
The Impact of Tobacco Cigarettes, Vaping Products and Tobacco Heating Products on Oxidative Stress

 ,
,
Abstract
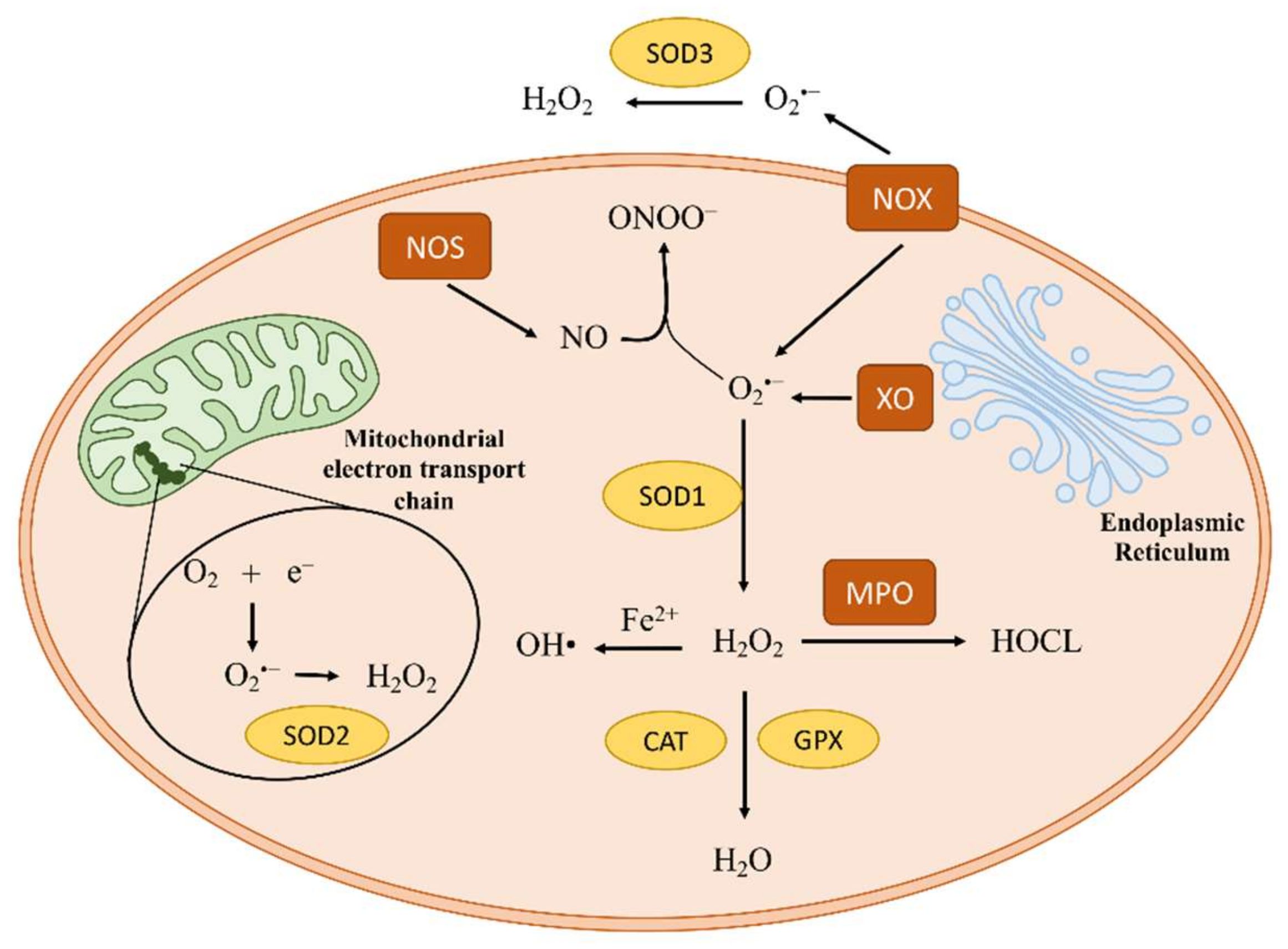
:1. Introduction
2. Oxidative Stress and Smoking/Vaping Related to Airway Diseases
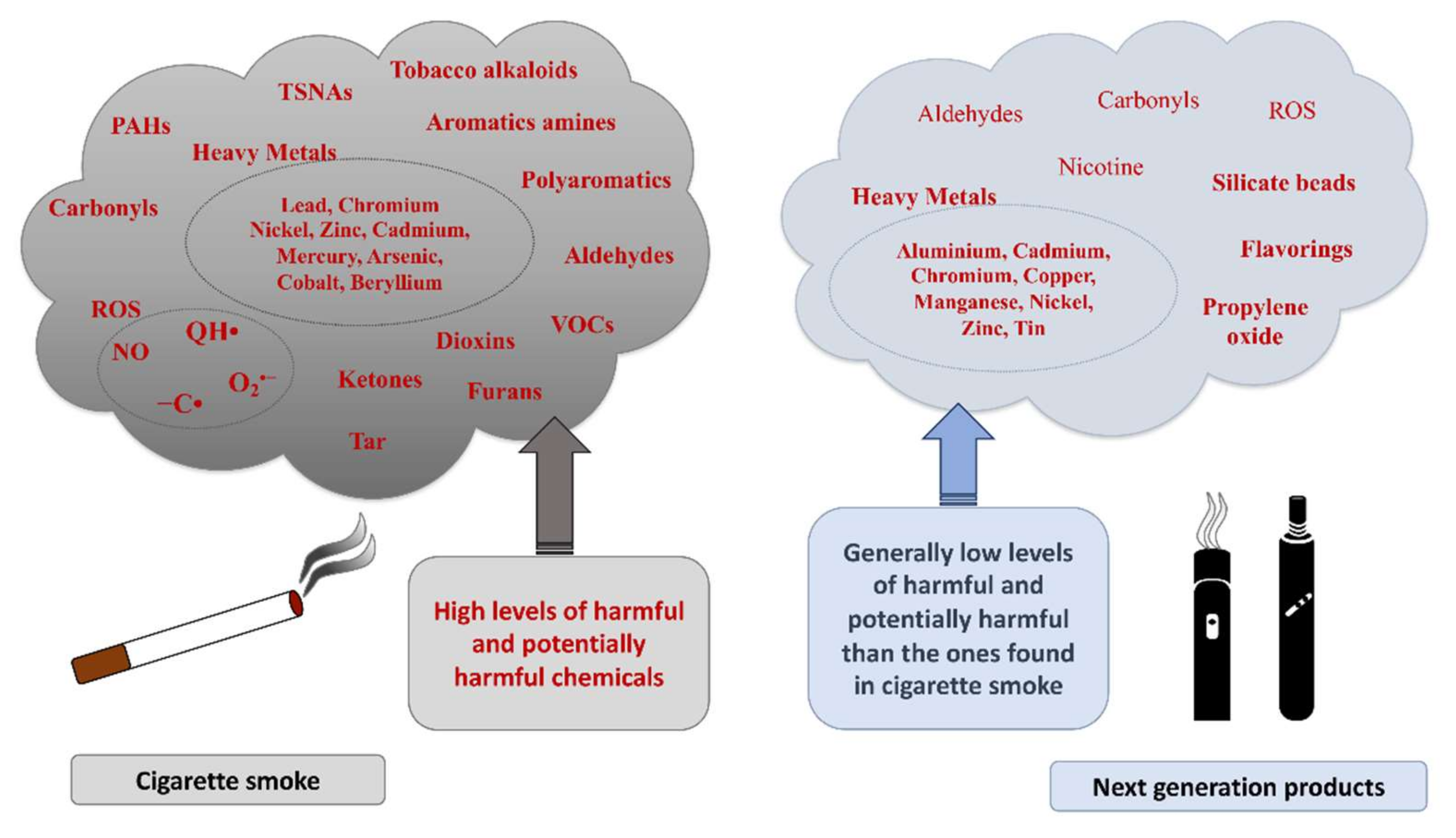
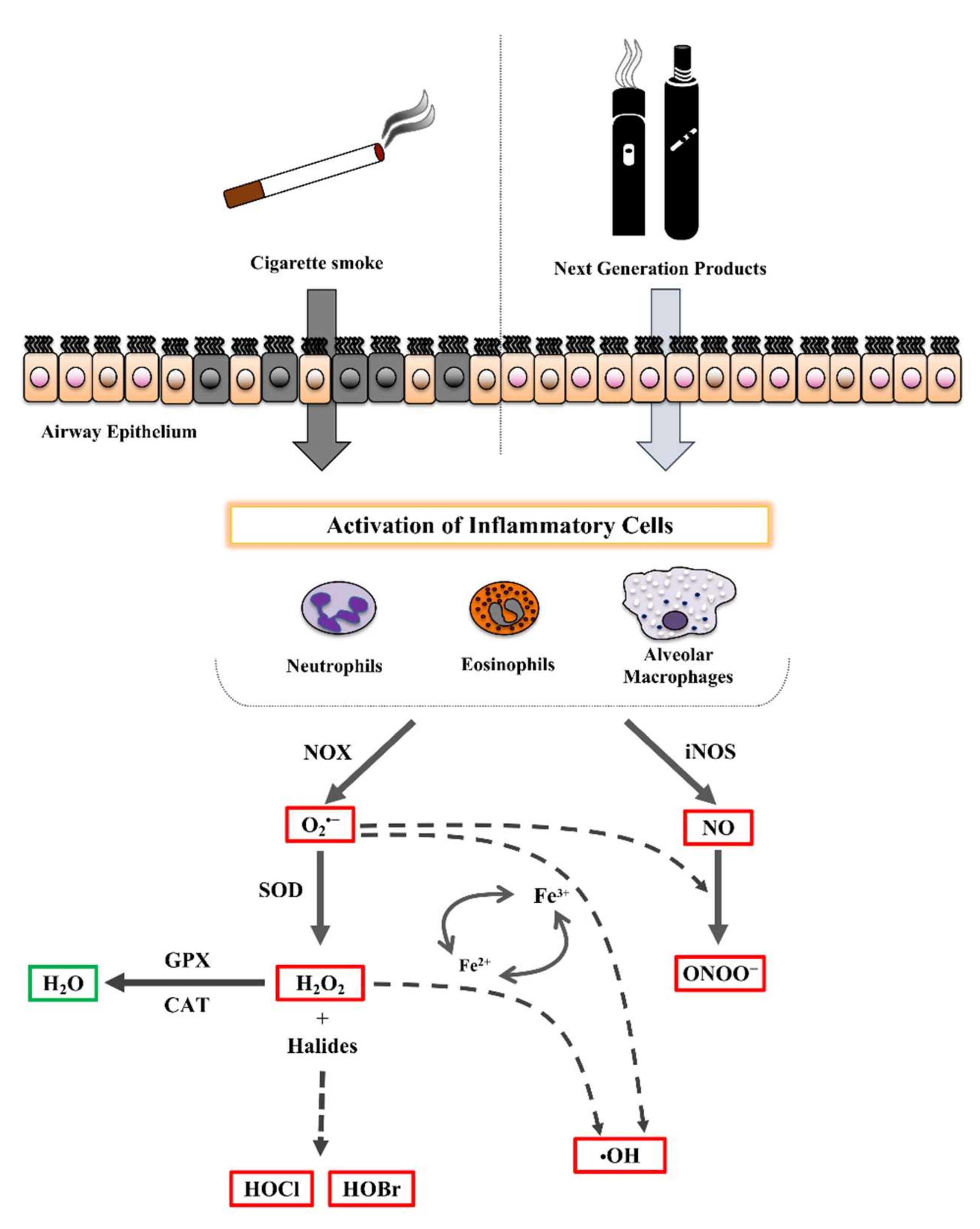
2.1. Cigarette Smoke Effect on Airway Diseases
2.2. Effect of NGPs on Airways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Study Findings | Product(s) Tested | Experimental Setup |
|---|---|---|---|
| Kirkham et al. [46] | Repeated low-micromolar exposure to car-bonyls (e.g., acrolein) from cigarette smoke leads to carbonyl adduct (modified pro-teins) accumulation over time in collagen type IV. Acrolein-modified proteins can ac-tivate macrophages, such as oxidative burst and the release of MCP-1, independently of other stimuli. | Tobacco smoke | In vitro |
| Plaschke et al. [47] | Smoking was found to be a risk factor for onset of asthma in adults. | Tobacco smoke | Human study |
| Rasmussen et al. [48] | Smoking is an independent risk factor for the development of asthma-like symptoms during adolescence. | Tobacco smoke | Human study |
| Kim et al. [49] | Active smoking may play an important role in the development of asthma and bronchial hyper-responsiveness among the elderly. | Tobacco smoke | Human study |
| Polosa et al. [50] | Cigarette smoking is an important predictor of asthma severity and poor asthma control. | Tobacco smoke | Human study |
| Polosa et al. [72] | Cigarette smoking is an important independent risk factor for the development of new asthma cases in adults with allergic rhinitis. | Tobacco smoke | Human study |
| Emma et al. [78] | Cigarette smoking in severe asthma patients causes greater systemic oxidative stress. Moreover, active smoking in asthmatic subjects can lead to inhibition of NOS2 mRNA expression in pulmonary cells (bronchial brushing) by negative feedback, possibly due to the high level of NO contained in cigarette smoke. | Tobacco smoke | Human study |
| Takahashi et al. [80] | Increased colony stimulating factor (CSF) 2 protein levels, xenobiotic metabolism, oxidative stress, and endoplasmic reticulum stress in the respiratory tract (bronchial brushing, biopsies, and sputum cells) have been observed in asthmatics who currently smoke. In former asthmatic smokers, there is a predominant neutrophilic inflammation and loss of epithelial barrier function. | Tobacco smoke | Human study |
| Lerner et al. [82] | Exposure to e-cigarette aerosols/juices sustains oxidative and inflammatory responses in lung cells (human airway epithelial cells and human lung fibroblasts) and pulmonary tissues in C57BL/6J mice. | NGP aerosols | In vitro |
| Sussan et al. [83] | Mice exposed to e-cigarette aerosol showed significantly impaired pulmonary bacterial clearance, compared to air-exposed mice. This defective bacterial clearance was partially due to reduced phagocytosis by alveolar macrophages. | NGP aerosols | In vivo |
| Shivalingappa et al. [85] | E-cigarette vapor exposure induces proteostasis/autophagy impairment, leading to oxidative stress, apoptosis, and senescence. | NGP aerosols | In vitro |
| Scheffler et al. [86] | In an in vitro model of human bronchial epithelial cells exposed to e-cigarette aerosols with different concentrations of nicotine and tobacco cigarette smoke, authors observed toxicological effects induced by smoke and aerosol, whereas the nicotine concentration did not have an effect on the cell viability. | NGPs aerosol Tobacco smoke | In vitro |
| Taylor et al. [88] | Concentration-dependent oxidative stress, intracellular generation of oxidant species, reduced GSH:GSSG, increased transcriptional activation of ARE, increased Caspase 3/7 activity, and strong decrease in viability were observed in human bronchial epithelial cells following exposure to cigarette smoke AqE. No cellular stress responses were detected following exposure to e-cigarette AqE. | NGP aerosols | In vitro |
| Malinska et al. [89] | Tobacco cigarette total particulate matter (TPM) had a stronger effect on oxidative phosphorylation, gene expression, and proteins involved in oxidative stress than TPM from a tobacco heating product (THS2.2) in a model of human bronchial epithelial cells. | NGP aerosols | In vitro |
| Moses et al. [90] | E-cigarette aerosol can induce gene expression changes in bronchial airway epithelium in vitro, some of which are shared with tobacco cigarette smoke. These changes were generally less pronounced than the effects of tobacco cigarette exposure and were more pronounced in e-cigarette products containing nicotine than those without nicotine. | NGP aerosols | In vitro |
| Jabba et al. [91] | Reaction products formed in e-liquids between flavor aldehydes and solvent chemicals have differential toxicological properties from their parent flavor aldehydes and may contribute to the health effects of e-cigarette aerosols in the respiratory systems of e-cigarette users. | NGP aerosols | In vitro |
3. Oxidative Stress and Smoking/Vaping Related to Cardiovascular Diseases
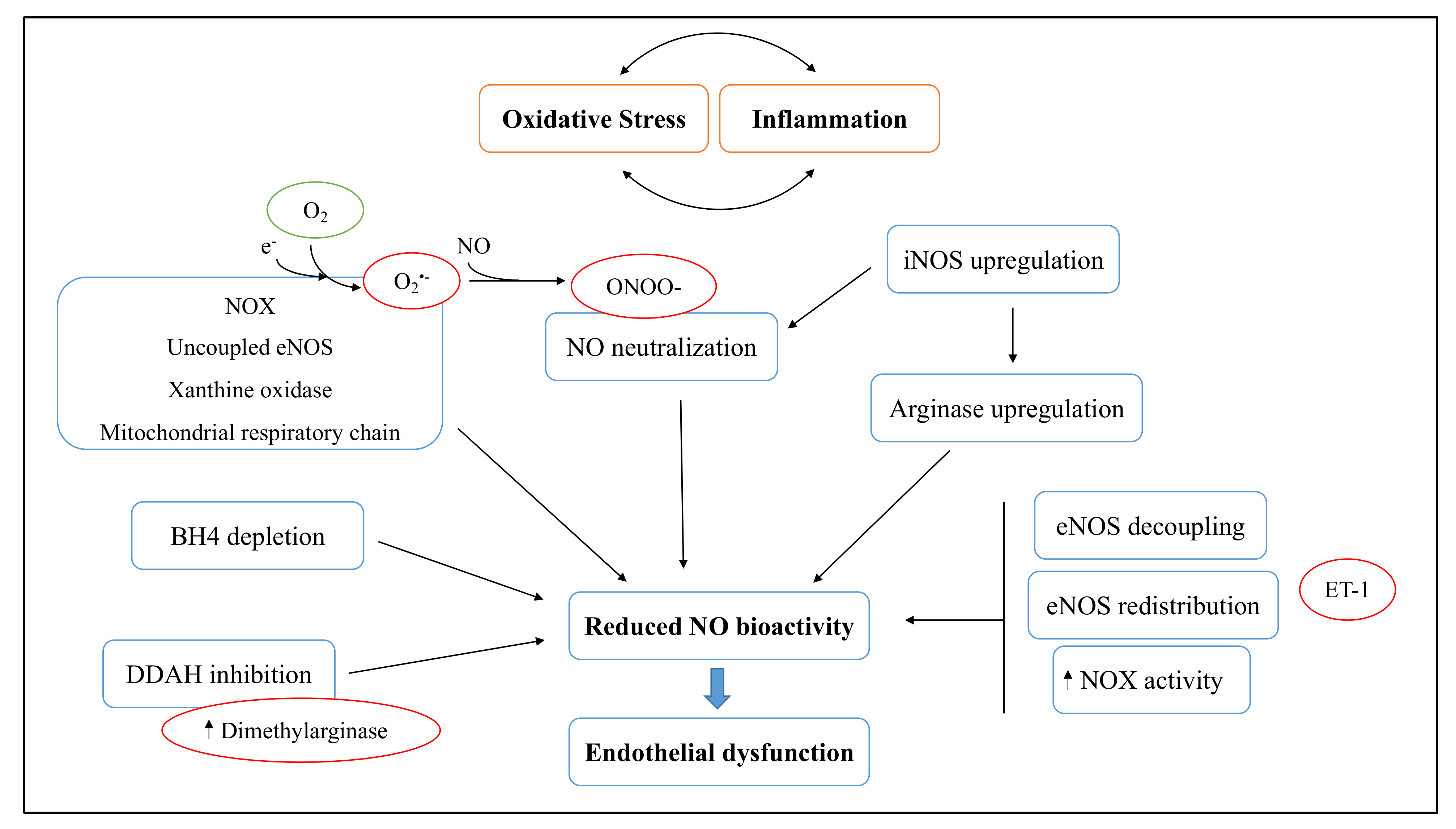
3.1. Cigarette Smoke Effect on Endothelial Dysfunction
3.2. Effects of NGPs on Oxidative Stress-Related Endothelial Dysfunction
| Author | Study Findings | Product(s) Tested | Experimental Setup |
|---|---|---|---|
| Barua et al. [112] | The study confirmed that oxidative stress plays a central role in smoking-mediated dysfunction of NO biosynthesis in human coronary artery endothelial cells. | Tobacco smoke | In vitro |
| Celermajer et al. [114] | Both active and passive smoking impaired endothelium-dependent arterial dilatation, suggesting early arterial damage in healthy young adults. | Tobacco smoke | Human study |
| Zeiher et al. [115] | The impairment of coronary arterial vasodilator function is associated with long-term cigarette smoking. | Tobacco smoke | Human study |
| Raveendran et al. [117] | Cigarette smoke extract induced human aortic endothelial cells’ (HAECs) apoptosis, but endogenous NO production reduced the cigarette smoking-induced apoptosis. | Tobacco smoke | In vitro |
| Jaimes et al. [118] | Thiol-reactive stable compounds in cigarette smoke increase endothelial O2•− through NADPH oxidase activation, thereby reducing NO bioactivity and resulting in endothelial dysfunction. | Tobacco smoke | In vitro |
