
Fatal Epileptic Seizures in Mice Having Compromised Glutathione and Ascorbic Acid Biosynthesis

Abstract
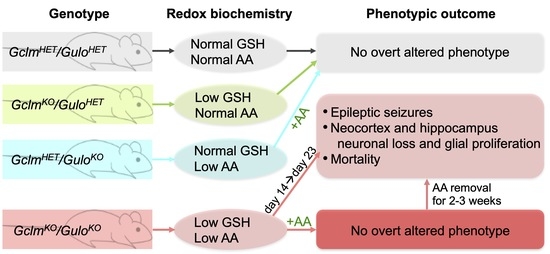
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals
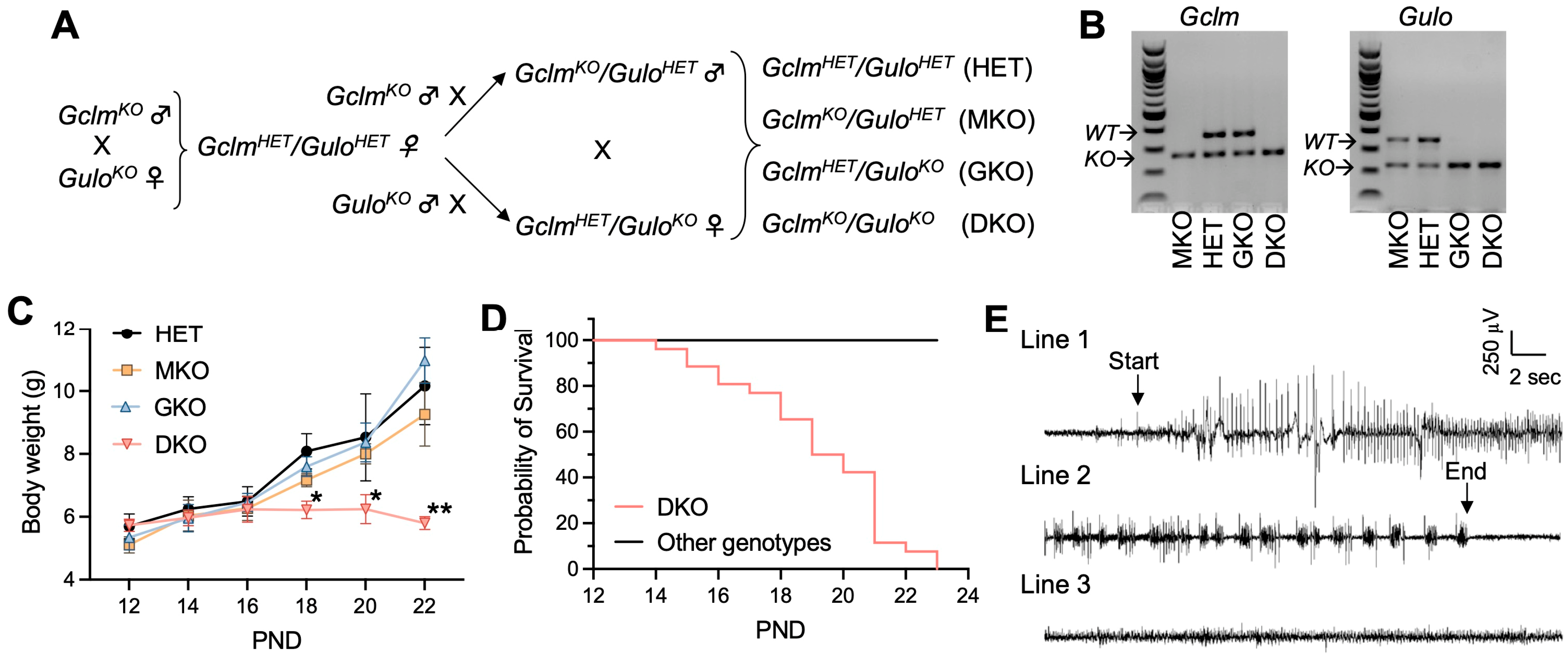
2.3. Generation of GclmKO/GuloKO DKO Mice and Ascorbic Acid Supplementation
2.4. Electroencephalography (EEG) Recording
2.5. Histological and Immunohistochemical Analyses
2.6. Biochemical Measurements of Redox Molecules
2.7. Measurements of Hematocrit and Plasma Glucose and Lipid Profile
2.8. Statistics
3. Results
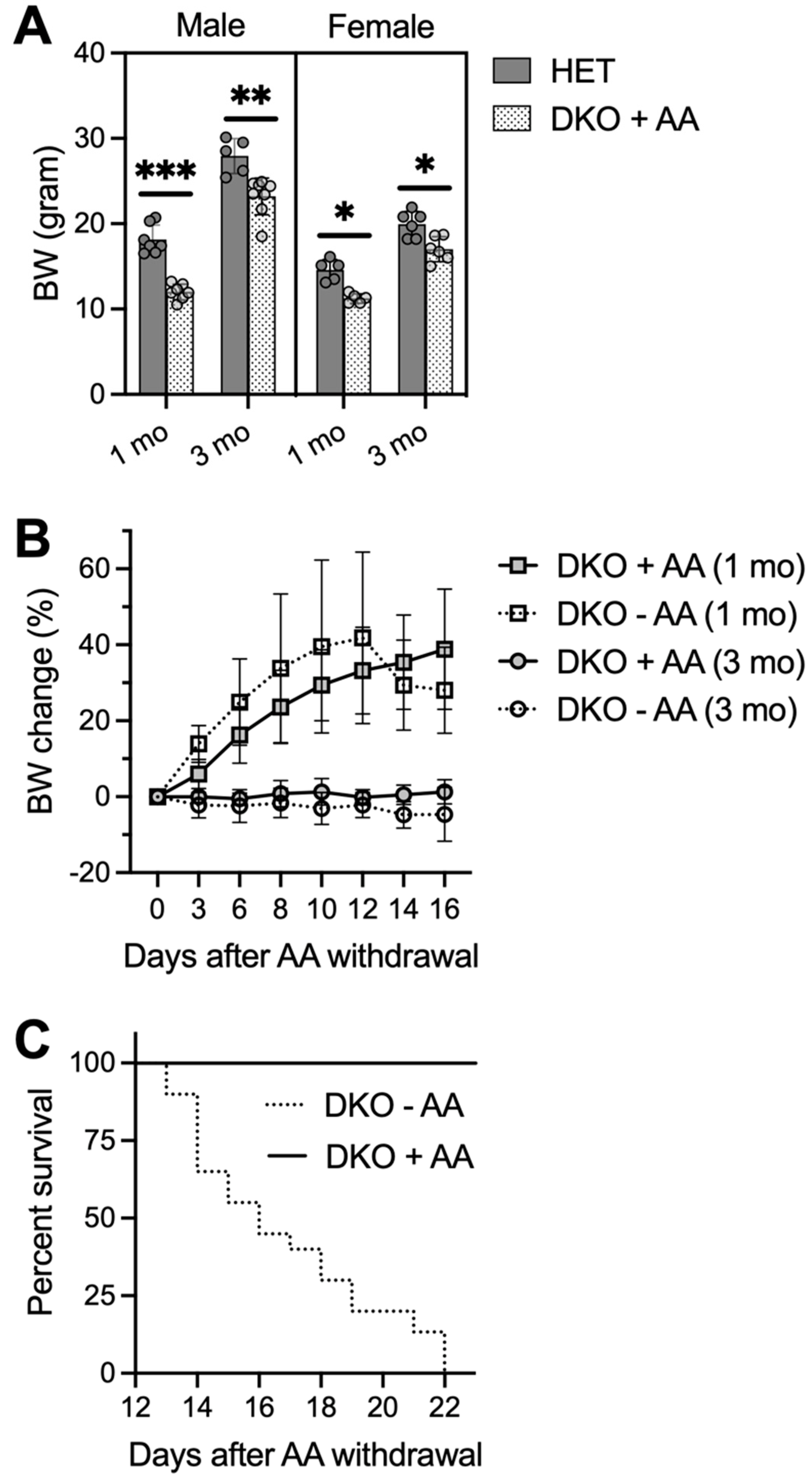
3.1. Concomitant Deficiency in GSH and AA Biosynthesis Leads to Growth Retardation, Spontaneous Seizures, and Premature Death
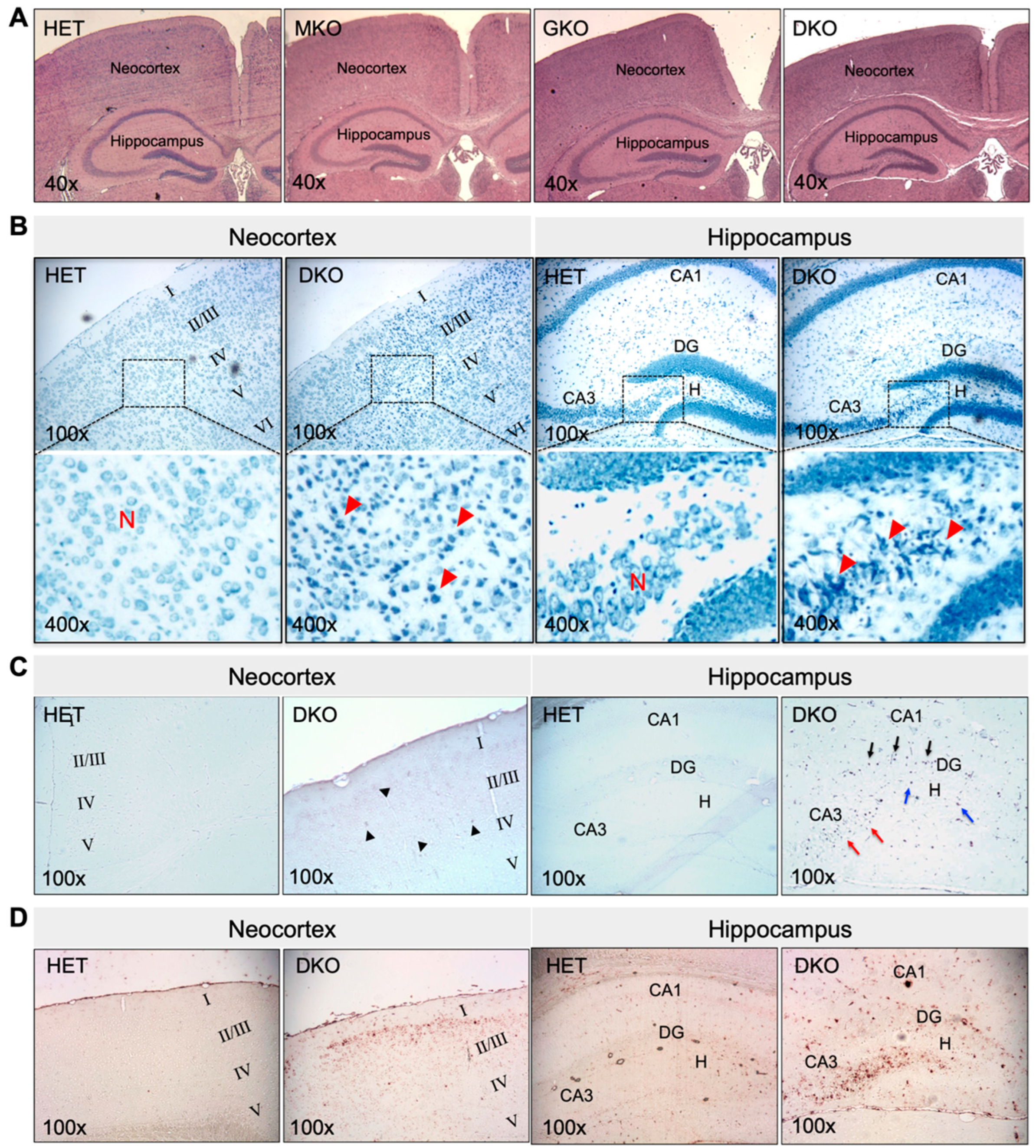
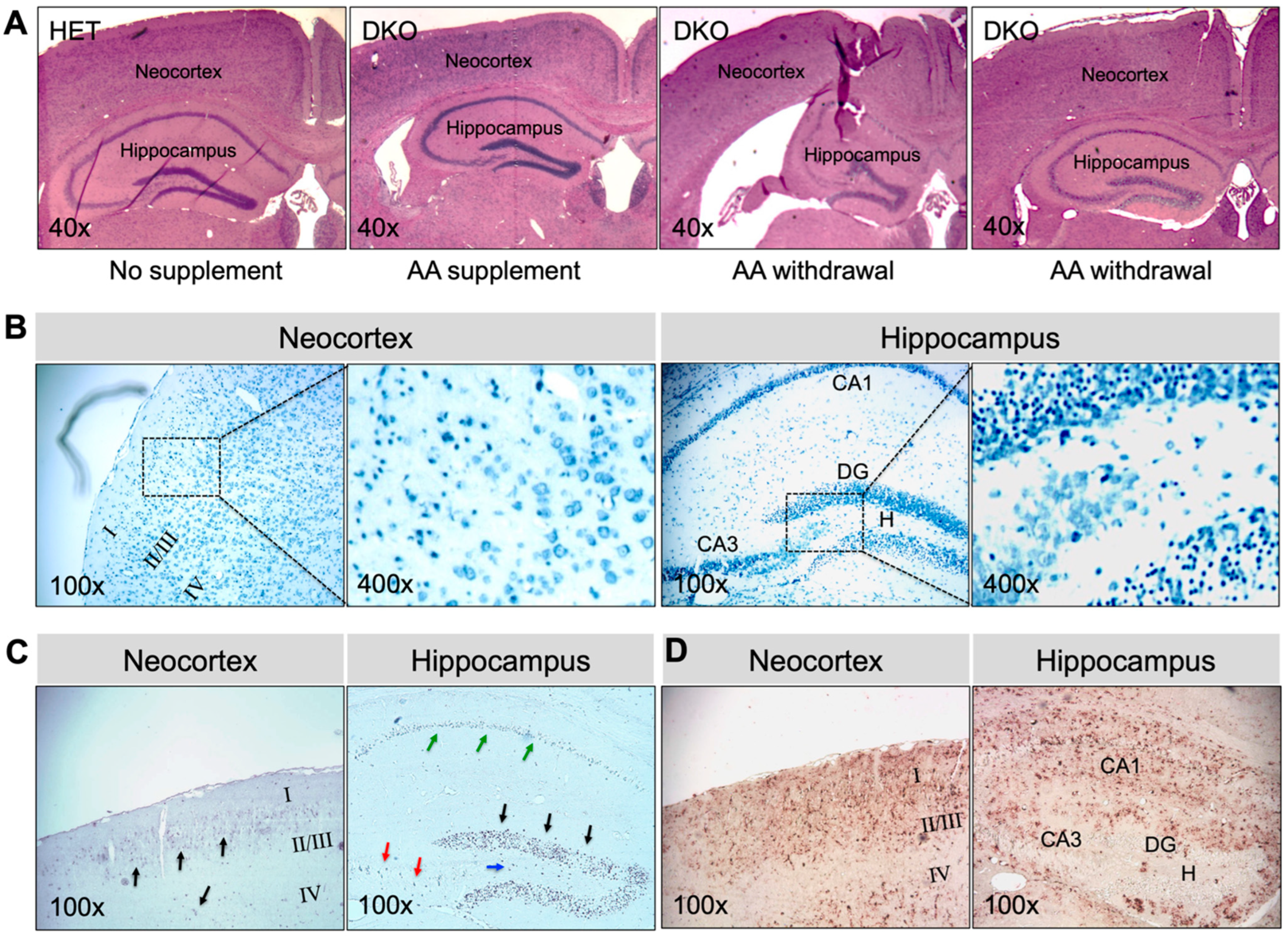
3.2. DKO Mice Show Neuronal Loss and Glial Proliferation in the Neocortex and Hippocampus
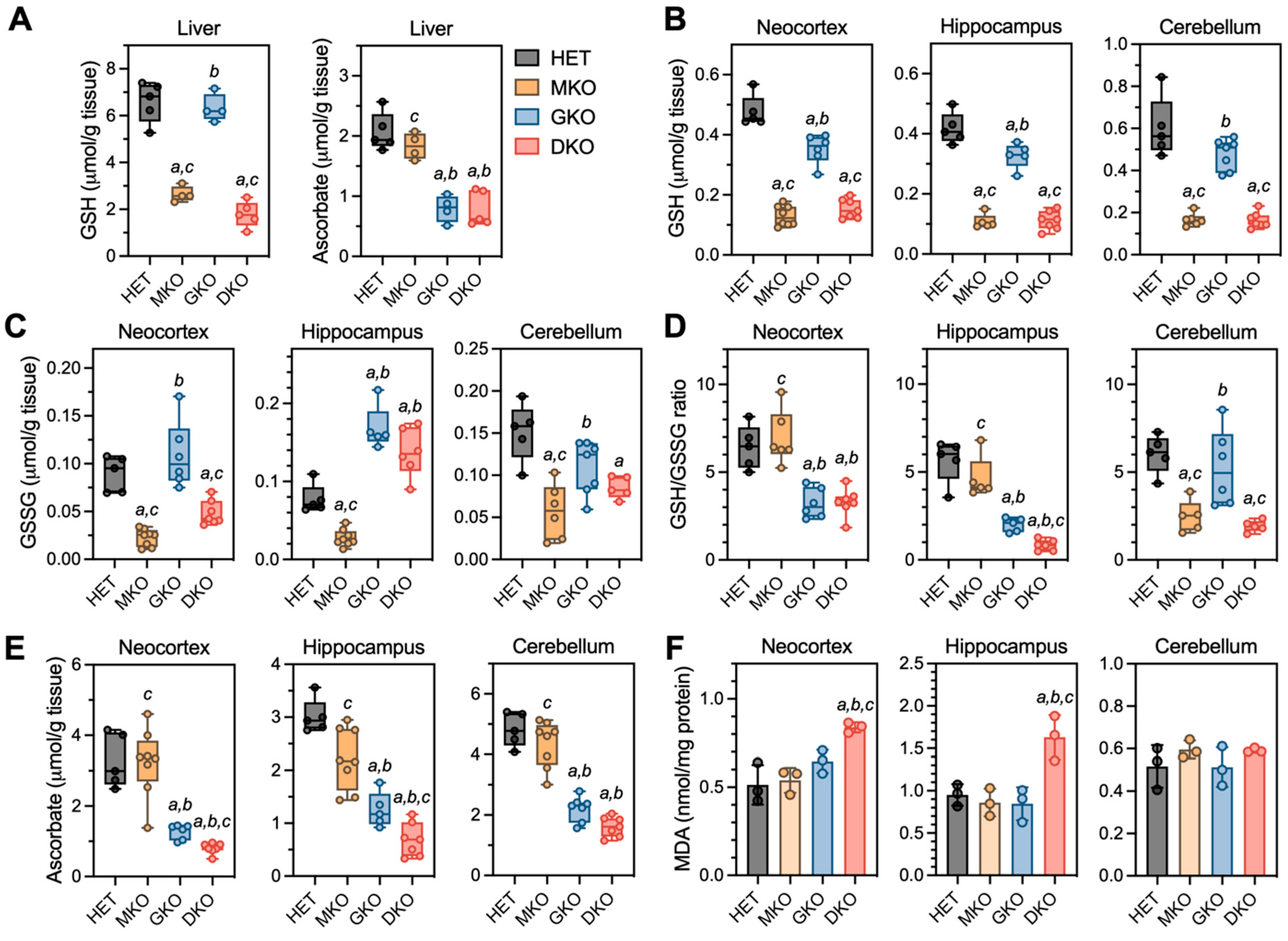
3.3. Brains of Young DKO Mice Show Region-Specific GSH and AA Deficiencies and Lipid Peroxidation
3.4. AA Rescues DKO Mice, but Removal of AA in Rescued DKO Mice Results in Spontaneous Seizures and Premature Death
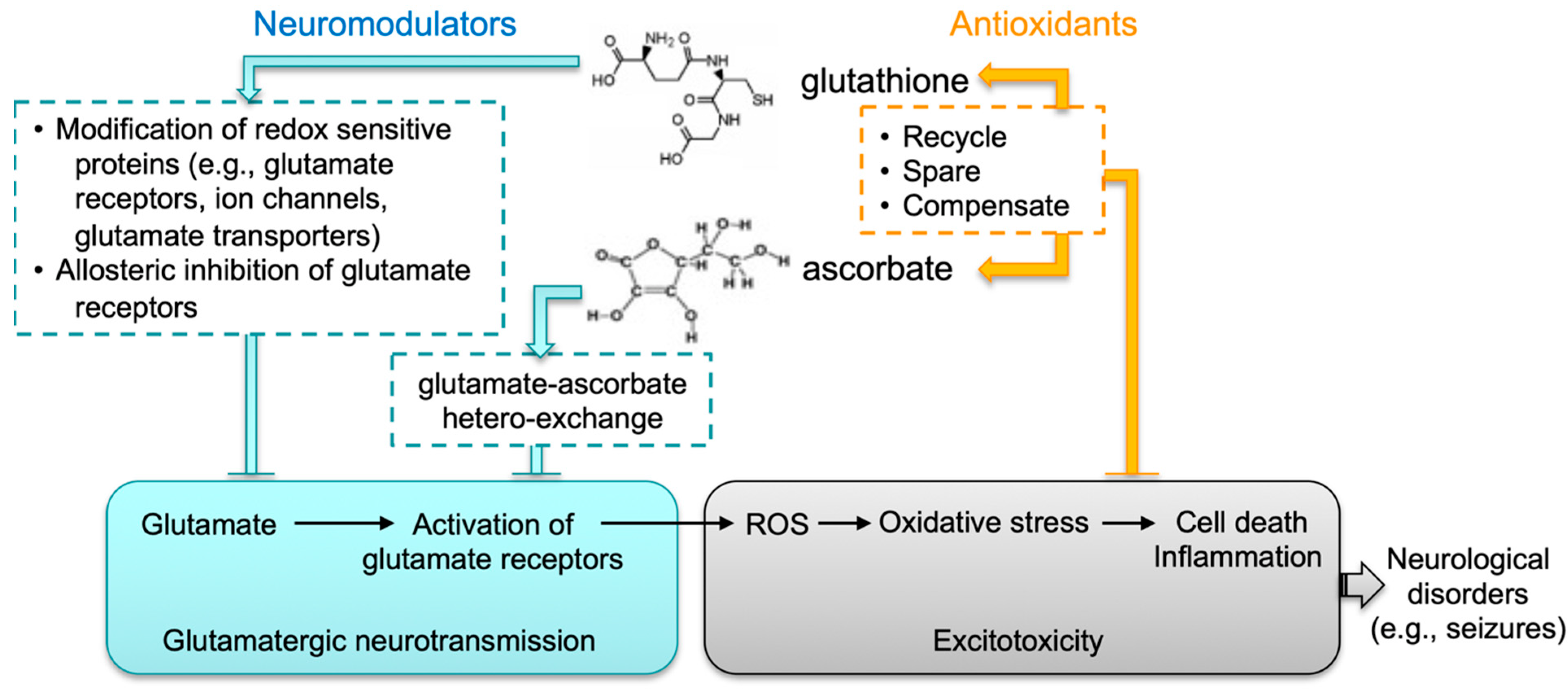
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Dwivedi, D.; Megha, K.; Mishra, R.; Mandal, P.K. Glutathione in Brain: Overview of Its Conformations, Functions, Biochemical Characteristics, Quantitation and Potential Therapeutic Role in Brain Disorders. Neurochem. Res. 2020, 45, 1461–1480. [Google Scholar] [CrossRef]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Wilson, J.X. Regulation of vitamin C transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Rivas, C.I.; Zuniga, F.A.; Salas-Burgos, A.; Mardones, L.; Ormazabal, V.; Vera, J.C. Vitamin C transporters. J. Physiol. Biochem. 2008, 64, 357–375. [Google Scholar] [CrossRef]
