
Celastrol: A Promising Agent Fighting against Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Celastrol against Metabolic Disorders
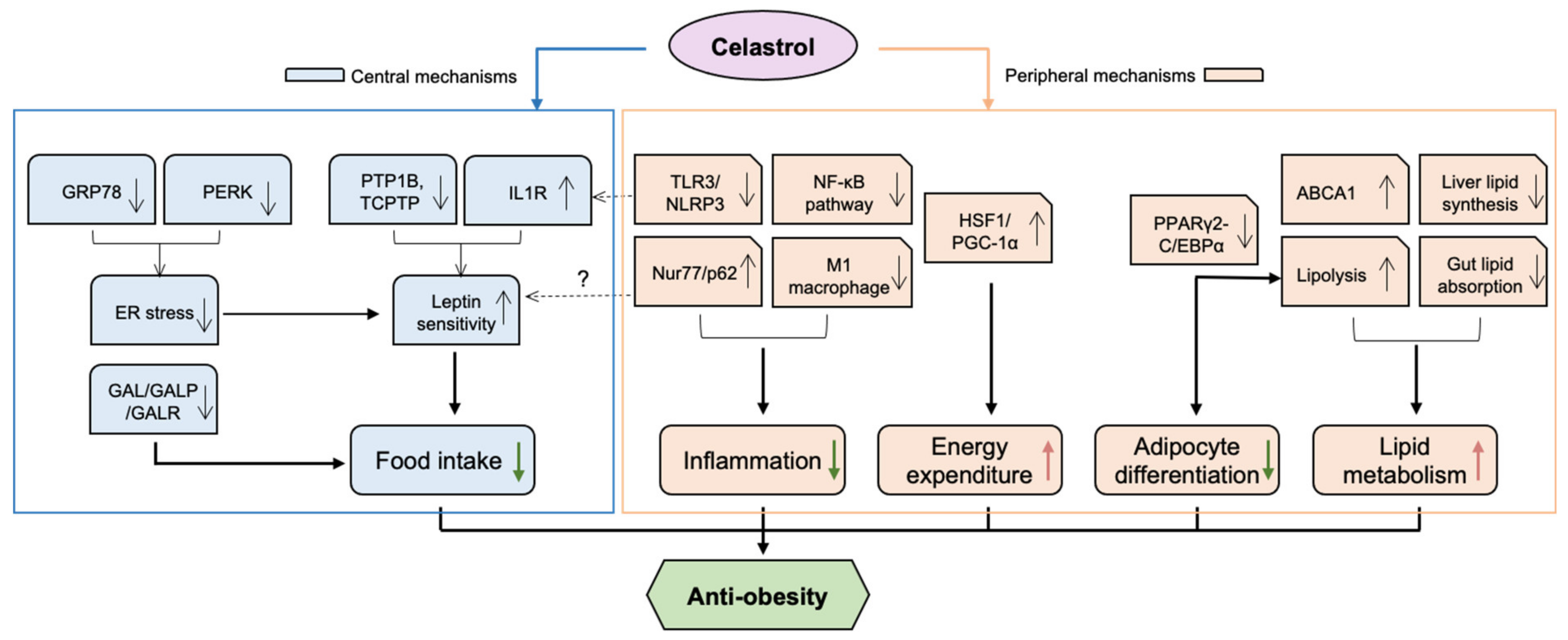
2.1. Obesity
2.1.1. Anti-Obesity Effect of Celastrol through Central Mechanisms
A Leptin Sensitiser
An Endoplasmic Reticulum (ER) Stress Suppressor
Downregulation of Galanin (GAL)
2.1.2. Anti-Obesity Effect of Celastrol through Peripheral Mechanisms
Modulation of Lipid Metabolism
A Heat Shock Factor 1 (HSF1) Activator
Inhibition of Adipocyte Differentiation
Anti-Inflammatory Activities
2.2. Diabetes Mellitus
3. Celastrol against Atherosclerosis
4. Celastrol against Cerebrovascular Injury
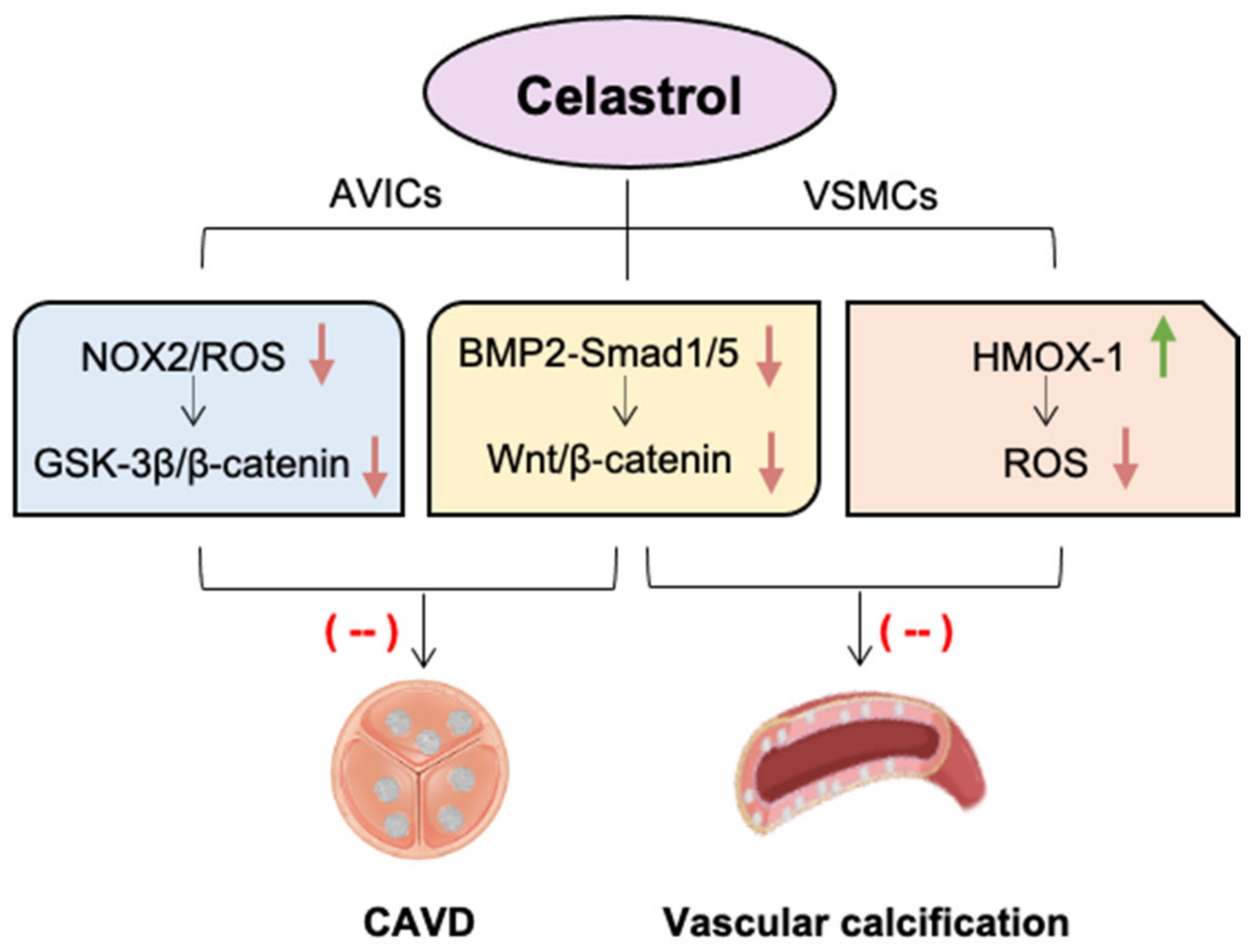
5. Celastrol against Valvular and Vascular Calcification
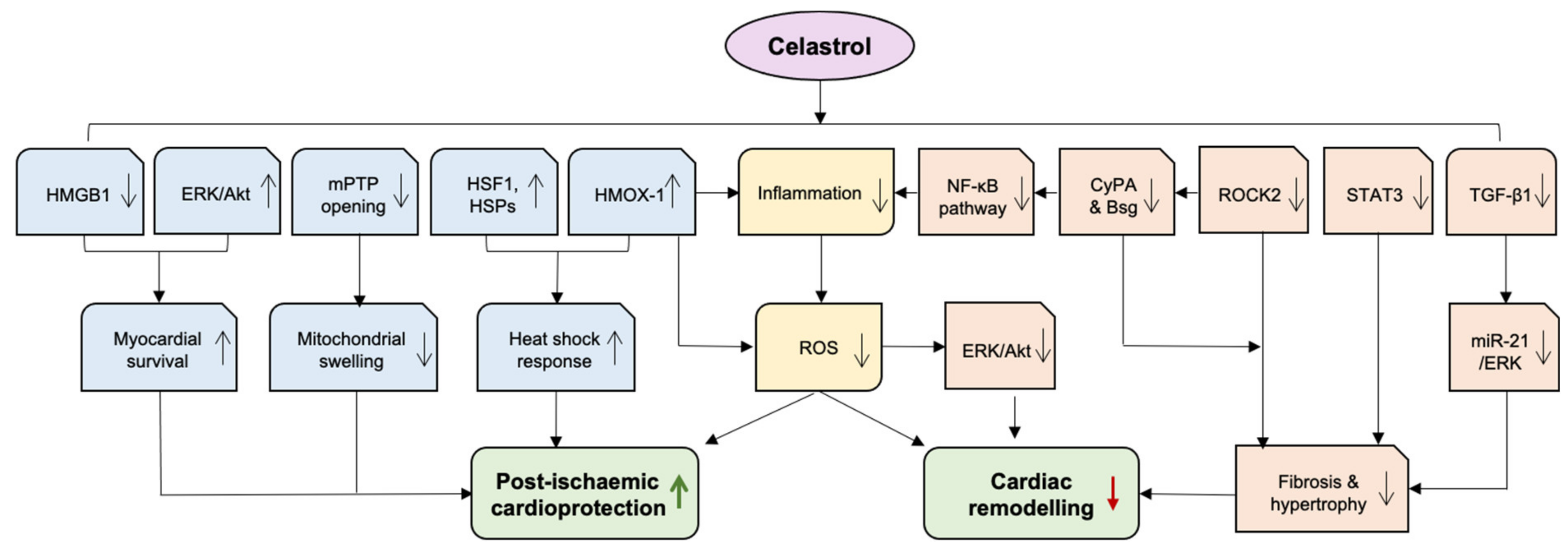
6. Celastrol against Heart Failure
6.1. Mitigating Cardiac Remodelling
6.2. Post-Ischaemic Cardioprotection
7. Challenges of Celastrol and Strategies
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Chen, S.R.; Dai, Y.; Zhao, J.; Lin, L.; Wang, Y.; Wang, Y. A Mechanistic Overview of Triptolide and Celastrol, Natural Products from Tripterygium wilfordii Hook F. Front. Pharmacol. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Liu, B.; Xu, H. Celastrol: Progresses in structure-modifications, structure-activity relationships, pharmacology and toxicology. Eur. J. Med. Chem. 2020, 189, 112081. [Google Scholar] [CrossRef]
- Yousefian, M.; Shakour, N.; Hosseinzadeh, H.; Hayes, A.W.; Hadizadeh, F.; Karimi, G. The natural phenolic compounds as modulators of NADPH oxidases in hypertension. Phytomedicine 2019, 55, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Chan, Y.; Chellappan, D.K.; Madheswaran, T.; Zeeshan, F.; Chan, Y.L.; Collet, T.; Gupta, G.; Oliver, B.G.; Wark, P.; et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed. Pharmacother. 2019, 109, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Feng, Y.; He, W.; Xu, W.; Xu, W.; Yang, H.; Li, X. Celastrol in metabolic diseases: Progress and application prospects. Pharmacol. Res. 2021, 167, 105572. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Ong, P.S.; Wang, L.; Goel, A.; Ding, L.; Li-Ann Wong, A.; Ho, P.C.; Sethi, G.; Xiang, X.; Goh, B.C. Celastrol in cancer therapy: Recent developments, challenges and prospects. Cancer Lett. 2021, 521, 252–267. [Google Scholar] [CrossRef]
- Shi, J.; Li, J.; Xu, Z.; Chen, L.; Luo, R.; Zhang, C.; Gao, F.; Zhang, J.; Fu, C. Celastrol: A Review of Useful Strategies Overcoming its Limitation in Anticancer Application. Front. Pharmacol. 2020, 11, 558741. [Google Scholar] [CrossRef]
- Piche, M.E.; Tchernof, A.; Despres, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Kouhpayeh, H. Clinical features predicting COVID-19 mortality risk. Eur. J. Transl. Myol. 2022, 32, 10268. [Google Scholar] [CrossRef]
- Florencio, L.L.; Fernandez-de-Las-Penas, C. Long COVID: Systemic inflammation and obesity as therapeutic targets. Lancet Respir. Med. 2022, 10, 726–727. [Google Scholar] [CrossRef]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 2007, 4, 18. [Google Scholar] [CrossRef]
- Zhang, Y.; Chua, S., Jr. Leptin Function and Regulation. Compr. Physiol. 2017, 8, 351–369. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Leptin resistance: Underlying mechanisms and diagnosis. Diabetes Metab. Syndr. Obes. 2019, 12, 191–198. [Google Scholar] [CrossRef]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef]
- Saito, K.; Davis, K.C.; Morgan, D.A.; Toth, B.A.; Jiang, J.; Singh, U.; Berglund, E.D.; Grobe, J.L.; Rahmouni, K.; Cui, H. Celastrol Reduces Obesity in MC4R Deficiency and Stimulates Sympathetic Nerve Activity Affecting Metabolic and Cardiovascular Functions. Diabetes 2019, 68, 1210–1220. [Google Scholar] [CrossRef]