| Kayyali et al. [119] | Cigarette smoke condensate induced xanthine oxidase mRNA expression and xanthine oxidase gene promoter activity. | Tobacco smoke | In vitro |
| Talukder et al. [120] | Exposure of C57BL/6J mice to cigarette smoke for 32 weeks led to blunted weight gain, hypertension, endothelial dysfunction, leukocyte activation with ROS generation, decreased NO bioavailability, and mild cardiac hypertrophy. | Tobacco smoke | In vivo, mouse model |
| van den Berg et al. [123] | The transcription factor NF-kB was increased in the peripheral blood mononuclear cells of smokers compared to non-smokers, confirming the role of this biomarker in smoke-induced inflammation. | Tobacco smoke | Human study |
| Orosz et al. [124] | Results from this study suggest that water-soluble components of cigarette smoke activate the vascular NAD(P)H oxidase with increased production of O2•− and consequently H2O2. NAD(P)H oxidase-derived H2O2 activates NF-kB, leading to pro-inflammatory alterations in vascular phenotype. | Tobacco smoke | In vivo, rat model |
| Miyaura et al. [127] | Cigarette smoke extract inhibits, in a dose-dependent manner, the activity of PAF-acetylhydrolase, an important enzyme that regulates the degradation of the vascular pro-inflammatory platelet-activating factor (PAF). | Tobacco smoke | In vitro–ex vivo |
| Imaizumi et al. [128] | The platelet-activating factor-like lipid(s) (PAF-LL) were detected in LDL and HDL plasma lipoproteins, and their levels were significantly increased in smokers after smoking, contributing to atherosclerosis. | Tobacco smoke | Human study |
| Kangavari et al. [132] | Cigarette smoking increases markers of inflammation, including macrophage immunoreactivity (CD68 expression) and MMP-12, and tissue destruction in atherosclerotic plaques (TIMP-1) in smokers compared to non-smokers. | Tobacco smoke | In vitro–ex vivo |
| Huang et al. [133] | Active cigarette smoking status was positively associated with increased matrix metalloproteinase-12 (MMP-12), growth/differentiation factor 15 (GDF-15), urokinase plasminogen activator surface receptor (uPAR), TNF-related apoptosis-inducing ligand receptor 2 (TRAIL-R2), lectin-like oxidized LDL receptor 1 (LOX-1), hepatocyte growth factor (HGF), matrix metalloproteinase-10 (MMP-10), and matrix metalloproteinase-1 (MMP-1). Negative association with active smoking was reported for endothelial cell-specific molecule 1 (ESM-1) and interleukin-27 subunit alpha (IL27-A). All these results suggest the interference of smoking with the atherosclerosis process. | Tobacco smoke | Human study |
| Teasdale et al. [134] | Cigarette smoke extract induced the activation of NRF2 and upregulation of cytochrome p450 in human coronary artery endothelial cells. However, e-cigarette extract did not induce NRF2 nuclear activation, or the upregulation of cytochrome p450. | NGP aerosols | In vitro |
| Anderson et al. [135] | E-cigarette aerosol induced reactive oxygen species, DNA damage, and cell death in human umbilical vein endothelial cells. However, the effects of e-cigarette aerosols were lower than those of cigarette smoke applied at the same nicotine concentration. | NGP aerosols | In vitro |
| Carnevale et al. [136] | Significant increase in soluble NOX2-derived peptide and 8-iso-prostaglandin F2α and a significant decrease in nitric oxide bioavailability, vitamin E levels, and FMD were observed in e-cigarette (dual users) and traditional cigarette consumers. However, e-cigarettes seemed to have a lesser impact. | NGP aerosols | Human study |
| Chaumont et al. [137] | Sham vaping and vaping without nicotine were not associated with modification of cardiovascular parameters or oxidative stress. Instead, vaping with nicotine resulted in modification of cardiovascular parameters and increased plasma myeloperoxidase. | NGP aerosols | Human study |
| Ikonomidis et al. [138] | Aortic stiffness, assessed by pulse wave velocity (PWV) and augmentation index (AIX75), exhaled CO concentration, and oxidative stress, assessed by malondialdehyde (MDA) plasma concentrations, were reduced in smokers who switched to e-cigarettes after 1 month of use. | NGP aerosols | Human study |
| Espinoza-Derout et al. [139] | E-cigarette with 2.4% nicotine decreased left ventricular fractional shortening and ejection fraction, induced changes in genes associated with metabolism, circadian rhythm, and inflammation, and also induced ultrastructural abnormalities of cardiomyocytes in ApoE−/− mice compared to controls (saline). Additionally, increased oxidative stress and mitochondrial DNA mutations were observed in mice treated with e-cigarettes (2.4%). | NGP aerosols | In vivo, mouse model |
| Farsalinos et al. [140] | The release of toxic aldehydes is associated with the generation of dry puffs. Under realistic conditions, e-cigarettes emit minimal aldehydes/g liquid at both low and high power. | NGP aerosols | In vitro |
| Kuntic et al. [141] | E-cigarette vapor exposure, particularly acrolein, increases vascular, cerebral, and pulmonary oxidative stress via a NOX-2-dependent mechanism in mice. | NGP aerosols | In vivo, mouse model |
| Lee et al. [142] | Exposure to cinnamon-flavored e-cigarette vapor led to significantly decreased cell viability, increased reactive oxygen species (ROS) levels, Caspase 3/7 activity, low-density lipoprotein uptake, activation of oxidative stress-related pathway, and impaired tube formation and migration, confirming endothelial dysfunction. | NGP aerosols | In vitro |
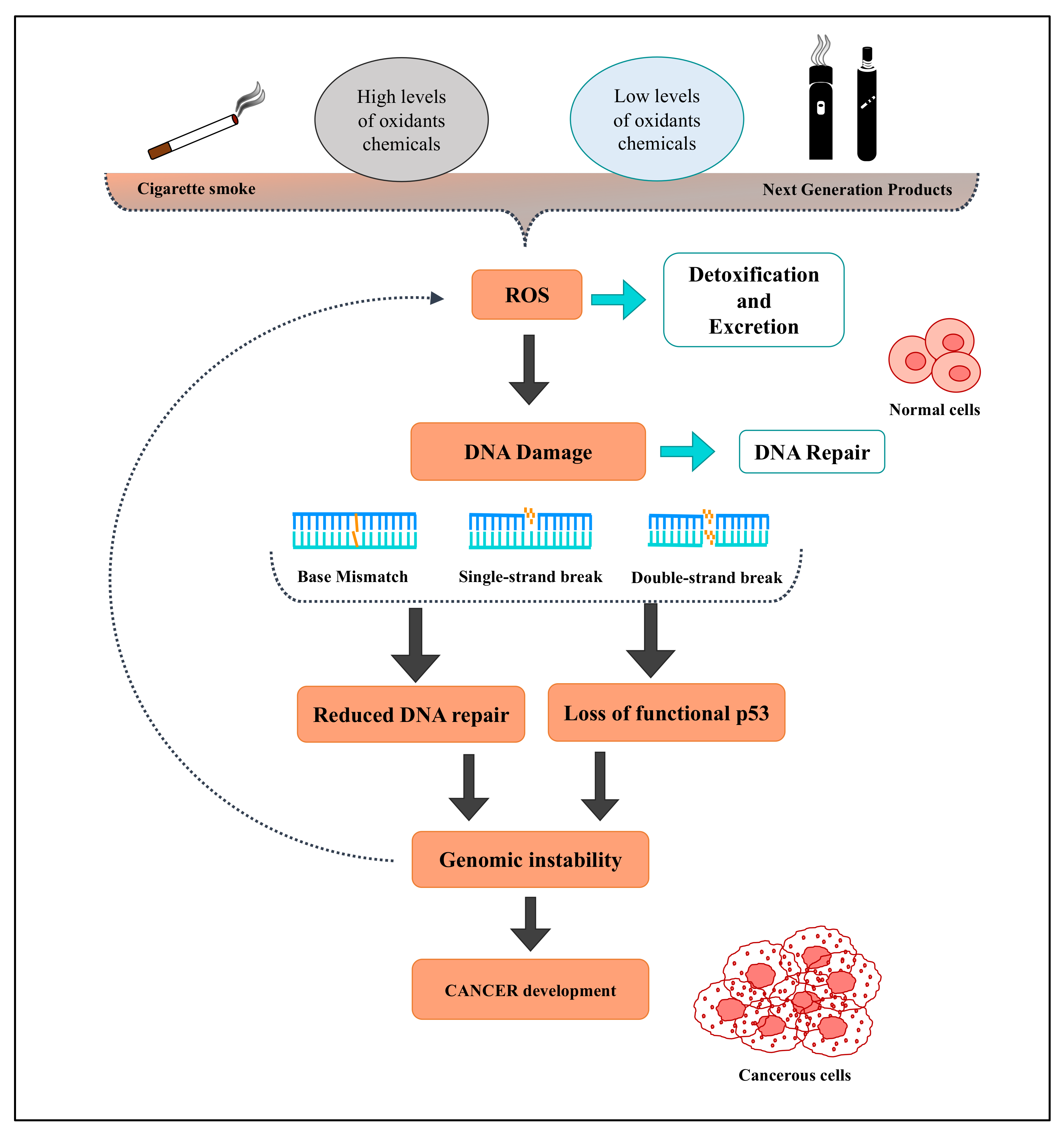
4. Oxidative Stress and Smoking/Vaping Related to Tumors
4.1. Cigarette Smoke Effects on Cancer Development
4.2. Effects of NGPs on Oxidative Stress-Related Carcinogenesis
| Author | Study Findings | Product(s) Tested | Experimental Setup |
|---|---|---|---|
| Leanderson et al. [162] | This study demonstrates the ability of cigarette smoke condensate to generate hydrogen peroxide and to hydroxylate deoxyguanosine (dG) residues in isolated DNA from calf thymus to 8-hydroxydeoxyguanosine (8-OHdG). It seems that hydroquinone and catechol may be responsible for the ability of cigarette smoke to cause 8-OHdG formation in DNA, and that the oxidative DNA damage is due to the action of hydroxyl radicals formed during the dissociation of hydrogen peroxide. Moreover, the hydrogen peroxide in cigarette smoke is generated via the autooxidation of hydroquinone and catechol. | Tobacco smoke | In vitro |