- Nishikimi, M.; Yagi, K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am. J. Clin. Nutr. 1991, 54, 1203S–1208S. [Google Scholar] [CrossRef]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of vitamin C homeostasis during deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef]
- Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Distribution of vitamin C is tissue specific with early saturation of the brain and adrenal glands following differential oral dose regimens in guinea pigs. Br. J. Nutr. 2015, 113, 1539–1549. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Figueroa-Mendez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M.; Tu, Y.J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef]
- Sucher, N.J.; Lipton, S.A. Redox modulatory site of the NMDA receptor-channel complex: Regulation by oxidized glutathione. J. Neurosci. Res. 1991, 30, 582–591. [Google Scholar] [CrossRef]
- Volterra, A.; Trotti, D.; Floridi, S.; Racagni, G. Reactive oxygen species inhibit high-affinity glutamate uptake: Molecular mechanism and neuropathological implications. Ann. N. Y. Acad. Sci. 1994, 738, 153–162. [Google Scholar] [CrossRef]
- Amato, A.; Connolly, C.N.; Moss, S.J.; Smart, T.G. Modulation of neuronal and recombinant GABAA receptors by redox reagents. J. Physiol. 1999, 517 Pt 1, 35–50. [Google Scholar] [CrossRef]
- Nelson, M.T.; Joksovic, P.M.; Su, P.; Kang, H.W.; Van Deusen, A.; Baumgart, J.P.; David, L.S.; Snutch, T.P.; Barrett, P.Q.; Lee, J.H.; et al. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J. Neurosci. 2007, 27, 12577–12583. [Google Scholar] [CrossRef]
- Raju, K.; Doulias, P.T.; Evans, P.; Krizman, E.N.; Jackson, J.G.; Horyn, O.; Daikhin, Y.; Nissim, I.; Yudkoff, M.; Nissim, I.; et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal. 2015, 8, ra68. [Google Scholar] [CrossRef] [Green Version]
- Ballaz, S.J.; Rebec, G.V. Neurobiology of vitamin C: Expanding the focus from antioxidant to endogenous neuromodulator. Pharmacol. Res. 2019, 146, 104321. [Google Scholar] [CrossRef] [PubMed]