- Ma, X.; Xu, L.; Alberobello, A.T.; Gavrilova, O.; Bagattin, A.; Skarulis, M.; Liu, J.; Finkel, T.; Mueller, E. Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1alpha Transcriptional Axis. Cell Metab. 2015, 22, 695–708. [Google Scholar] [CrossRef]
- Feng, X.; Guan, D.; Auen, T.; Choi, J.W.; Salazar Hernandez, M.A.; Lee, J.; Chun, H.; Faruk, F.; Kaplun, E.; Herbert, Z.; et al. IL1R1 is required for celastrol’s leptin-sensitization and antiobesity effects. Nat. Med. 2019, 25, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, R.; Fantuzzi, G.; Fuller, J.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C. IL-1 beta mediates leptin induction during inflammation. Am. J. Physiol. 1998, 274, R204–R208. [Google Scholar] [CrossRef]
- Feng, X.; Guan, D.; Auen, T.; Choi, J.W.; Salazar-Hernandez, M.A.; Faruk, F.; Copps, K.D.; Ozcan, U. Lipocalin 2 Does Not Play A Role in Celastrol-Mediated Reduction in Food Intake and Body Weight. Sci. Rep. 2019, 9, 12809. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, E.; Schmidt, S.; Dodd, G.T.; Pfuhlmann, K.; Simonds, S.E.; Lenhart, D.; Geerlof, A.; Schriever, S.C.; De Angelis, M.; Schramm, K.W.; et al. Celastrol Promotes Weight Loss in Diet-Induced Obesity by Inhibiting the Protein Tyrosine Phosphatases PTP1B and TCPTP in the Hypothalamus. J. Med. Chem. 2018, 61, 11144–11157. [Google Scholar] [CrossRef] [PubMed]
- Pfuhlmann, K.; Schriever, S.C.; Baumann, P.; Kabra, D.G.; Harrison, L.; Mazibuko-Mbeje, S.E.; Contreras, R.E.; Kyriakou, E.; Simonds, S.E.; Tiganis, T.; et al. Celastrol-Induced Weight Loss Is Driven by Hypophagia and Independent From UCP1. Diabetes 2018, 67, 2456–2465. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Clement, K.; Durand, E.; Hercberg, S.; Guy-Grand, B.; Froguel, P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Investig. 2000, 106, 253–262. [Google Scholar] [CrossRef]
- Ye, T.; Meng, X.; Zhai, Y.; Xie, W.; Wang, R.; Sun, G.; Sun, X. Gastrodin Ameliorates Cognitive Dysfunction in Diabetes Rat Model via the Suppression of Endoplasmic Reticulum Stress and NLRP3 Inflammasome Activation. Front. Pharmacol. 2018, 9, 1346. [Google Scholar] [CrossRef]
- Xiao, Y.; Deng, Y.; Yuan, F.; Xia, T.; Liu, H.; Li, Z.; Liu, Z.; Ying, H.; Liu, Y.; Zhai, Q.; et al. ATF4/ATG5 Signaling in Hypothalamic Proopiomelanocortin Neurons Regulates Fat Mass via Affecting Energy Expenditure. Diabetes 2017, 66, 1146–1158. [Google Scholar] [CrossRef]
- He, Z.; Lieu, L.; Dong, Y.; Afrin, S.; Chau, D.; Kabahizi, A.; Wallace, B.; Cao, J.; Hwang, E.S.; Yao, T.; et al. PERK in POMC neurons connects celastrol with metabolism. JCI Insight 2021, 6, e145306. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Luo, D.; Fan, N.; Zhang, X.; Ngo, F.Y.; Zhao, J.; Zhao, W.; Huang, M.; Li, D.; Wang, Y.; Rong, J. Covalent inhibition of endoplasmic reticulum chaperone GRP78 disconnects the transduction of ER stress signals to inflammation and lipid accumulation in diet-induced obese mice. Elife 2022, 11, 72182. [Google Scholar] [CrossRef] [PubMed]
- Marcos, P.; Covenas, R. Neuropeptidergic Control of Feeding: Focus on the Galanin Family of Peptides. Int. J. Mol. Sci. 2021, 22, 2544. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; He, B.; Yu, M.; Shi, M.; Zhu, Y.; Zhang, Z.; Bo, P. Treatment with celastrol protects against obesity through suppression of galanin-induced fat intake and activation of PGC-1alpha/GLUT4 axis-mediated glucose consumption. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shi, C.; Yang, X.; Yang, M.; Sun, H.; Wang, C. Celastrol suppresses obesity process via increasing antioxidant capacity and improving lipid metabolism. Eur. J. Pharmacol. 2014, 744, 52–58. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Q.; Xiao, X.; Yang, R.; Hu, D.; Zhu, X.; Gonzalez, F.J.; Li, F. Modulation of Lipid Metabolism by Celastrol. J. Proteome Res. 2019, 18, 1133–1144. [Google Scholar] [CrossRef]