| Asami et al. [163] | Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine in human lung, obtained by surgical lobectomy or pneumonectomy. | Tobacco smoke | Human study |
| Huang et al. [164] | This study showed that cigarette combustion will produce a high concentration of ROS and they are mainly in the gaseous phase of smoke (PM2.5). These ROS come from the combustion process and not from the tobacco leaves. There is no effective means of eliminating ROS from mainstream smoke, regardless of whether a cigarette filter contains active charcoal. | Tobacco smoke | In vitro |
| Valavanidis et al. [165] | Results from this work show that aqueous cigarette tar (ACT) solutions can generate adducts with DNA nucleobases, particularly the mutagenic 8-hydroxy-2′-deoxyguanosine. Moreover, synergistic effects in the generation of HO• with environmental respirable particles (asbestos fibers, coal dust, etc.) and ambient particulate matter (PM), such as PM(10), PM(2.5), and diesel exhaust particles (DEP), were observed. It seems that the semiquinone radical system has the potential for redox recycling and oxidative action. | Tobacco smoke | In vitro |
| Cao et al. [170] | In this study, authors reported a higher level of 8-OHdG expression and secretion in airways of lung cancer patients than that of non-cancer controls; 8-OHdG expression was associated with the TNM stage. Additionally, cigarette smoke-induced oxidative DNA damage response was observed in bronchial epithelial cells in vitro and in vivo. These findings underline the importance of smoking in oxidative DNA damage response of lung cancer patients and also suggest 8-OHdG as a potential diagnostic biomarker for lung cancer. | Tobacco smoke | Human study/in vitro study |
| Advani et al. [171] | This study reports original observations on long-term (12 months) cigarette smoke effects in the H292 cell line, on miRNA expression profiling, and quantitative proteomic analysis. Authors identified 112 upregulated and 147 downregulated miRNAs (by twofold) in cigarette smoke-treated H292 cells. Moreover, they identified 303 proteins overexpressed and 112 proteins downregulated (by twofold). Moreover, 39 miRNA target pairs (proven targets) were differentially expressed in response to chronic cigarette smoke exposure. Gene ontology analysis of the target proteins revealed enrichment of proteins in biological processes driving metabolism, cell communication, and nucleic acid metabolism. Pathway analysis revealed the enrichment of phagosome maturation, antigen presentation pathway, nuclear factor erythroid 2-related factor 2-mediated oxidative stress response, and cholesterol biosynthesis pathways in cigarette smoke-exposed cells. | Tobacco smoke | In vitro |
| Goniewicz et al. [172] | E-cigarettes deliver nicotine by an aerosol, which was found to contain some toxic substances (carbonyls, volatile organic compounds, nitrosamines, and heavy metals). However, the levels of toxicants were 9–450 times lower than in cigarette smoke and were, in many cases, comparable with trace amounts found in the reference product (a medicinal nicotine inhaler; Nicorette® inhalator) and in blank samples. | NGP aerosols | In vitro |
| Takahashi et al. [173] | This study demonstrates that emission levels for selected cigarette smoke constituents, so-called “Hoffmann analytes”, and in vitro toxicity (measured by in vitro bacterial reverse mutation, micronucleus and neutral red uptake assays) of aerosol from a novel tobacco vapor product (NTV) were substantially lower than in 3R4F cigarette smoke or absent. The authors did not detect any measurable genotoxicity or cytotoxicity. | Tobacco smoke NGP aerosols | In vitro |
| Schaller et al. [174] | The chemical composition, in vitro genotoxicity, and cytotoxicity of the mainstream aerosol from the tobacco heating system 2.2 (THS2.2), a THP, were compared with those of the mainstream smoke from the 3R4F reference cigarette. The aerosol from THS2.2, compared with 3R4F smoke, showed a significant reduction of more than 90% for the majority of the analyzed harmful and potentially harmful constituents (HPHCs), while the mass median aerodynamic diameter of the aerosol remained similar, even under extreme puffing regimen. A reduction of around 90% was also observed when comparing the cytotoxicity determined by the neutral red uptake assay and the mutagenic potency in the mouse lymphoma assay. The THS2.2 aerosol was not mutagenic in the Ames assay. When using puffing regimens that were more intense than the standard Health Canada Intense (HCI) machine smoking conditions, the HPHC yields remained lower than when smoking the 3R4F reference cigarette with the HCI regimen. | NGP aerosols | In vitro |
| Jaccard et al. [175] | In this study, it is demonstrated that the aerosol from a THP, the tobacco heating system 2.2 (THS2.2), has a mean reduction of around 90% on average across a broad range of harmful and potentially harmful constituents (HPHC) compared against the levels of HPHC of cigarettes representative of selected markets, well in line with the reduction observed against 3R4F reference cigarette smoke constituents in previous studies. | NGP aerosols Tobacco smoke | In vitro |
| Saffari et al. [176] | In this study, particles generated by e-cigarettes showed a 10-fold decrease in the total emission of particulate elements compared to normal cigarette smoke. Nevertheless, specific metals (e.g., Ni and Ag) displayed a higher emission rate from e-cigarette devices (not from e-liquid). Organic species in e-cigarette aerosol showed lower emission rates compared to tobacco cigarette smoke. Moreover, polycyclic aromatic hydrocarbons (PAHs) from e-cigarette aerosol were non-detectable, while substantial emission of these species was observed from tobacco cigarettes. | NGP aerosols Tobacco smoke | In vitro |
| Ganapathy et al. [177] | This study shows that exposure of human oral and lung epithelial cells to e-cigarette aerosol extracts suppressed the cellular antioxidant defenses and led to significant DNA damage. Overall, e-cigarette aerosol extracts induced significantly less DNA damage than mainstream smoke extracts, as measured by q-PADDA. However, the levels of oxidative DNA damage were similar or slightly higher after exposure to e-cigarette aerosol compared to mainstream smoke extracts. | NGP aerosols Tobacco smoke | In vitro |
