. Pharmacol. 2021, 264, 395–423. [Google Scholar] [CrossRef]
- Ojima, A.; Matsui, T.; Nishino, Y.; Nakamura, N.; Yamagishi, S. Empagliflozin, an Inhibitor of Sodium-Glucose Cotransporter 2 Exerts Anti-Inflammatory and Antifibrotic Effects on Experimental Diabetic Nephropathy Partly by Suppressing AGEs-Receptor Axis. Horm. Metab. Res. 2015, 47, 686–692. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.; Bhanumathy, K.K. AGE-RAGE Axis in the Pathophysiology of Chronic Lower Limb Ischemia and a Novel Strategy for Its Treatment. Int. J. Angiol. 2020, 29, 156–167. [Google Scholar] [CrossRef]
- Prasad, K. Pathophysiology and Medical Treatment of Carotid Artery Stenosis. Int. J. Angiol. 2015, 24, 158–172. [Google Scholar] [CrossRef] [Green Version]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimer Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef]
- Tam, H.L.; Shiu, S.W.M.; Wong, Y.; Chow, W.S.; Betteridge, D.J.; Tan, K.C.B. Effects of Atorvastatin on Serum Soluble Receptors for Advanced Glycation End-Products in Type 2 Diabetes. Atherosclerosis 2010, 209, 173–177. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorpe, S.R.; Thallas-Bonke, V.; Pete, J.; Thomas, M.C.; Deemer, E.R.; Bassal, S.; El-Osta, A.; Long, D.M.; Panagiotopoulos, S.; et al. Modulation of Soluble Receptor for Advanced Glycation End Products by Angiotensin-Converting Enzyme-1 Inhibition in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2363–2372. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.C.B.; Chow, W.S.; Tso, A.W.K.; Xu, A.; Tse, H.F.; Hoo, R.L.C.; Betteridge, D.J.; Lam, K.S.L. Thiazolidinedione Increases Serum Soluble Receptor for Advanced Glycation End-Products in Type 2 Diabetes. Diabetologia 2007, 50, 1819–1825. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, T.; Yan, S.F.; du Yan, S.; Belov, D.; Rong, L.L.; Sousa, M.; Andrassy, M.; Marso, S.P.; Duda, S.; Arnold, B.; et al. Central Role of RAGE-Dependent Neointimal Expansion in Arterial Restenosis. J. Clin. Investig. 2003, 111, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- CERF, O.; METRO, F. Tailing of Survival Curves of Bacillus Licheniformis Spores Treated with Hydrogen Peroxide. J. Appl. Bacteriol. 1977, 42, 405–415. [Google Scholar] [CrossRef]
- Little, W.C.; Zile, M.R.; Kitzman, D.W.; Hundley, W.G.; O’Brien, T.X.; Degroof, R.C. The Effect of Alagebrium Chloride (ALT-711), a Novel Glucose Cross-Link Breaker, in the Treatment of Elderly Patients with Diastolic Heart Failure. J. Card. Fail. 2005, 11, 191–195. [Google Scholar] [CrossRef]
- Fujimoto, N.; Hastings, J.L.; Carrick-Ranson, G.; Shafer, K.M.; Shibata, S.; Bhella, P.S.; Abdullah, S.M.; Barkley, K.W.; Adams-Huet, B.; Boyd, K.N.; et al. Cardiovascular Effects of 1 Year of Alagebrium and Endurance Exercise Training in Healthy Older Individuals. Circ. Heart Fail. 2013, 6, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized Trial of an Inhibitor of Formation of Advanced Glycation End Products in Diabetic Nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef]
- Freedman, B.I.; Wuerth, J.P.; Cartwright, K.; Bain, R.P.; Dippe, S.; Hershon, K.; Mooradian, A.D.; Spinowitz, B.S. Design and Baseline Characteristics for the Aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control. Clin. Trials 1999, 20, 493–510. [Google Scholar] [CrossRef]
- Stirban, A.; Negrean, M.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Götting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Benfotiamine Prevents Macro- and Microvascular Endothelial Dysfunction and Oxidative Stress Following a Meal Rich in Advanced Glycation End Products in Individuals with Type 2 Diabetes. Diabetes Care 2006, 29, 2064–2071. [Google Scholar] [CrossRef] [Green Version]
- Elbarbary, N.S.; Ismail, E.A.R.; El-Naggar, A.R.; Hamouda, M.H.; El-Hamamsy, M. The Effect of 12 Weeks Carnosine Supplementation on Renal Functional Integrity and Oxidative Stress in Pediatric Patients with Diabetic Nephropathy: A Randomized Placebo-Controlled Trial. Pediatr. Diabetes 2018, 19, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Inagaki, Y.; Okamoto, T.; Amano, S.; Koga, K.; Takeuchi, M. Advanced Glycation End Products Inhibit de Novo Protein Synthesis and Induce TGF-Beta Overexpression in Proximal Tubular Cells. Kidney Int. 2003, 63, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of Pyridoxamine in Combined Phase 2 Studies of Patients with Type 1 and Type 2 Diabetes and Overt Nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Alam, S.S.; Riaz, S.; Larkin, J.R.; Akhtar, M.W.; Shafi, T.; Thornalley, P.J. High-Dose Thiamine Therapy for Patients with Type 2 Diabetes and Microalbuminuria: A Randomised, Double-Blind Placebo-Controlled Pilot Study. Diabetologia 2009, 52, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šebeková, K.; Gazdíková, K.; Syrová, D.; Blažíček, P.; Schinzel, R.; Heidland, A.; Spustová, V.; Dzúrik, R. Effects of Ramipril in Nondiabetic Nephropathy: Improved Parameters of Oxidatives Stress and Potential Modulation of Advanced Glycation End Products. J. Hum. Hypertens. 