- Hua, H.; Zhang, Y.; Zhao, F.; Chen, K.; Wu, T.; Liu, Q.; Huang, S.; Zhang, A.; Jia, Z. Celastrol inhibits intestinal lipid absorption by reprofiling the gut microbiota to attenuate high-fat diet-induced obesity. iScience 2021, 24, 102077. [Google Scholar] [CrossRef]
- Hu, W.; Wang, L.; Du, G.; Guan, Q.; Dong, T.; Song, L.; Xia, Y.; Wang, X. Effects of Microbiota on the Treatment of Obesity with the Natural Product Celastrol in Rats. Diabetes Metab. J. 2020, 44, 747–763. [Google Scholar] [CrossRef]
- Xu, S.; Lyu, L.; Zhu, H.; Huang, X.; Xu, W.; Xu, W.; Feng, Y.; Fan, Y. Serum Metabolome Mediates the Antiobesity Effect of Celastrol-Induced Gut Microbial Alterations. J. Proteome Res. 2021, 20, 4840–4851. [Google Scholar] [CrossRef]
- Xu, L.; Ma, X.; Bagattin, A.; Mueller, E. The transcriptional coactivator PGC1alpha protects against hyperthermic stress via cooperation with the heat shock factor HSF1. Cell Death Dis. 2016, 7, e2102. [Google Scholar] [CrossRef]
- Bost, F.; Aouadi, M.; Caron, L.; Binetruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 2005, 87, 51–56. [Google Scholar] [CrossRef]
- Choi, S.K.; Park, S.; Jang, S.; Cho, H.H.; Lee, S.; You, S.; Kim, S.H.; Moon, H.S. Cascade regulation of PPARgamma(2) and C/EBPalpha signaling pathways by celastrol impairs adipocyte differentiation and stimulates lipolysis in 3T3-L1 adipocytes. Metabolism 2016, 65, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Feve, B.; Claes, A.; Viengchareun, S.; Lombes, M.; Zennaro, M.C. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007, 21, 2185–2194. [Google Scholar] [CrossRef]
- Hong, W.; Park, J.; Yun, W.; Kang, P.J.; Son, D.; Jang, J.; Kim, I.Y.; You, S. Inhibitory effect of celastrol on adipogenic differentiation of human adipose-derived stem cells. Biochem. Biophys. Res. Commun. 2018, 507, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage polarization in obesity and type 2 diabetes: Weighing down our understanding of macrophage function? Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef]
- Luo, D.; Guo, Y.; Cheng, Y.; Zhao, J.; Wang, Y.; Rong, J. Natural product celastrol suppressed macrophage M1 polarization against inflammation in diet-induced obese mice via regulating Nrf2/HO-1, MAP kinase and NF-kappaB pathways. Aging 2017, 9, 2069–2082. [Google Scholar] [CrossRef]
- Zhao, J.; Luo, D.; Zhang, Z.; Fan, N.; Wang, Y.; Nie, H.; Rong, J. Celastrol-loaded PEG-PCL nanomicelles ameliorate inflammation, lipid accumulation, insulin resistance and gastrointestinal injury in diet-induced obese mice. J. Control. Release 2019, 310, 188–197. [Google Scholar] [CrossRef]
- Abu Bakar, M.H.; Shariff, K.A.; Tan, J.S.; Lee, L.K. Celastrol attenuates inflammatory responses in adipose tissues and improves skeletal muscle mitochondrial functions in high fat diet-induced obese rats via upregulation of AMPK/SIRT1 signaling pathways. Eur. J. Pharmacol. 2020, 883, 173371. [Google Scholar] [CrossRef]
- Lee, M.K.; Yvan-Charvet, L.; Masters, S.L.; Murphy, A.J. The modern interleukin-1 superfamily: Divergent roles in obesity. Semin. Immunol. 2016, 28, 441–449. [Google Scholar] [CrossRef]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef]
- Yang, X.; Wu, F.; Li, L.; Lynch, E.C.; Xie, L.; Zhao, Y.; Fang, K.; Li, J.; Luo, J.; Xu, L.; et al. Celastrol alleviates metabolic disturbance in high-fat diet-induced obese mice through increasing energy expenditure by ameliorating metabolic inflammation. Phytother. Res. 2021, 35, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, R.; Chen, H.Z.; Xiao, Q.; Wang, W.J.; He, J.P.; Li, X.X.; Yu, X.W.; Li, L.; Wang, P.; et al. Enhancement of hypothalamic STAT3 acetylation by nuclear receptor Nur77 dictates leptin sensitivity. Diabetes 2015, 64, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Veum, V.L.; Dankel, S.N.; Gjerde, J.; Nielsen, H.J.; Solsvik, M.H.; Haugen, C.; Christensen, B.J.; Hoang, T.; Fadnes, D.J.; Busch, C.; et al. The nuclear receptors NUR77, NURR1 and NOR1 in obesity and during fat loss. Int. J. Obes. 2012, 36, 1195–1202. [Google Scholar] [CrossRef]