| Breheny et al. [178] | This study assessed the toxicological and biological responses of aerosols from both hybrid and heated tobacco products (HTPs) using in vitro test methods, which were outlined as part of a framework to substantiate the risk reduction potential of novel tobacco and nicotine products. All the THPs tested demonstrated significantly reduced responses in in vitro assays (evaluating mutagenicity, genotoxicity, cytotoxicity, tumor promotion, oxidative stress, and endothelial dysfunction) when compared to 3R4F tobacco cigarette smoke. | NGP aerosols Tobacco smoke | In vitro |
| Tang et al. [179] | In this study, it was found that mice exposed to e-cigarette aerosol for 54 weeks developed lung adenocarcinomas (9 of 40 mice, 22.5%) and bladder urothelial hyperplasia (23 of 40 mice, 57.5%). | NGP aerosols | In vivo, mouse model |
| Canistro et al. [180] | This study demonstrates the co-mutagenic and cancer-initiating effects of e-cigarette aerosol in a rat lung model. The authors found that e-cigarettes have a powerful booster effect on phase I carcinogen-bioactivating enzymes, including activators of polycyclic aromatic hydrocarbons (PAHs), and increase oxygen free radical production and DNA oxidation to 8-hydroxy-2′-deoxyguanosine. | NGP aerosols | In vivo, rat model |
| Cirillo et al. [181] | This study showed that the manipulation of e-cig resistance influences the carbonyl production from non-nicotine vapor and the oxidative and inflammatory status in a rat model. Sprague Dawley rats were exposed to e-cig aerosol generated under a voltage setting of 3.5 with different resistances from 1.5 to 0.25 Ohm for 28-days; the authors found a perturbation of the antioxidant and phase II enzymes and a disorganization of alveolar and bronchial epithelium in 0.25 Ohm group. | NGP aerosols | In vivo, rat model |
| Song et al. [182] | In this study, authors conducted a cross-sectional analysis of bronchoalveolar lavage and bronchial brushings from 73 subjects (42 never smokers, 15 e-cig users, and 16 smokers). Lung inflammation (by cell counts), cytokines, genome-wide gene expression, and DNA methylation were assessed. Inflammatory cell counts and cytokines from the e-cigarette users showed values intermediate between smokers and never smokers, with levels for most of the biomarkers more similar to never smokers. For differential gene expression (for smoking-related pathways) and DNA methylation, e-cigarette users were also more similar to never smokers. | NGP aerosols Tobacco smoke | In vivo |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Kemp, M.; Go, Y.M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef]
- Sacerdoti, D.; Colombrita, C.; Ghattas, M.H.; Ismaeil, E.F.; Scapagnini, G.; Bolognesi, M.; Li Volti, G.; Abraham, N.G. Heme oxygenase-1 transduction in endothelial cells causes downregulation of monocyte chemoattractant protein-1 and of genes involved in inflammation and growth. Cell. Mol. Biol. (Noisy-le-grand) 2005, 51, 363–370. [Google Scholar]
- Kushida, T.; LiVolti, G.; Goodman, A.I.; Abraham, N.G. TNF-alpha-mediated cell death is attenuated by retrovirus delivery of human heme oxygenase-1 gene into human microvessel endothelial cells. Transplant. Proc. 2002, 34, 2973–2978. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging. 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Acquaviva, R.; Lanteri, R.; Li Destri, G.; Caltabiano, R.; Vanella, L.; Lanzafame, S.; Di Cataldo, A.; Li Volti, G.; Di Giacomo, C. Beneficial effects of rutin and L-arginine coadministration in a rat model of liver ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G664–G670. [Google Scholar] [CrossRef]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef]
- Yanbaeva, D.G.; Dentener, M.A.; Creutzberg, E.C.; Wesseling, G.; Wouters, E.F. Systemic effects of smoking. Chest 2007, 131, 1557–1566. [Google Scholar] [CrossRef]
- Ientile, R.; Campisi, A.; Raciti, G.; Caccamo, D.; Curro, M.; Cannavo, G.; Li Volti, G.; Macaione, S.; Vanella, A. Cystamine inhibits transglutaminase and caspase-3 cleavage in glutamate-exposed astroglial cells. J. Neurosci. Res. 2003, 74, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Seagrave, J. Oxidative mechanisms in tobacco smoke-induced emphysema. J. Toxicol. Environ. Health A 2000, 61, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Li Volti, G. Heme oxygenase-1 nuclear translocation regulates bortezomibinduced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef]
- Zuo, L.; Otenbaker, N.P.; Rose, B.A.; Salisbury, K.S. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013, 56, 57–63. [Google Scholar] [CrossRef]
- Colombo, G.; Clerici, M.; Giustarini, D.; Portinaro, N.M.; Aldini, G.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Pathophysiology of tobacco smoke exposure: Recent insights from comparative and redox proteomics. Mass Spectrom. Rev. 2014, 33, 183–218. [Google Scholar] [CrossRef] [PubMed]
- Rom, O.; Kaisari, S.; Aizenbud, D.; Reznick, A.Z. Identification of possible cigarette smoke constituents responsible for muscle catabolism. J. Muscle Res. Cell Motil. 2012, 33, 199–208. [Google Scholar] [CrossRef]
- Rom, O.; Avezov, K.; Aizenbud, D.; Reznick, A.Z. Cigarette smoking and inflammation revisited. Respir. Physiol. Neurobiol. 2013, 187, 5–10. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2010. [Google Scholar]
- U.S. Public Health Service. 2008 PHS Guideline Update Panel, Liaisons and Staff: Treating tobacco use and dependence: 2008 update U.S. Public Health Service Clinical Practice Guideline executive summary. Respir. Care 2008, 53, 1217–1222. [Google Scholar]
- World Health Organization. WHO Report on the Global Tobacco Epidemic, 2017: Monitoring Tobacco Use and Prevention Policies; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- U.S. Department of Health and Human Services. The Health Consequences of Smoking: 50 Years of Progress. A Report of the Surgeon General; Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, USA, 2014; Volume 2021. [Google Scholar]
- Farsalinos, K. Electronic cigarettes: An aid in smoking cessation, or a new health hazard? Ther. Adv. Respir. Dis. 2018, 12, 1753465817744960. [Google Scholar] [CrossRef]
- Fagerstrom, K.O.; Bridgman, K. Tobacco harm reduction: The need for new products that can compete with cigarettes. Addict. Behav. 2014, 39, 507–511. [Google Scholar] [CrossRef] [PubMed]