- Moretti, M.; Rodrigues, A.L.S. Functional role of ascorbic acid in the central nervous system: A focus on neurogenic and synaptogenic processes. Nutr. Neurosci. 2022, 25, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Orselli, S.M.; Rex, T.S. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic. Biol. Med. 1994, 17, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qu, Z.C.; May, J.M. GSH is required to recycle ascorbic acid in cultured liver cell lines. Antioxid. Redox Signal. 2001, 3, 1089–1097. [Google Scholar] [CrossRef]
- Ehrhart, J.; Zeevalk, G.D. Cooperative interaction between ascorbate and glutathione during mitochondrial impairment in mesencephalic cultures. J. Neurochem. 2003, 86, 1487–1497. [Google Scholar] [CrossRef]
- Shin, E.J.; Suh, S.K.; Lim, Y.K.; Jhoo, W.K.; Hjelle, O.P.; Ottersen, O.P.; Shin, C.Y.; Ko, K.H.; Kim, W.K.; Kim, D.S.; et al. Ascorbate attenuates trimethyltin-induced oxidative burden and neuronal degeneration in the rat hippocampus by maintaining glutathione homeostasis. Neuroscience 2005, 133, 715–727. [Google Scholar] [CrossRef]
- Dalton, T.P.; Dieter, M.Z.; Yang, Y.; Shertzer, H.G.; Nebert, D.W. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: Embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem. Biophys. Res. Commun. 2000, 279, 324–329. [Google Scholar] [CrossRef]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002, 277, 49446–49452. [Google Scholar] [CrossRef]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar] [CrossRef]
- Chen, Y.; Dong, H.; Thompson, D.C.; Shertzer, H.G.; Nebert, D.W.; Vasiliou, V. Glutathione defense mechanism in liver injury: Insights from animal models. Food Chem. Toxicol. 2013, 60, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of chronic glutathione deficiency on the behavioral phenotype of Gclm-/- knockout mice. Neurotoxicol. Teratol. 