- Hu, M.; Luo, Q.; Alitongbieke, G.; Chong, S.; Xu, C.; Xie, L.; Chen, X.; Zhang, D.; Zhou, Y.; Wang, Z.; et al. Celastrol-Induced Nur77 Interaction with TRAF2 Alleviates Inflammation by Promoting Mitochondrial Ubiquitination and Autophagy. Mol. Cell 2017, 66, 141–153.e6. [Google Scholar] [CrossRef]
- Peng, S.Z.; Chen, X.H.; Chen, S.J.; Zhang, J.; Wang, C.Y.; Liu, W.R.; Zhang, D.; Su, Y.; Zhang, X.K. Phase separation of Nur77 mediates celastrol-induced mitophagy by promoting the liquidity of p62/SQSTM1 condensates. Nat. Commun. 2021, 12, 5989. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. SIRT1 in Type 2 Diabetes: Mechanisms and Therapeutic Potential. Diabetes Metab. J. 2013, 37, 315–325. [Google Scholar] [CrossRef]
- Nesto, R. C-reactive protein, its role in inflammation, Type 2 diabetes and cardiovascular disease, and the effects of insulin-sensitizing treatment with thiazolidinediones. Diabet. Med. 2004, 21, 810–817. [Google Scholar] [CrossRef]
- Newman, J.D.; Schwartzbard, A.Z.; Weintraub, H.S.; Goldberg, I.J.; Berger, J.S. Primary Prevention of Cardiovascular Disease in Diabetes Mellitus. J. Am. Coll. Cardiol. 2017, 70, 883–893. [Google Scholar] [CrossRef]
- Taylor, S.I.; Yazdi, Z.S.; Beitelshees, A.L. Pharmacological treatment of hyperglycemia in type 2 diabetes. J. Clin. Investig. 2021, 131, e142243. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J. Insulin resistance as the core defect in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3G–10G. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, M.H.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Han, S.Y.; et al. Celastrol, an NF-kappaB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS ONE 2013, 8, e62068. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Ge, H.Y.; Gu, Y.J.; Cao, F.F.; Yang, C.X.; Uzan, G.; Peng, B.; Zhang, D.H. Celastrol reverses palmitic acid (PA)-caused TLR4-MD2 activation-dependent insulin resistance via disrupting MD2-related cellular binding to PA. J. Cell. Physiol. 2018, 233, 6814–6824. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, B.H.; Kim, N.D.; Lee, J.Y. Celastrol blocks binding of lipopolysaccharides to a Toll-like receptor4/myeloid differentiation factor2 complex in a thiol-dependent manner. J. Ethnopharmacol. 2015, 172, 254–260. [Google Scholar] [CrossRef]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef]
- Jang, P.G.; Namkoong, C.; Kang, G.M.; Hur, M.W.; Kim, S.W.; Kim, G.H.; Kang, Y.; Jeon, M.J.; Kim, E.H.; Lee, M.S.; et al. NF-kappaB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. J. Biol. Chem. 2010, 285, 9706–9715. [Google Scholar] [CrossRef]
- Abu Bakar, M.H.; Tan, J.S. Improvement of mitochondrial function by celastrol in palmitate-treated C2C12 myotubes via activation of PI3K-Akt signaling pathway. Biomed. Pharmacother. 2017, 93, 903–912. [Google Scholar] [CrossRef]
- Bjorkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Bai, W.; Li, S.; Zhang, Y.; Han, Y.; Gu, Y.; Meng, G.; Xie, L.; Wang, J.; Xiao, Y.; et al. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS ONE 2013, 8, e65477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, C.; Jin, X.P.; Weng, S.X.; Fan, L.L.; Zheng, Z.; Li, W.L.; Wang, F.; Wang, W.F.; Hu, X.F.; et al. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model. Int. J. Clin. Exp. Med. 2014, 7, 1684–1691. [Google Scholar] [PubMed]
- Allen, S.D.; Liu, Y.G.; Kim, T.; Bobbala, S.; Yi, S.; Zhang, X.; Choi, J.; Scott, E.A. Celastrol-loaded PEG-b-PPS nanocarriers as an anti-inflammatory treatment for atherosclerosis. Biomater. Sci. 2019, 7, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, J.Y.; He, P.P.; Yu, X.H.; Tang, C.K. Resistin: Potential biomarker and therapeutic target in atherosclerosis. Clin. Chim. Acta 2021, 512, 84–91. [Google Scholar] [CrossRef]