- PMI. Heets and Heatsticks. Available online: https://www.pmi.com/glossary-section/glossary/heets-and-heatsticks (accessed on 5 July 2022).
- Kauneliene, V.; Meisutovic-Akhtarieva, M.; Martuzevicius, D. A review of the impacts of tobacco heating system on indoor air quality versus conventional pollution sources. Chemosphere 2018, 206, 568–578. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Etter, J.F.; Farsalinos, K.; Hajek, P.; le Houezec, J.; McRobbie, H. A critique of a World Health Organization-commissioned report and associated paper on electronic cigarettes. Addiction 2014, 109, 2128–2134. [Google Scholar] [CrossRef] [PubMed]
- Middlekauff, H.R. COUNTERPOINT: Does the Risk of Electronic Cigarettes Exceed Potential Benefits? No. Chest 2015, 148, 582–584. [Google Scholar] [CrossRef]
- Etter, J.F. Should electronic cigarettes be as freely available as tobacco? Yes. BMJ 2013, 346, f3845. [Google Scholar] [CrossRef]
- Salomone, F.; Li Volti, G.; Vitaglione, P.; Morisco, F.; Fogliano, V.; Zappala, A.; Palmigiano, A.; Garozzo, D.; Caporaso, N.; D’Argenio, G.; et al. Coffee enhances the expression of chaperones and antioxidant proteins in rats with nonalcoholic fatty liver disease. Transl. Res. 2014, 163, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Schraufnagel, D.E.; Blasi, F.; Drummond, M.B.; Lam, D.C.; Latif, E.; Rosen, M.J.; Sansores, R.; Van Zyl-Smit, R.; Forum of International Respiratory, S. Electronic cigarettes. A position statement of the forum of international respiratory societies. Am. J. Respir. Crit. Care Med. 2014, 190, 611–618. [Google Scholar] [CrossRef]
- Chapman, S. Should electronic cigarettes be as freely available as tobacco cigarettes? No. BMJ 2013, 346, f3840. [Google Scholar] [CrossRef]
- Avdalovic, M.V.; Murin, S. POINT: Does the Risk of Electronic Cigarettes Exceed Potential Benefits? Yes. Chest 2015, 148, 580–582. [Google Scholar] [CrossRef]
- Ferkol, T.W.; Farber, H.J.; La Grutta, S.; Leone, F.T.; Marshall, H.M.; Neptune, E.; Pisinger, C.; Vanker, A.; Wisotzky, M.; Zabert, G.E.; et al. Electronic cigarette use in youths: A position statement of the Forum of International Respiratory Societies. Eur. Respir. J. 2018, 51, 1800278. [Google Scholar] [CrossRef]
- Crystal, R.G. Oxidants and respiratory tract epithelial injury: Pathogenesis and strategies for therapeutic intervention. Am. J. Med. 1991, 91, 39S–44S. [Google Scholar] [CrossRef]
- Holguin, F. Oxidative stress in airway diseases. Ann. Am. Thorac. Soc. 2013, 10, S150–S157. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Crapo, J.D.; Raivio, K.O. Generation and disposal of reactive oxygen metabolites in the lung. Lab. Invest. 1995, 73, 3–19. [Google Scholar] [PubMed]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Asthma: Implications for Mitochondria-Targeted Antioxidant Therapeutics. Pharmaceuticals (Basel) 2011, 4, 429–456. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit. Care Med. 2003, 167, 1600–1619. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Role of free radicals and catalytic metal ions in human disease: An overview. Methods Enzymol. 1990, 186, 1–85. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef]
- Dweik, R.A. Nitric oxide, hypoxia, and superoxide: The good, the bad, and the ugly! Thorax 2005, 60, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Spooner, G.; Ffoulkes-Jones, C.; Calvez, R. Cigarette smoke triggers macrophage adhesion and activation: Role of lipid peroxidation products and scavenger receptor. Free Radic. Biol. Med. 2003, 35, 697–710. [Google Scholar] [CrossRef]
- Plaschke, P.P.; Janson, C.; Norrman, E.; Bjornsson, E.; Ellbjar, S.; Jarvholm, B. Onset and remission of allergic rhinitis and asthma and the relationship with atopic sensitization and smoking. Am. J. Respir. Crit. Care Med. 2000, 162, 920–924. [Google Scholar] [CrossRef]
- Rasmussen, F.; Siersted, H.C.; Lambrechtsen, J.; Hansen, H.S.; Hansen, N.C. Impact of airway lability, atopy, and tobacco smoking on the development of asthma-like symptoms in asymptomatic teenagers. Chest 2000, 117, 1330–1335. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, S.H.; Tak, Y.J.; Jee, Y.K.; Lee, B.J.; Kim, S.H.; Park, H.W.; Jung, J.W.; Bahn, J.W.; Chang, Y.S.; et al. High prevalence of current asthma and active smoking effect among the elderly. Clin. Exp. Allergy 2002, 32, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Polosa, R.; Russo, C.; Caponnetto, P.; Bertino, G.; Sarva, M.; Antic, T.; Mancuso, S.; Al-Delaimy, W.K. Greater severity of new onset asthma in allergic subjects who smoke: A 10-year longitudinal study. Respir. Res. 2011, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Kleniewska, P.; Pawliczak, R. The participation of oxidative stress in the pathogenesis of bronchial asthma. Biomed Pharm. 2017, 94, 100–108. [Google Scholar] [CrossRef]
- Kettle, A.J.; Turner, R.; Gangell, C.L.; Harwood, D.T.; Khalilova, I.S.; Chapman, A.L.; Winterbourn, C.C.; Sly, P.D.; Arest, C.F. Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur. Respir. J. 2014, 44, 122–129. [Google Scholar] [CrossRef]
- Ziady, A.G.; Hansen, J. Redox balance in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 113–123. [Google Scholar] [CrossRef]
- Wood, L.G.; Garg, M.L.; Simpson, J.L.; Mori, T.A.; Croft, K.D.; Wark, P.A.; Gibson, P.G. Induced sputum 8-isoprostane concentrations in inflammatory airway diseases. Am. J. Respir. Crit. Care Med. 2005, 171, 426–430. [Google Scholar] [CrossRef]
- Bargagli, E.; Olivieri, C.; Bennett, D.; Prasse, A.; Muller-Quernheim, J.; Rottoli, P. Oxidative stress in the pathogenesis of diffuse lung diseases: A review. Respir. Med. 2009, 103, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Faner, R.; Rojas, M.; Macnee, W.; Agusti, A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, D.J.R.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants (Basel) 2017, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Page, C.; O’Shaughnessy, B.; Barnes, P. Pathogenesis of COPD and Asthma. Handb. Exp. Pharmacol. 2017, 237, 1–21. [Google Scholar] [CrossRef]
- MacNee, W. Pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 258–266, discussion 290–251. [Google Scholar] [CrossRef]
- Corradi, M.; Pignatti, P.; Manini, P.; Andreoli, R.; Goldoni, M.; Poppa, M.; Moscato, G.; Balbi, B.; Mutti, A. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur. Respir. J. 2004, 24, 1011–1017. [Google Scholar] [CrossRef]