2012, 34, 450–457. [Google Scholar] [CrossRef]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of vitamin C deficiency during postnatal development on adult behavior: Functional phenotype of Gulo-/- knockout mice. Genes Brain Behav. 2012, 11, 269–277. [Google Scholar] [CrossRef]
- Nakamura, B.N.; Fielder, T.J.; Hoang, Y.D.; Lim, J.; McConnachie, L.A.; Kavanagh, T.J.; Luderer, U. Lack of maternal glutamate cysteine ligase modifier subunit (Gclm) decreases oocyte glutathione concentrations and disrupts preimplantation development in mice. Endocrinology 2011, 152, 2806–2815. [Google Scholar] [CrossRef]
- Graham, D.L.; Herring, N.R.; Schaefer, T.L.; Holland, K.D.; Vorhees, C.V.; Williams, M.T. Electroencephalographic and convulsive effects of binge doses of (+)-methamphetamine, 5-methoxydiisopropyltryptamine, and (±)-3,4-methylenedioxymethamphetamine in rats. Open Neuropsychopharmacol. J. 2012, 5, 1–8. [Google Scholar] [CrossRef]
- Castro, O.W.; Santos, V.R.; Pun, R.Y.; McKlveen, J.M.; Batie, M.; Holland, K.D.; Gardner, M.; Garcia-Cairasco, N.; Herman, J.P.; Danzer, S.C. Impact of corticosterone treatment on spontaneous seizure frequency and epileptiform activity in mice with chronic epilepsy. PLoS ONE 2012, 7, e46044. [Google Scholar] [CrossRef]
- Senft, A.P.; Dalton, T.P.; Shertzer, H.G. Determining glutathione and glutathione disulfide using the fluorescence probe o-phthalaldehyde. Anal. Biochem. 2000, 280, 80–86. [Google Scholar] [CrossRef]
- Zannoni, V.; Lynch, M.; Goldstein, S.; Sato, P. A rapid micromethod for the determination of ascorbic acid in plasma and tissues. Biochem. Med. 1974, 11, 41–48. [Google Scholar] [CrossRef]
- Meister, A. Glutathione, ascorbate, and cellular protection. Cancer Res. 1994, 54, 1969s–1975s. [Google Scholar]
- Jacob, R.A. The integrated antioxidant system. Nutr. Res. 1995, 15, 755–766. [Google Scholar] [CrossRef]