- Zhu, Y.; Wan, N.; Shan, X.; Deng, G.; Xu, Q.; Ye, H.; Sun, Y. Celastrol targets adenylyl cyclase-associated protein 1 to reduce macrophages-mediated inflammation and ameliorates high fat diet-induced metabolic syndrome in mice. Acta Pharm. Sin. B 2021, 11, 1200–1212. [Google Scholar] [CrossRef]
- Kang, S.W.; Kim, M.S.; Kim, H.S.; Kim, Y.; Shin, D.; Park, J.H.; Kang, Y.H. Celastrol attenuates adipokine resistin-associated matrix interaction and migration of vascular smooth muscle cells. J. Cell. Biochem. 2013, 114, 398–408. [Google Scholar] [CrossRef]
- Xu, X.J.; Zhao, W.B.; Feng, S.B.; Sun, C.; Chen, Q.; Ni, B.; Hu, H.Y. Celastrol alleviates angiotensin IImediated vascular smooth muscle cell senescence via induction of autophagy. Mol. Med. Rep. 2017, 16, 7657–7664. [Google Scholar] [CrossRef]
- Shi, Y.; Jiang, S.; Zhao, T.; Gong, Y.; Liao, D.; Qin, L. Celastrol suppresses lipid accumulation through LXRalpha/ABCA1 signaling pathway and autophagy in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2020, 532, 466–474. [Google Scholar] [CrossRef]
- Li, M.; Liu, X.; He, Y.; Zheng, Q.; Wang, M.; Wu, Y.; Zhang, Y.; Wang, C. Celastrol attenuates angiotensin II mediated human umbilical vein endothelial cells damage through activation of Nrf2/ERK1/2/Nox2 signal pathway. Eur. J. Pharmacol. 2017, 797, 124–133. [Google Scholar] [CrossRef]
- Lu, C.; Yu, X.; Zuo, K.; Zhang, X.; Cao, C.; Xu, J.; Wang, S.; Tang, T.; Ye, M.; Pei, E.; et al. Tripterine treatment improves endothelial progenitor cell function via integrin-linked kinase. Cell. Physiol. Biochem. 2015, 37, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.D.; Atkinson, T.M.; Lindner, J.R. Platelets and von Willebrand factor in atherogenesis. Blood 2017, 129, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Straub, A.; Tian, Z.; Bassler, N.; Cheng, J.; Peter, K. Celastrol, a triterpene extracted from Tripterygium wilfordii Hook F, inhibits platelet activation. J. Cardiovasc. Pharmacol. 2009, 54, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, D.; Zhang, X.; Liu, Z.; Zhang, X.; Dong, L.; Xing, Y.; Wang, C.; Qiao, H.; Zhu, C.; et al. Protective effect of celastrol in rat cerebral ischemia model: Down-regulating p-JNK, p-c-Jun and NF-kappaB. Brain Res. 2012, 1464, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Liu, X.; Zhang, D.; Wang, Y.; Hu, X.; Xu, F.; Jin, M.; Cao, F.; Xu, L. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an IL-33/ST2 axis-mediated microglia/macrophage M2 polarization. J. Neuroinflamm. 2018, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, M.; Luo, Y.; Wang, H.; Huang, H.; Peng, Z.; Li, M.; Fei, H.; Luo, W.; Yang, J. Lipidomic Profiling of Ipsilateral Brain and Plasma after Celastrol Post-Treatment in Transient Middle Cerebral Artery Occlusion Mice Model. Molecules 2021, 26, 4124. [Google Scholar] [CrossRef]
- Chen, M.; Liu, M.; Luo, Y.; Cao, J.; Zeng, F.; Yang, L.; Yang, J.; Tao, T.; Jiang, Y. Celastrol Protects against Cerebral Ischemia/Reperfusion Injury in Mice by Inhibiting Glycolysis through Targeting HIF-1alpha/PDK1 Axis. Oxid Med. Cell. Longev. 2022, 2022, 7420507. [Google Scholar] [CrossRef]
- Liu, J.; Guo, X.; Yang, L.; Tao, T.; Cao, J.; Hong, Z.; Zeng, F.; Lu, Y.; Lin, C.; Qin, Z. Effect of Celastrol on LncRNAs and mRNAs Profiles of Cerebral Ischemia-Reperfusion Injury in Transient Middle Cerebral Artery Occlusion Mice Model. Front. Neurosci. 2022, 16, 889292. [Google Scholar] [CrossRef]
- Zhang, B.; Zhong, Q.; Chen, X.; Wu, X.; Sha, R.; Song, G.; Zhang, C.; Chen, X. Neuroprotective Effects of Celastrol on Transient Global Cerebral Ischemia Rats via Regulating HMGB1/NF-kappaB Signaling Pathway. Front. Neurosci. 2020, 14, 847. [Google Scholar] [CrossRef]
- Liu, D.D.; Luo, P.; Gu, L.; Zhang, Q.; Gao, P.; Zhu, Y.; Chen, X.; Guo, Q.; Zhang, J.; Ma, N.; et al. Celastrol exerts a neuroprotective effect by directly binding to HMGB1 protein in cerebral ischemia-reperfusion. J. Neuroinflamm. 2021, 18, 174. [Google Scholar] [CrossRef]