- Singh, S.; Verma, S.K.; Kumar, S.; Ahmad, M.K.; Nischal, A.; Singh, S.K.; Dixit, R.K. Evaluation of Oxidative Stress and Antioxidant Status in Chronic Obstructive Pulmonary Disease. Scand. J. Immunol. 2017, 85, 130–137. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Ilumets, H.; Myllarniemi, M.; Sovijarvi, A.; Rytila, P. 8-Isoprostane as a marker of oxidative stress in nonsymptomatic cigarette smokers and COPD. Eur. Respir. J. 2007, 29, 51–55. [Google Scholar] [CrossRef]
- Brindicci, C.; Ito, K.; Torre, O.; Barnes, P.J.; Kharitonov, S.A. Effects of aminoguanidine, an inhibitor of inducible nitric oxide synthase, on nitric oxide production and its metabolites in healthy control subjects, healthy smokers, and COPD patients. Chest 2009, 135, 353–367. [Google Scholar] [CrossRef]
- Bernardo, I.; Bozinovski, S.; Vlahos, R. Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol. Ther. 2015, 155, 60–79. [Google Scholar] [CrossRef]
- Vaguliene, N.; Zemaitis, M.; Lavinskiene, S.; Miliauskas, S.; Sakalauskas, R. Local and systemic neutrophilic inflammation in patients with lung cancer and chronic obstructive pulmonary disease. BMC Immunol. 2013, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Mediators of chronic obstructive pulmonary disease. Pharmacol. Rev. 2004, 56, 515–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Hao, B.; Ma, A.; He, J.; Liu, X.; Chen, J. The Expression of NOX4 in Smooth Muscles of Small Airway Correlates with the Disease Severity of COPD. Biomed. Res. Int. 2016, 2016, 2891810. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Caramori, G.; Ito, K.; Capelli, A.; Brun, P.; Abatangelo, G.; Papi, A.; Chung, K.F.; Adcock, I.; Barnes, P.J.; et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2005, 116, 1028–1035. [Google Scholar] [CrossRef]
- Ichinose, M.; Sugiura, H.; Yamagata, S.; Koarai, A.; Shirato, K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 2000, 162, 701–706. [Google Scholar] [CrossRef]
- Domej, W.; Oettl, K.; Renner, W. Oxidative stress and free radicals in COPD--implications and relevance for treatment. Int. J. Chron. Obstruct. Pulmon. Dis. 2014, 9, 1207–1224. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, X.; Hu, D. Mitochondrial alterations during oxidative stress in chronic obstructive pulmonary disease. Int J Chron. Obstruct. Pulmon. Dis. 2017, 12, 1153–1162. [Google Scholar] [CrossRef]
- Cantin, A.M. Cystic Fibrosis Transmembrane Conductance Regulator. Implications in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S2), S150–S155. [Google Scholar] [CrossRef]
- Polosa, R.; Knoke, J.D.; Russo, C.; Piccillo, G.; Caponnetto, P.; Sarva, M.; Proietti, L.; Al-Delaimy, W.K. Cigarette smoking is associated with a greater risk of incident asthma in allergic rhinitis. J. Allergy Clin. Immunol. 2008, 121, 1428–1434. [Google Scholar] [CrossRef]
- Sorrenti, V.; Mazza, F.; Campisi, A.; Vanella, L.; Li Volti, G.; Di Giacomo, C. High glucose-mediated imbalance of nitric oxide synthase and dimethylarginine dimethylaminohydrolase expression in endothelial cells. Curr. Neurovasc. Res. 2006, 3, 49–54. [Google Scholar] [CrossRef]
- Lange, P.; Parner, J.; Vestbo, J.; Schnohr, P.; Jensen, G. A 15-year follow-up study of ventilatory function in adults with asthma. N. Engl. J. Med. 1998, 339, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Haselkorn, T.; Borish, L.; Rasouliyan, L.; Chipps, B.E.; Wenzel, S.E. Risk factors associated with persistent airflow limitation in severe or difficult-to-treat asthma: Insights from the TENOR study. Chest 2007, 132, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, G.A.; Vollema, E.M.; Weersink, E.J.; Reinartz, S.M.; de Nijs, S.B.; Bel, E.H. Predictors for the development of progressive severity in new-onset adult asthma. J. Allergy Clin. Immunol. 2014, 134, 1051–1056-e1052. [Google Scholar] [CrossRef]
- Emma, R.; Bansal, A.T.; Kolmert, J.; Wheelock, C.E.; Dahlen, S.E.; Loza, M.J.; De Meulder, B.; Lefaudeux, D.; Auffray, C.; Dahlen, B.; et al. Enhanced oxidative stress in smoking and ex-smoking severe asthma in the U-BIOPRED cohort. PLoS ONE 2018, 13, e0203874. [Google Scholar] [CrossRef]
- Al Obaidi, A.H.; Al Samarai, A.M. Biochemical markers as a response guide for steroid therapy in asthma. J. Asthma. 2008, 45, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Pavlidis, S.; Ng Kee Kwong, F.; Hoda, U.; Rossios, C.; Sun, K.; Loza, M.; Baribaud, F.; Chanez, P.; Fowler, S.J.; et al. Sputum proteomics and airway cell transcripts of current and ex-smokers with severe asthma in U-BIOPRED: An exploratory analysis. Eur. Respir. J. 2018, 51, 1702173. [Google Scholar] [CrossRef]
- Carpagnano, G.E.; Lacedonia, D.; Malerba, M.; Palmiotti, G.A.; Cotugno, G.; Carone, M.; Foschino-Barbaro, M.P. Analysis of mitochondrial DNA alteration in new phenotype ACOS. BMC Pulm. Med. 2016, 16, 31. [Google Scholar] [CrossRef]
- Lerner, C.A.; Sundar, I.K.; Yao, H.; Gerloff, J.; Ossip, D.J.; McIntosh, S.; Robinson, R.; Rahman, I. Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS ONE 2015, 10, e0116732. [Google Scholar] [CrossRef]
- Sussan, T.E.; Gajghate, S.; Thimmulappa, R.K.; Ma, J.; Kim, J.H.; Sudini, K.; Consolini, N.; Cormier, S.A.; Lomnicki, S.; Hasan, F.; et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS ONE 2015, 10, e0116861. [Google Scholar] [CrossRef]
- CORESTA. CORESTA recommended method no 81. Routine analytical machine for e-cigarette aerosol generation and collection–definitions and standard conditions. 2015. Available online: https://www.coresta.org/sites/default/files/technical_documents/main/CRM_81.pdf (accessed on 5 July 2022).
- Shivalingappa, P.C.; Hole, R.; Westphal, C.V.; Vij, N. Airway Exposure to E-Cigarette Vapors Impairs Autophagy and Induces Aggresome Formation. Antioxid. Redox. Signal. 2016, 24, 186–204. [Google Scholar] [CrossRef]
- Scheffler, S.; Dieken, H.; Krischenowski, O.; Forster, C.; Branscheid, D.; Aufderheide, M. Evaluation of E-cigarette liquid vapor and mainstream cigarette smoke after direct exposure of primary human bronchial epithelial cells. Int. J. Environ. Res. Public Health 2015, 12, 3915–3925. [Google Scholar] [CrossRef] [PubMed]
- ISO/TR 19478-2:2015; ISO and Health Canada Intense Smoking Parameters—Part 2: Examination of Factors Contributing to Variability in the Routine Measurement of TPM, Water and NFDPM Smoke Yields of Cigarettes. ISO: Geneva, Switzerland, 2015.