- Chainy, G.B.N.; Sahoo, D.K. Hormones and oxidative stress: An overview. Free Radic. Res. 2020, 54, 1–26. [Google Scholar] [CrossRef]
- Fritschy, J.M. Epilepsy, E/I Balance and GABA(A) Receptor Plasticity. Front. Mol. Neurosci. 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonansco, C.; Fuenzalida, M. Plasticity of Hippocampal Excitatory-Inhibitory Balance: Missing the Synaptic Control in the Epileptic Brain. Neural Plast. 2016, 2016, 8607038. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.; White, H.S. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, S.; Raisuddin, S.; Parvez, S. Glutamate Excitotoxicity and Oxidative Stress in Epilepsy: Modulatory Role of Melatonin. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 365–374. [Google Scholar] [CrossRef]
- Eid, T.; Gruenbaum, S.E.; Dhaher, R.; Lee, T.W.; Zhou, Y.; Danbolt, N.C. The Glutamate-Glutamine Cycle in Epilepsy. Adv. Neurobiol. 2016, 13, 351–400. [Google Scholar] [CrossRef]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef]
- Oja, S.S.; Janaky, R.; Varga, V.; Saransaari, P. Modulation of glutamate receptor functions by glutathione. Neurochem. Int. 2000, 37, 299–306. [Google Scholar] [CrossRef]
- Janáky, R.; Varga, V.; Jenei, Z.; Saransaari, P.; Oja, S.S. Glutathione and glutathione derivatives: Possible modulators of ionotropic glutamate receptors. In Glutathione in the Nervous System; CRC Press: Boca Raton, FL, USA, 2018; pp. 163–196. [Google Scholar]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef]
- Domith, I.; Socodato, R.; Portugal, C.C.; Munis, A.F.; Duarte-Silva, A.T.; Paes-de-Carvalho, R. Vitamin C modulates glutamate transport and NMDA receptor function in the retina. J. Neurochem. 2018, 144, 408–420. [Google Scholar] [CrossRef]
- Patsoukis, N.; Zervoudakis, G.; Georgiou, C.D.; Angelatou, F.; Matsokis, N.A.; Panagopoulos, N.T. Thiol redox state and lipid and protein oxidation in the mouse striatum after pentylenetetrazol-induced epileptic seizure. Epilepsia 2005, 46, 1205–1211. [Google Scholar] [CrossRef]