- North, K.; Slayden, A.; Mysiewicz, S.; Bukiya, A.; Dopico, A. Celastrol Dilates and Counteracts Ethanol-Induced Constriction of Cerebral Arteries. J. Pharmacol. Exp. Ther. 2020, 375, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cai, Y.; Yu, M.; Sun, J.; Cai, J.; Li, J.; Qin, B.; Ying, G.; Chen, T.; Shen, Y.; et al. Celastrol protects against early brain injury after subarachnoid hemorrhage in rats through alleviating blood-brain barrier disruption and blocking necroptosis. Aging 2021, 13, 16816–16833. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular calcification: Mechanisms and clinical ramifications. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stenslokken, K.O.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef]
- Vossen, L.M.; Kroon, A.A.; Schurgers, L.J.; de Leeuw, P.W. Pharmacological and Nutritional Modulation of Vascular Calcification. Nutrients 2019, 12, 100. [Google Scholar] [CrossRef]
- Branchetti, E.; Sainger, R.; Poggio, P.; Grau, J.B.; Patterson-Fortin, J.; Bavaria, J.E.; Chorny, M.; Lai, E.; Gorman, R.C.; Levy, R.J.; et al. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e66–e74. [Google Scholar] [CrossRef]
- Greenberg, H.Z.E.; Zhao, G.; Shah, A.M.; Zhang, M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc. Res. 2022, 118, 1433–1451. [Google Scholar] [CrossRef]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Liberman, M.; Bassi, E.; Martinatti, M.K.; Lario, F.C.; Wosniak, J., Jr.; Pomerantzeff, P.M.; Laurindo, F.R. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 463–470. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.V.; Mokas, S.; Lariviere, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef]
- Jaquet, V.; Marcoux, J.; Forest, E.; Leidal, K.G.; McCormick, S.; Westermaier, Y.; Perozzo, R.; Plastre, O.; Fioraso-Cartier, L.; Diebold, B.; et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011, 164, 507–520. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Pan, Y.; Wang, X.; Ding, Y.; Zhou, C.; Shah, A.M.; Zhao, G.; Zhang, M. Celastrol Alleviates Aortic Valve Calcification Via Inhibition of NADPH Oxidase 2 in Valvular Interstitial Cells. JACC Basic Transl. Sci. 2020, 5, 35–49. [Google Scholar] [CrossRef]
- Su, Z.; Zong, P.; Chen, J.; Yang, S.; Shen, Y.; Lu, Y.; Yang, C.; Kong, X.; Sheng, Y.; Sun, W. Celastrol attenuates arterial and valvular calcification via inhibiting BMP2/Smad1/5 signalling. J. Cell. Mol. Med. 2020, 24, 12476–12490. [Google Scholar] [CrossRef]
- Yang, X.; Chen, A.; Liang, Q.; Dong, Q.; Fu, M.; Liu, X.; Wang, S.; Li, Y.; Ye, Y.; Lan, Z.; et al. Up-regulation of heme oxygenase-1 by celastrol alleviates oxidative stress and vascular calcification in chronic kidney disease. Free Radic. Biol. Med. 2021, 172, 530–540. [Google Scholar] [CrossRef]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Nigro, P.; Zeidan, A.; Soe, N.N.; Jaffre, F.; Oikawa, M.; O’Dell, M.R.; Cui, Z.; Menon, P.; Lu, Y.; et al. Cyclophilin A promotes cardiac hypertrophy in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1116–1123. [Google Scholar] [CrossRef]
- Sunamura, S.; Satoh, K.; Kurosawa, R.; Ohtsuki, T.; Kikuchi, N.; Elias-Al-Mamun, M.; Shimizu, T.; Ikeda, S.; Suzuki, K.; Satoh, T.; et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E7129–E7138. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Gibbs, J.S.; Wachter, R.; De Marco, T.; Vonk-Noordegraaf, A.; Vachiery, J.L. Left ventricular heart failure and pulmonary hypertension. Eur. Heart J. 2016, 37, 942–954. [Google Scholar] [CrossRef]
- Xue, C.; Sowden, M.; Berk, B.C. Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1138–1146. [Google Scholar] [CrossRef]