- Taylor, M.; Carr, T.; Oke, O.; Jaunky, T.; Breheny, D.; Lowe, F.; Gaca, M. E-cigarette aerosols induce lower oxidative stress in vitro when compared to tobacco smoke. Toxicol. Mech. Methods 2016, 26, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Malinska, D.; Szymanski, J.; Patalas-Krawczyk, P.; Michalska, B.; Wojtala, A.; Prill, M.; Partyka, M.; Drabik, K.; Walczak, J.; Sewer, A.; et al. Assessment of mitochondrial function following short- and long-term exposure of human bronchial epithelial cells to total particulate matter from a candidate modified-risk tobacco product and reference cigarettes. Food Chem. Toxicol. 2018, 115, 1–12. [Google Scholar] [CrossRef]
- Moses, E.; Wang, T.; Corbett, S.; Jackson, G.R.; Drizik, E.; Perdomo, C.; Perdomo, C.; Kleerup, E.; Brooks, D.; O’Connor, G.; et al. Molecular Impact of Electronic Cigarette Aerosol Exposure in Human Bronchial Epithelium. Toxicol. Sci. 2017, 155, 248–257. [Google Scholar] [CrossRef]
- Jabba, S.V.; Diaz, A.N.; Erythropel, H.C.; Zimmerman, J.B.; Jordt, S.E. Chemical Adducts of Reactive Flavor Aldehydes Formed in E-Cigarette Liquids Are Cytotoxic and Inhibit Mitochondrial Function in Respiratory Epithelial Cells. Nicotine Tob. Res. 2020, 22, S25–S34. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef]
- Busse, R.; Luckhoff, A.; Bassenge, E. Endothelium-derived relaxant factor inhibits platelet activation. Naunyn Schmiedebergs Arch. Pharmacol. 1987, 336, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Alheid, U.; Frolich, J.C.; Forstermann, U. Endothelium-derived relaxing factor from cultured human endothelial cells inhibits aggregation of human platelets. Thromb. Res. 1987, 47, 561–571. [Google Scholar] [CrossRef]
- Zeiher, A.M.; Fisslthaler, B.; Schray-Utz, B.; Busse, R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ. Res. 1995, 76, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Investig. 1989, 83, 1774–1777. [Google Scholar] [CrossRef]
- Nakaki, T.; Nakayama, M.; Kato, R. Inhibition by nitric oxide and nitric oxide-producing vasodilators of DNA synthesis in vascular smooth muscle cells. Eur. J. Pharmacol. 1990, 189, 347–353. [Google Scholar] [CrossRef]
- Hogan, M.; Cerami, A.; Bucala, R. Advanced glycosylation endproducts block the antiproliferative effect of nitric oxide. Role in the vascular and renal complications of diabetes mellitus. J. Clin. Investig. 1992, 90, 1110–1115. [Google Scholar] [CrossRef]
- Forstermann, U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 338–349. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Rodriguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef]
- Sydow, K.; Munzel, T. ADMA and oxidative stress. Atheroscler. Suppl. 2003, 4, 41–51. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Sun, X.; Kumar, S.; Sharma, S.; Aggarwal, S.; Lu, Q.; Gross, C.; Rafikova, O.; Lee, S.G.; Dasarathy, S.; Hou, Y.; et al. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappala, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollnau, H.; Wendt, M.; Szocs, K.; Lassegue, B.; Schulz, E.; Oelze, M.; Li, H.; Bodenschatz, M.; August, M.; Kleschyov, A.L.; et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002, 90, E58–E65. [Google Scholar] [CrossRef] [PubMed]
- Nickenig, G.; Harrison, D.G. The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part I: Oxidative stress and atherogenesis. Circulation 2002, 105, 393–396. [Google Scholar] [CrossRef]
- Rahman, M.M.; Laher, I. Structural and functional alteration of blood vessels caused by cigarette smoking: An overview of molecular mechanisms. Curr. Vasc. Pharmacol. 2007, 5, 276–292. [Google Scholar] [CrossRef]
- Barua, R.S.; Ambrose, J.A.; Srivastava, S.; DeVoe, M.C.; Eales-Reynolds, L.J. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: An in vitro demonstration in human coronary artery endothelial cells. Circulation 2003, 107, 2342–2347. [Google Scholar] [CrossRef]
- Csiszar, A.; Podlutsky, A.; Wolin, M.S.; Losonczy, G.; Pacher, P.; Ungvari, Z. Oxidative stress and accelerated vascular aging: Implications for cigarette smoking. Front. Biosci. Landmark 2009, 14, 3128–3144. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Adams, M.R.; Clarkson, P.; Robinson, J.; McCredie, R.; Donald, A.; Deanfield, J.E. Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. N. Engl. J. Med. 1996, 334, 150–154. [Google Scholar] [CrossRef]
- Zeiher, A.M.; Schachinger, V.; Minners, J. Long-term cigarette smoking impairs endothelium-dependent coronary arterial vasodilator function. Circulation 1995, 92, 1094–1100. [Google Scholar] [CrossRef]
- Csordas, A.; Bernhard, D. The biology behind the atherothrombotic effects of cigarette smoke. Nat. Rev. Cardiol. 2013, 10, 219–230. [Google Scholar] [CrossRef]
- Raveendran, M.; Wang, J.; Senthil, D.; Wang, J.; Utama, B.; Shen, Y.; Dudley, D.; Zhang, Y.; Wang, X.L. Endogenous nitric oxide activation protects against cigarette smoking induced apoptosis in endothelial cells. FEBS Lett. 2005, 579, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, E.A.; DeMaster, E.G.; Tian, R.X.; Raij, L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1031–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayyali, U.S.; Budhiraja, R.; Pennella, C.M.; Cooray, S.; Lanzillo, J.J.; Chalkley, R.; Hassoun, P.M. Upregulation of xanthine oxidase by tobacco smoke condensate in pulmonary endothelial cells. Toxicol. Appl. Pharmacol. 2003, 188, 59–68. [Google Scholar] [CrossRef]
- Talukder, M.A.; Johnson, W.M.; Varadharaj, S.; Lian, J.; Kearns, P.N.; El-Mahdy, M.A.; Liu, X.; Zweier, J.L. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H388–H396. [Google Scholar] [CrossRef]
- Lander, H.M.; Milbank, A.J.; Tauras, J.M.; Hajjar, D.P.; Hempstead, B.L.; Schwartz, G.D.; Kraemer, R.T.; Mirza, U.A.; Chait, B.T.; Burk, S.C.; et al. Redox regulation of cell signalling. Nature 1996, 381, 380–381. [Google Scholar] [CrossRef]
- Flohe, L.; Brigelius-Flohe, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox regulation of NF-kappa B activation. Free Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- Van den Berg, R.; Haenen, G.R.; van den Berg, H.; Bast, A. Nuclear factor-kappaB activation is higher in peripheral blood mononuclear cells of male smokers. Environ. Toxicol. Pharmacol. 2001, 9, 147–151. [Google Scholar] [CrossRef]
- Orosz, Z.; Csiszar, A.; Labinskyy, N.; Smith, K.; Kaminski, P.M.; Ferdinandy, P.; Wolin, M.S.; Rivera, A.; Ungvari, Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: Role of NAD(P)H oxidase activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H130–H139. [Google Scholar] [CrossRef]
- Csiszar, A.; Smith, K.E.; Koller, A.; Kaley, G.; Edwards, J.G.; Ungvari, Z. Regulation of bone morphogenetic protein-2 expression in endothelial cells: Role of nuclear factor-kappaB activation by tumor necrosis factor-alpha, H2O2, and high intravascular pressure. Circulation 2005, 111, 2364–2372. [Google Scholar] [CrossRef]
- Wissler, R.W. New insights into the pathogenesis of atherosclerosis as revealed by PDAY. Pathobiological Determinants of Atherosclerosis in Youth. Atherosclerosis 1994, 108, S3–S20. [Google Scholar] [CrossRef]
- Miyaura, S.; Eguchi, H.; Johnston, J.M. Effect of a cigarette smoke extract on the metabolism of the proinflammatory autacoid, platelet-activating factor. Circ. Res. 1992, 70, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Satoh, K.; Yoshida, H.; Kawamura, Y.; Hiramoto, M.; Takamatsu, S. Effect of cigarette smoking on the levels of platelet-activating factor-like lipid(s) in plasma lipoproteins. Atherosclerosis 1991, 87, 47–55. [Google Scholar] [CrossRef]
- Perlstein, T.S.; Lee, R.T. Smoking, metalloproteinases, and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 250–256. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 2007, 17, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Falk, E.; Badimon, J.J.; Fernandez-Ortiz, A.; Mailhac, A.; Villareal-Levy, G.; Fallon, J.T.; Regnstrom, J.; Fuster, V. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation 1995, 92, 1565–1569. [Google Scholar]
- Kangavari, S.; Matetzky, S.; Shah, P.K.; Yano, J.; Chyu, K.Y.; Fishbein, M.C.; Cercek, B. Smoking increases inflammation and metalloproteinase expression in human carotid atherosclerotic plaques. J. Cardiovasc. Pharmacol. Ther. 2004, 9, 291–298. [Google Scholar] [CrossRef]
- Huang, B.; Svensson, P.; Arnlov, J.; Sundstrom, J.; Lind, L.; Ingelsson, E. Effects of cigarette smoking on cardiovascular-related protein profiles in two community-based cohort studies. Atherosclerosis 2016, 254, 52–58. [Google Scholar] [CrossRef]
- Teasdale, J.E.; Newby, A.C.; Timpson, N.J.; Munafo, M.R.; White, S.J. Cigarette smoke but not electronic cigarette aerosol activates a stress response in human coronary artery endothelial cells in culture. Drug. Alcohol. Depend. 2016, 163, 256–260. [Google Scholar] [CrossRef]
- Anderson, C.; Majeste, A.; Hanus, J.; Wang, S. E-Cigarette Aerosol Exposure Induces Reactive Oxygen Species, DNA Damage, and Cell Death in Vascular Endothelial Cells. Toxicol. Sci. 2016, 154, 332–340. [Google Scholar] [CrossRef]
- Carnevale, R.; Sciarretta, S.; Violi, F.; Nocella, C.; Loffredo, L.; Perri, L.; Peruzzi, M.; Marullo, A.G.; De Falco, E.; Chimenti, I.; et al. Acute Impact of Tobacco vs Electronic Cigarette Smoking on Oxidative Stress and Vascular Function. Chest 2016, 150, 606–612. [Google Scholar] [CrossRef]
- Chaumont, M.; de Becker, B.; Zaher, W.; Culie, A.; Deprez, G.; Melot, C.; Reye, F.; Van Antwerpen, P.; Delporte, C.; Debbas, N.; et al. Differential Effects of E-Cigarette on Microvascular Endothelial Function, Arterial Stiffness and Oxidative Stress: A Randomized Crossover Trial. Sci. Rep. 2018, 8, 10378. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Vlastos, D.; Kourea, K.; Kostelli, G.; Varoudi, M.; Pavlidis, G.; Efentakis, P.; Triantafyllidi, H.; Parissis, J.; Andreadou, I.; et al. Electronic Cigarette Smoking Increases Arterial Stiffness and Oxidative Stress to a Lesser Extent Than a Single Conventional Cigarette: An Acute and Chronic Study. Circulation 2018, 137, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Derout, J.; Hasan, K.M.; Shao, X.M.; Jordan, M.C.; Sims, C.; Lee, D.L.; Sinha, S.; Simmons, Z.; Mtume, N.; Liu, Y.; et al. Chronic intermittent electronic cigarette exposure induces cardiac dysfunction and atherosclerosis in apolipoprotein-E knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H445–H459. [Google Scholar] [CrossRef]
- Farsalinos, K.E.; Kistler, K.A.; Pennington, A.; Spyrou, A.; Kouretas, D.; Gillman, G. Aldehyde levels in e-cigarette aerosol: Findings from a replication study and from use of a new-generation device. Food Chem. Toxicol. 2018, 111, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Kuntic, M.; Oelze, M.; Steven, S.; Kroller-Schon, S.; Stamm, P.; Kalinovic, S.; Frenis, K.; Vujacic-Mirski, K.; Bayo Jimenez, M.T.; Kvandova, M.; et al. Short-term e-cigarette vapour exposure causes vascular oxidative stress and dysfunction: Evidence for a close connection to brain damage and a key role of the phagocytic NADPH oxidase (NOX-2). Eur. Heart J. 2020, 41, 2472–2483. [Google Scholar] [CrossRef]
- Lee, W.H.; Ong, S.G.; Zhou, Y.; Tian, L.; Bae, H.R.; Baker, N.; Whitlatch, A.; Mohammadi, L.; Guo, H.; Nadeau, K.C.; et al. Modeling Cardiovascular Risks of E-Cigarettes With Human-Induced Pluripotent Stem Cell-Derived Endothelial Cells. J. Am. Coll. Cardiol. 2019, 73, 2722–2737. [Google Scholar] [CrossRef]
- Benz, C.C.; Yau, C. Ageing, oxidative stress and cancer: Paradigms in parallax. Nat. Rev. Cancer 2008, 8, 875–879. [Google Scholar] [CrossRef]
- Oberley, L.W. Free radicals and diabetes. Free Radic. Biol. Med. 1988, 5, 113–124. [Google Scholar] [CrossRef]
- Okamoto, K.; Toyokuni, S.; Uchida, K.; Ogawa, O.; Takenewa, J.; Kakehi, Y.; Kinoshita, H.; Hattori-Nakakuki, Y.; Hiai, H.; Yoshida, O. Formation of 8-hydroxy-2’-deoxyguanosine and 4-hydroxy-2-nonenal-modified proteins in human renal-cell carcinoma. Int. J. Cancer 1994, 58, 825–829. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, P.; Anathy, V.; Burch, P.M.; Weirather, K.; Lambeth, J.D.; Heintz, N.H. Redox-dependent expression of cyclin D1 and cell proliferation by Nox1 in mouse lung epithelial cells. Antioxid. Redox. Signal. 2006, 8, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug. Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Hecht, S.S. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002, 3, 461–469. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 207. [Google Scholar] [CrossRef]
- Xu, D.; Rovira, I.I.; Finkel, T. Oxidants painting the cysteine chapel: Redox regulation of PTPs. Dev. Cell 2002, 2, 251–252. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- WHO. Tobacco or health: A global status report. 1997. Available online: https://apps.who.int/iris/handle/10665/41922 (accessed on 5 July 2022).