- Stark, D.T.; Bazan, N.G. Synaptic and extrasynaptic NMDA receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection. J. Neurosci. 2011, 31, 13710–13721. [Google Scholar] [CrossRef]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef]
- Crowe, S.L.; Tsukerman, S.; Gale, K.; Jorgensen, T.J.; Kondratyev, A.D. Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J. Neurosci. 2011, 31, 7648–7656. [Google Scholar] [CrossRef]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed. Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef]
- Sun, H.; Li, X.; Guo, Q.; Liu, S. Research progress on oxidative stress regulating different types of neuronal death caused by epileptic seizures. Neurol. Sci. 2022, 43, 6279–6298. [Google Scholar] [CrossRef]
- Chauhan, P.; Philip, S.E.; Chauhan, G.; Mehra, S. The Anatomical Basis of Seizures. In Epilepsy; Czuczwar, S.J., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar] [CrossRef]
- Ambrogini, P.; Torquato, P.; Bartolini, D.; Albertini, M.C.; Lattanzi, D.; Di Palma, M.; Marinelli, R.; Betti, M.; Minelli, A.; Cuppini, R.; et al. Excitotoxicity, neuroinflammation and oxidant stress as molecular bases of epileptogenesis and epilepsy-derived neurodegeneration: The role of vitamin E. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1098–1112. [Google Scholar] [CrossRef]
- Thyrion, L.; Portelli, J.; Raedt, R.; Glorieux, G.; Larsen, L.E.; Sprengers, M.; Van Lysebettens, W.; Carrette, E.; Delbeke, J.; Vonck, K.; et al. Disruption, but not overexpression of urate oxidase alters susceptibility to pentylenetetrazole- and pilocarpine-induced seizures in mice. Epilepsia 2016, 57, e146–e150. [Google Scholar] [CrossRef]