- Kurosawa, R.; Satoh, K.; Nakata, T.; Shindo, T.; Kikuchi, N.; Satoh, T.; Siddique, M.A.H.; Omura, J.; Sunamura, S.; Nogi, M.; et al. Identification of Celastrol as a Novel Therapeutic Agent for Pulmonary Arterial Hypertension and Right Ventricular Failure Through Suppression of Bsg (Basigin)/CyPA (Cyclophilin A). Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1205–1217. [Google Scholar] [CrossRef]
- Harhous, Z.; Booz, G.W.; Ovize, M.; Bidaux, G.; Kurdi, M. An Update on the Multifaceted Roles of STAT3 in the Heart. Front. Cardiovasc. Med. 2019, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Luo, W.; Khan, Z.A.; Wu, G.; Xuan, L.; Shan, P.; Lin, K.; Chen, T.; Wang, J.; Hu, X.; et al. Celastrol Attenuates Angiotensin II-Induced Cardiac Remodeling by Targeting STAT3. Circ. Res. 2020, 126, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Wu, G.; Song, Y.; Wang, L.; Tu, L.; Zhang, L.; Zhang, C. Celastrol-Induced Suppression of the MiR-21/ERK Signalling Pathway Attenuates Cardiac Fibrosis and Dysfunction. Cell. Physiol. Biochem. 2016, 38, 1928–1938. [Google Scholar] [CrossRef]
- Yu, X.; Tao, W.; Jiang, F.; Li, C.; Lin, J.; Liu, C. Celastrol attenuates hypertension-induced inflammation and oxidative stress in vascular smooth muscle cells via induction of heme oxygenase-1. Am. J. Hypertens. 2010, 23, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Multicenter Postinfarction Research, G. Risk stratification and survival after myocardial infarction. N. Engl. J. Med. 1983, 309, 331–336. [Google Scholar] [CrossRef]
- Cahill, T.J.; Kharbanda, R.K. Heart failure after myocardial infarction in the era of primary percutaneous coronary intervention: Mechanisms, incidence and identification of patients at risk. World J. Cardiol. 2017, 9, 407–415. [Google Scholar] [CrossRef]
- Nair, S.P.; Sharma, R.K. Heat shock proteins and their expression in primary murine cardiac cell populations during ischemia and reperfusion. Mol. Cell. Biochem. 2020, 464, 21–26. [Google Scholar] [CrossRef]
- Kupatt, C.; Dessy, C.; Hinkel, R.; Raake, P.; Daneau, G.; Bouzin, C.; Boekstegers, P.; Feron, O. Heat shock protein 90 transfection reduces ischemia-reperfusion-induced myocardial dysfunction via reciprocal endothelial NO synthase serine 1177 phosphorylation and threonine 495 dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1435–1441. [Google Scholar] [CrossRef]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160530. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Zhong, C.B.; Wang, X.B. Heat shock protein 70: A promising therapeutic target for myocardial ischemia-reperfusion injury. J. Cell. Physiol. 2019, 234, 1190–1207. [Google Scholar] [CrossRef]
- Der Sarkissian, S.; Cailhier, J.F.; Borie, M.; Stevens, L.M.; Gaboury, L.; Mansour, S.; Hamet, P.; Noiseux, N. Celastrol protects ischaemic myocardium through a heat shock response with up-regulation of haeme oxygenase-1. Br. J. Pharmacol. 2014, 171, 5265–5279. [Google Scholar] [CrossRef] [PubMed]
- Aceros, H.; Der Sarkissian, S.; Borie, M.; Stevens, L.M.; Mansour, S.; Noiseux, N. Celastrol-type HSP90 modulators allow for potent cardioprotective effects. Life Sci. 2019, 227, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail. Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef]
- Tong, S.; Zhang, L.; Joseph, J.; Jiang, X. Celastrol pretreatment attenuates rat myocardial ischemia/reperfusion injury by inhibiting high mobility group box 1 protein expression via the PI3K/Akt pathway. Biochem. Biophys. Res. Commun. 2018, 497, 843–849. [Google Scholar] [CrossRef]
- Li, X.; Wu, N.; Zou, L.; Jia, D. Protective effect of celastrol on myocardial ischemia-reperfusion injury. Anatol. J. Cardiol. 2017, 18, 384–390. [Google Scholar] [CrossRef]