2003, 17, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Cuccurullo, C.; Iezzi, A.; Fazia, M.L.; de Cesare, D.; di Francesco, A.; Muraro, R.; Bei, R.; Ucchino, S.; Spigonardo, F.; Chiarelli, F.; et al. Suppression of RAGE as a Basis of Simvastatin-Dependent Plaque Stabilization in Type 2 Diabetes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2716–2723. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sato, E.; Fujiwara, N.; Kawagoe, Y.; Takeuchi, M.; Maeda, S.; Yamagishi, S.I. Atorvastatin Reduces Proteinuria in Non-Diabetic Chronic Kidney Disease Patients Partly via Lowering Serum Levels of Advanced Glycation End Products (AGEs). Oxid. Med. Cell. Longev. 2010, 3, 304–307. [Google Scholar] [CrossRef] [Green Version]
- Komiya, N.; Hirose, H.; Saisho, Y.; Saito, I.; Itoh, H. Effects of 12-Month Valsartan Therapy on Glycation and Oxidative Stress Markers in Type 2 Diabetic Subjects with Hypertension. Int. Heart J. 2008, 49, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Mizuno, K.; Takahashi, M.; Miura, Y.; Watanabe, T. Suppression of Advanced Glycation and Lipoxidation End Products by Angiotensin II Type-1 Receptor Blocker Candesartan in Type 2 Diabetic Patients with Essential Hypertension. Fukushima J. Med. Sci. 2013, 59, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Metformin Inhibits Advanced Glycation End Products (AGEs)-Induced Renal Tubular Cell Injury by Suppressing Reactive Oxygen Species Generation via Reducing Receptor for AGEs (RAGE) Expression. Horm. Metab. Res. 2012, 44, 891–895. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, Y.; Jin, X.; Chen, C.; Lu, Y.; Liu, L.; Shen, C. Metformin Inhibits Advanced Glycation End Products-Induced Inflammatory Response in Murine Macrophages Partly through AMPK Activation and RAGE/NF κ B Pathway Suppression. J. Diabetes Res. 2016, 2016, 4847812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afsar, B.; Afsar, R.E. Sodium-Glucose Cotransporter Inhibitors and Kidney Fibrosis: Review of the Current Evidence and Related Mechanisms. Pharmacol. Rep. 2023, 75, 44–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Chang, J.; Liu, J.; Liu, Y.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble RAGE Attenuates Myocardial I/R Injuries via FoxO3-Bnip3 Pathway. Cell. Mol. Life Sci. 2022, 79, 269. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Cho, S.; Seo, M.; Lee, B.S.; Jang, Y.; Lim, S.; Park, S. Soluble RAGE Attenuates Ang II-Induced Arterial Calcification via Inhibiting AT1R-HMGB1-RAGE Axis. Atherosclerosis 2022, 346, 53–62. [Google Scholar] [CrossRef]
- Scavello, F.; Zeni, F.; Milano, G.; Macrì, F.; Castiglione, S.; Zuccolo, E.; Scopece, A.; Pezone, G.; Tedesco, C.C.; Nigro, P.; et al. Soluble Receptor for Advanced Glycation End-Products Regulates Age-Associated Cardiac Fibrosis. Int. J. Biol. Sci. 2021, 17, 2399–2416. [Google Scholar] [CrossRef]
- Oz Gul, O.; Tuncel, E.; Yilmaz, Y.; Ulukaya, E.; Gul, C.B.; Kiyici, S.; Oral, A.Y.; Guclu, M.; Ersoy, C.; Imamoglu, S. Comparative Effects of Pioglitazone and Rosiglitazone on Plasma Levels of Soluble Receptor for Advanced Glycation End Products in Type 2 Diabetes Mellitus Patients. Metabolism 2010, 59, 64–69. [Google Scholar] [CrossRef]
- Gada, E.; Owens, A.W.; Gore, M.O.; See, R.; Abdullah, S.M.; Ayers, C.R.; Rohatgi, A.; Khera, A.; de Lemos, J.A.; McGuire, D.K. Discordant Effects of Rosiglitazone on Novel Inflammatory Biomarkers. Am. Heart J. 2013, 165, 609–614. [Google Scholar] [CrossRef]
- Poojary, M.M.; Zhang, W.; Greco, I.; de Gobba, C.; Olsen, K.; Lund, M.N. Liquid Chromatography Quadrupole-Orbitrap Mass Spectrometry for the Simultaneous Analysis of Advanced Glycation End Products and Protein-Derived Cross-Links in Food and Biological Matrices. J. Chromatogr. A 2020, 1615, 460767. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dozio, E.; Caldiroli, L.; Molinari, P.; Castellano, G.; Delfrate, N.W.; Romanelli, M.M.C.; Vettoretti, S. Accelerated AGEing: The Impact of Advanced Glycation End Products on the Prognosis of Chronic Kidney Disease. Antioxidants 2023, 12, 584. https://doi.org/10.3390/antiox12030584
Dozio E, Caldiroli L, Molinari P, Castellano G, Delfrate NW, Romanelli MMC, Vettoretti S. Accelerated AGEing: The Impact of Advanced Glycation End Products on the Prognosis of Chronic Kidney Disease. Antioxidants. 2023; 12(3):584. https://doi.org/10.3390/antiox12030584
Chicago/Turabian StyleDozio, Elena, Lara Caldiroli, Paolo Molinari, Giuseppe Castellano, Nicholas Walter Delfrate, Massimiliano Marco Corsi Romanelli, and Simone Vettoretti. 2023. "Accelerated AGEing: The Impact of Advanced Glycation End Products on the Prognosis of Chronic Kidney Disease" Antioxidants 12, no. 3: 584. https://doi.org/10.3390/antiox12030584