- Silva-Adaya, D.; Perez-De La Cruz, V.; Herrera-Mundo, M.N.; Mendoza-Macedo, K.; Villeda-Hernandez, J.; Binienda, Z.; Ali, S.F.; Santamaria, A. Excitotoxic damage, disrupted energy metabolism, and oxidative stress in the rat brain: Antioxidant and neuroprotective effects of L-carnitine. J. Neurochem. 2008, 105, 677–689. [Google Scholar] [CrossRef]
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef]
- Grone, B.P.; Baraban, S.C. Animal models in epilepsy research: Legacies and new directions. Nat. Neurosci. 2015, 18, 339–343. [Google Scholar] [CrossRef]
- Marshall, G.F.; Gonzalez-Sulser, A.; Abbott, C.M. Modelling epilepsy in the mouse: Challenges and solutions. Dis. Model. Mech. 2021, 14, dmm.047449. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Ristoff, E.; Larsson, A. Patients with genetic defects in the gamma-glutamyl cycle. Chem. Biol. Interact. 1998, 111–112, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo (1)H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef]
- Schleicher, R.L.; Carroll, M.D.; Ford, E.S.; Lacher, D.A. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am. J. Clin. Nutr. 2009, 90, 1252–1263. [Google Scholar] [CrossRef]
- Carr, A.C.; Rowe, S. Factors Affecting Vitamin C Status and Prevalence of Deficiency: A Global Health Perspective. Nutrients 2020, 12, 1963. [Google Scholar] [CrossRef]
- Fletcher, B.D.; Flett, J.A.M.; Wickham, S.R.; Pullar, J.M.; Vissers, M.C.M.; Conner, T.S. Initial Evidence of Variation by Ethnicity in the Relationship between Vitamin C Status and Mental States in Young Adults. Nutrients 2021, 13, 792. [Google Scholar] [CrossRef]
- Johnston, C.S.; Thompson, L.L. Vitamin C status of an outpatient population. J. Am. Coll. Nutr. 1998, 17, 366–370. [Google Scholar] [CrossRef]
- Madruga de Oliveira, A.; Rondo, P.H.; Mastroeni, S.S.; Oliveira, J.M. Plasma concentrations of ascorbic acid in parturients from a hospital in Southeast Brazil. Clin. Nutr. 2008, 27, 228–232. [Google Scholar] [CrossRef]