- Sharma, S.; Mishra, R.; Walker, B.L.; Deshmukh, S.; Zampino, M.; Patel, J.; Anamalai, M.; Simpson, D.; Singh, I.S.; Kaushal, S.; et al. Celastrol, an oral heat shock activator, ameliorates multiple animal disease models of cell death. Cell Stress Chaperones 2015, 20, 185–201. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.Y.; Xu, M.J.; Wu, T.; Chu, J.H.; Liu, S.J.; Ju, W.Z. Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J. Ethnopharmacol. 2012, 144, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qin, J.; Ma, N.; Chou, X.; Wu, Z. Solid self-microemulsifying dispersible tablets of celastrol: Formulation development, charaterization and bioavailability evaluation. Int. J. Pharm. 2014, 472, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Cascão, R.; Carvalho, T.; Goncalves, J.; Moita, L.; Fonseca, J. AB0096 Efficacy and safety of oral administration of pure celastrol in aia rats. Ann. Rheum. Dis. 2017, 76, 1080. [Google Scholar]
- Yuan, Y.Y.; Gu, Z.P.; Shi, Q.X.; Qin, G.W.; Xu, R.S.; Cao, L. In vitro inhibition of celastrol on spermatozoa fertilization ability of guinea pig. Yao Xue Xue Bao 1995, 30, 331–335. [Google Scholar] [PubMed]
- Bai, J.P.; Shi, Y.L.; Fang, X.; Shi, Q.X. Effects of demethylzeylasteral and celastrol on spermatogenic cell Ca2+ channels and progesterone-induced sperm acrosome reaction. Eur. J. Pharmacol. 2003, 464, 9–15. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, C.; Wang, W.; Yuan, F.; He, T.; Chen, Y.; Wang, Q.; Huang, J. Integrated metabolomics and network toxicology to reveal molecular mechanism of celastrol induced cardiotoxicity. Toxicol. Appl. Pharmacol. 2019, 383, 114785. [Google Scholar] [CrossRef]
- Jin, C.; Wu, Z.; Wang, L.; Kanai, Y.; He, X. CYP450s-Activity Relations of Celastrol to Interact with Triptolide Reveal the Reasons of Hepatotoxicity of Tripterygium wilfordii. Molecules 2019, 24, 2162. [Google Scholar] [CrossRef]
- Wu, M.; Chen, W.; Yu, X.; Ding, D.; Zhang, W.; Hua, H.; Xu, M.; Meng, X.; Zhang, X.; Zhang, Y.; et al. Celastrol aggravates LPS-induced inflammation and injuries of liver and kidney in mice. Am. J. Transl. Res. 2018, 10, 2078–2086. [Google Scholar]
- Aqil, F.; Kausar, H.; Agrawal, A.K.; Jeyabalan, J.; Kyakulaga, A.H.; Munagala, R.; Gupta, R. Exosomal formulation enhances therapeutic response of celastrol against lung cancer. Exp. Mol. Pathol. 2016, 101, 12–21. [Google Scholar] [CrossRef]
- Song, J.; Shi, F.; Zhang, Z.; Zhu, F.; Xue, J.; Tan, X.; Zhang, L.; Jia, X. Formulation and evaluation of celastrol-loaded liposomes. Molecules 2011, 16, 7880–7892. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, T.; Zhou, X.; Liu, H.; Sun, H.; Ma, Z.; Wu, B. Enhancement of oral bioavailability of tripterine through lipid nanospheres: Preparation, characterization, and absorption evaluation. J. Pharm. Sci. 2014, 103, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, Y.; Luo, W.; Chen, S.; Lin, F.; Zhang, X.; Fan, S.; Shen, X.; Wang, Y.; Liang, G. Celastrol induces ROS-mediated apoptosis via directly targeting peroxiredoxin-2 in gastric cancer cells. Theranostics 2020, 10, 10290–10308. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W.; Bandyopadhyay, P.K. Model for how type I restriction enzymes select cleavage sites in DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 4677–4681. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Fujio, Y.; Nakanishi, T.; Itoh, N.; Yamamoto, Y.; Negoro, S.; Tanaka, K.; Kishimoto, T.; Kawase, I.; Azuma, J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc. Res. 2005, 65, 428–435. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Zhang, J.; Duan, X.; Zhao, G.; Zhang, M. Celastrol: A Promising Agent Fighting against Cardiovascular Diseases. Antioxidants 2022, 11, 1597. https://doi.org/10.3390/antiox11081597
Li Z, Zhang J, Duan X, Zhao G, Zhang M. Celastrol: A Promising Agent Fighting against Cardiovascular Diseases. Antioxidants. 2022; 11(8):1597. https://doi.org/10.3390/antiox11081597
Chicago/Turabian StyleLi, Zhexi, Jingyi Zhang, Xulei Duan, Guoan Zhao, and Min Zhang. 2022. "Celastrol: A Promising Agent Fighting against Cardiovascular Diseases" Antioxidants 11, no. 8: 1597. https://doi.org/10.3390/antiox11081597