- Levin, M.L.; Goldstein, H.; Gerhardt, P.R. Cancer and tobacco smoking; a preliminary report. J. Am. Med. Assoc. 1950, 143, 336–338. [Google Scholar] [CrossRef]
- Doll, R.; Hill, A.B. Smoking and carcinoma of the lung; preliminary report. Br. Med. J. 1950, 2, 739–748. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Tobacco Smoking; 92 832 1238 X; WHO: Geneva, Switzerland, 1986. [Google Scholar]
- International Agency for Research on Cancer (IARC). Tobacco smoke and involuntary smoking; WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Gandini, S.; Botteri, E.; Iodice, S.; Boniol, M.; Lowenfels, A.B.; Maisonneuve, P.; Boyle, P. Tobacco smoking and cancer: A meta-analysis. Int. J. Cancer 2008, 122, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Leanderson, P.; Tagesson, C. Cigarette smoke-induced DNA-damage: Role of hydroquinone and catechol in the formation of the oxidative DNA-adduct, 8-hydroxydeoxyguanosine. Chem. Biol. Interact. 1990, 75, 71–81. [Google Scholar] [CrossRef]
- Asami, S.; Manabe, H.; Miyake, J.; Tsurudome, Y.; Hirano, T.; Yamaguchi, R.; Itoh, H.; Kasai, H. Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis 1997, 18, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.F.; Lin, W.L.; Ma, Y.C. A study of reactive oxygen species in mainstream of cigarette. Indoor Air 2005, 15, 135–140. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Lowe, F.J.; Luettich, K.; Gregg, E.O. Lung cancer biomarkers for the assessment of modified risk tobacco products: An oxidative stress perspective. Biomarkers 2013, 18, 183–195. [Google Scholar] [CrossRef]
- Ichinose, T.; Yajima, Y.; Nagashima, M.; Takenoshita, S.; Nagamachi, Y.; Sagai, M. Lung carcinogenesis and formation of 8-hydroxy-deoxyguanosine in mice by diesel exhaust particles. Carcinogenesis 1997, 18, 185–192. [Google Scholar] [CrossRef]
- Kim, J.Y.; Mukherjee, S.; Ngo, L.C.; Christiani, D.C. Urinary 8-hydroxy-2’-deoxyguanosine as a biomarker of oxidative DNA damage in workers exposed to fine particulates. Environ. Health Perspect 2004, 112, 666–671. [Google Scholar] [CrossRef]
- Cao, C.; Lai, T.; Li, M.; Zhou, H.; Lv, D.; Deng, Z.; Ying, S.; Chen, Z.; Li, W.; Shen, H. Smoking-promoted oxidative DNA damage response is highly correlated to lung carcinogenesis. Oncotarget 2016, 7, 18919–18926. [Google Scholar] [CrossRef] [PubMed]
- Advani, J.; Subbannayya, Y.; Patel, K.; Khan, A.A.; Patil, A.H.; Jain, A.P.; Solanki, H.S.; Radhakrishnan, A.; Pinto, S.M.; Sahasrabuddhe, N.A.; et al. Long-Term Cigarette Smoke Exposure and Changes in MiRNA Expression and Proteome in Non-Small-Cell Lung Cancer. OMICS 2017, 21, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Goniewicz, M.L.; Knysak, J.; Gawron, M.; Kosmider, L.; Sobczak, A.; Kurek, J.; Prokopowicz, A.; Jablonska-Czapla, M.; Rosik-Dulewska, C.; Havel, C.; et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob. Control 2014, 23, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Kanemaru, Y.; Fukushima, T.; Eguchi, K.; Yoshida, S.; Miller-Holt, J.; Jones, I. Chemical analysis and in vitro toxicological evaluation of aerosol from a novel tobacco vapor product: A comparison with cigarette smoke. Regul. Toxicol. Pharmacol. 2018, 92, 94–103. [Google Scholar] [CrossRef]
- Schaller, J.P.; Keller, D.; Poget, L.; Pratte, P.; Kaelin, E.; McHugh, D.; Cudazzo, G.; Smart, D.; Tricker, A.R.; Gautier, L.; et al. Evaluation of the Tobacco Heating System 2.2. Part 2: Chemical composition, genotoxicity, cytotoxicity, and physical properties of the aerosol. Regul. Toxicol. Pharmacol. 2016, 81 (Suppl. S2), S27–S47. [Google Scholar] [CrossRef]
- Jaccard, G.; Tafin Djoko, D.; Moennikes, O.; Jeannet, C.; Kondylis, A.; Belushkin, M. Comparative assessment of HPHC yields in the Tobacco Heating System THS2.2 and commercial cigarettes. Regul. Toxicol. Pharmacol. 2017, 90, 1–8. [Google Scholar] [CrossRef]
- Saffari, A.; Daher, N.; Ruprecht, A.; De Marco, C.; Pozzi, P.; Boffi, R.; Hamad, S.H.; Shafer, M.M.; Schauer, J.J.; Westerdahl, D.; et al. Particulate metals and organic compounds from electronic and tobacco-containing cigarettes: Comparison of emission rates and secondhand exposure. Environ. Sci. Process Impacts 2014, 16, 2259–2267. [Google Scholar] [CrossRef]
- Ganapathy, V.; Manyanga, J.; Brame, L.; McGuire, D.; Sadhasivam, B.; Floyd, E.; Rubenstein, D.A.; Ramachandran, I.; Wagener, T.; Queimado, L. Electronic cigarette aerosols suppress cellular antioxidant defenses and induce significant oxidative DNA damage. PLoS ONE 2017, 12, e0177780. [Google Scholar] [CrossRef]
- Breheny, D.; Adamson, J.; Azzopardi, D.; Baxter, A.; Bishop, E.; Carr, T.; Crooks, I.; Hewitt, K.; Jaunky, T.; Larard, S.; et al. A novel hybrid tobacco product that delivers a tobacco flavour note with vapour aerosol (Part 2): In vitro biological assessment and comparison with different tobacco-heating products. Food Chem. Toxicol. 2017, 106, 533–546. [Google Scholar] [CrossRef]
- Tang, M.S.; Wu, X.R.; Lee, H.W.; Xia, Y.; Deng, F.M.; Moreira, A.L.; Chen, L.C.; Huang, W.C.; Lepor, H. Electronic-cigarette smoke induces lung adenocarcinoma and bladder urothelial hyperplasia in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 21727–21731. [Google Scholar] [CrossRef]
- Canistro, D.; Vivarelli, F.; Cirillo, S.; Babot Marquillas, C.; Buschini, A.; Lazzaretti, M.; Marchi, L.; Cardenia, V.; Rodriguez-Estrada, M.T.; Lodovici, M.; et al. E-cigarettes induce toxicological effects that can raise the cancer risk. Sci. Rep. 2017, 7, 2028. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, S.; Vivarelli, F.; Turrini, E.; Fimognari, C.; Burattini, S.; Falcieri, E.; Rocchi, M.B.L.; Cardenia, V.; Rodriguez-Estrada, M.T.; Paolini, M.; et al. The customizable e-cigarette resistance influences toxicological outcomes: Lung degeneration, inflammation and oxidative stress-induced in a rat model. Toxicol. Sci. 2019, 172, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Song, M.A.; Freudenheim, J.L.; Brasky, T.M.; Mathe, E.A.; McElroy, J.P.; Nickerson, Q.A.; Reisinger, S.A.; Smiraglia, D.J.; Weng, D.Y.; Ying, K.L.; et al. Biomarkers of Exposure and Effect in the Lungs of Smokers, Nonsmokers, and Electronic Cigarette Users. Cancer Epidemiol. Biomark. Prev. 2020, 29, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emma, R.; Caruso, M.; Campagna, D.; Pulvirenti, R.; Li Volti, G. The Impact of Tobacco Cigarettes, Vaping Products and Tobacco Heating Products on Oxidative Stress. Antioxidants 2022, 11, 1829. https://doi.org/10.3390/antiox11091829
Emma R, Caruso M, Campagna D, Pulvirenti R, Li Volti G. The Impact of Tobacco Cigarettes, Vaping Products and Tobacco Heating Products on Oxidative Stress. Antioxidants. 2022; 11(9):1829. https://doi.org/10.3390/antiox11091829
Chicago/Turabian StyleEmma, Rosalia, Massimo Caruso, Davide Campagna, Roberta Pulvirenti, and Giovanni Li Volti. 2022. "The Impact of Tobacco Cigarettes, Vaping Products and Tobacco Heating Products on Oxidative Stress" Antioxidants 11, no. 9: 1829. https://doi.org/10.3390/antiox11091829