- Yeh, M.Y.; Burnham, E.L.; Moss, M.; Brown, L.A. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am. J. Respir. Crit. Care Med. 2007, 176, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Chitty, K.M.; Lagopoulos, J.; Hickie, I.B.; Hermens, D.F. The impact of alcohol and tobacco use on in vivo glutathione in youth with bipolar disorder: An exploratory study. J. Psychiatr. Res. 2014, 55, 59–67. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype (N of Mice) | AA Supplement | Glu 1 | TG | PL | Chol | NEFA | Hct |
|---|---|---|---|---|---|---|---|
| (mg/dL) | (mEq/L) | (%) | |||||
| Pre-weanling mice (PND17-20) | |||||||
| HET (5) | none | 273 ± 21 | 88 ± 22 | 192 ± 20 | 115 ± 8 | 0.9 ± 0.2 | 49 ± 2 |
| MKO (4) | none | 277 ± 4 | 107 ± 31 | 207 ± 14 | 156 ± 11 | 1.2 ± 0.3 | 49 ± 2 |
| GKO (4) | none | 281 ± 10 | 77 ± 11 | 197 ± 18 | 145 ± 17 | 1.5 ± 0.4 | 48 ± 2 |
| DKO (5) | none | 223 ± 14 | 85 ± 14 | 195 ± 15 | 136 ± 18 | 0.7 ± 0.1 | 51 ± 3 |
| AA-rescued mice (1 or 3 mo) | |||||||
| DKO (8) | 1 g/L | 290 ± 36 | 49 ± 9 | 198 ± 39 | 94 ± 22 | 0.6 ± 0.1 | 47 ± 1.2 |
| DKO (9) | Removal 2 | 159 ± 85 † | 85 ± 42 | 269 ± 89 | 138 ± 35 † | 1.0 ± 0.4 | 49 ± 2.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Holland, K.D.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Fatal Epileptic Seizures in Mice Having Compromised Glutathione and Ascorbic Acid Biosynthesis. Antioxidants 2023, 12, 448. https://doi.org/10.3390/antiox12020448
Chen Y, Holland KD, Shertzer HG, Nebert DW, Dalton TP. Fatal Epileptic Seizures in Mice Having Compromised Glutathione and Ascorbic Acid Biosynthesis. Antioxidants. 2023; 12(2):448. https://doi.org/10.3390/antiox12020448
Chicago/Turabian StyleChen, Ying, Katherine D. Holland, Howard G. Shertzer, Daniel W. Nebert, and Timothy P. Dalton. 2023. "Fatal Epileptic Seizures in Mice Having Compromised Glutathione and Ascorbic Acid Biosynthesis" Antioxidants 12, no. 2: 448. https://doi.org/10.3390/antiox12020448