
Ginkgo biloba Extract Attenuates Light-Induced Photoreceptor Degeneration by Modulating CAV-1—Redoxosome Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Line and WHITE Light Illumination
2.3. Cell Viability and Apoptotic Assay
2.4. Oxidative Stress and NOX Activity Analysis
2.5. Mitochondrial Membrane Potential Analysis
2.6. Western Blot Analysis
2.7. Co-Immunoprecipitation Assay
2.8. Transcriptome Sequencing and Analysis
2.9. Animal and White Light Illumination
2.10. Electroretinography (ERG)
2.11. HE Staining and TUNEL Assay
2.12. Immunofluorescence Analysis
2.13. Statistical Analysis
3. Results
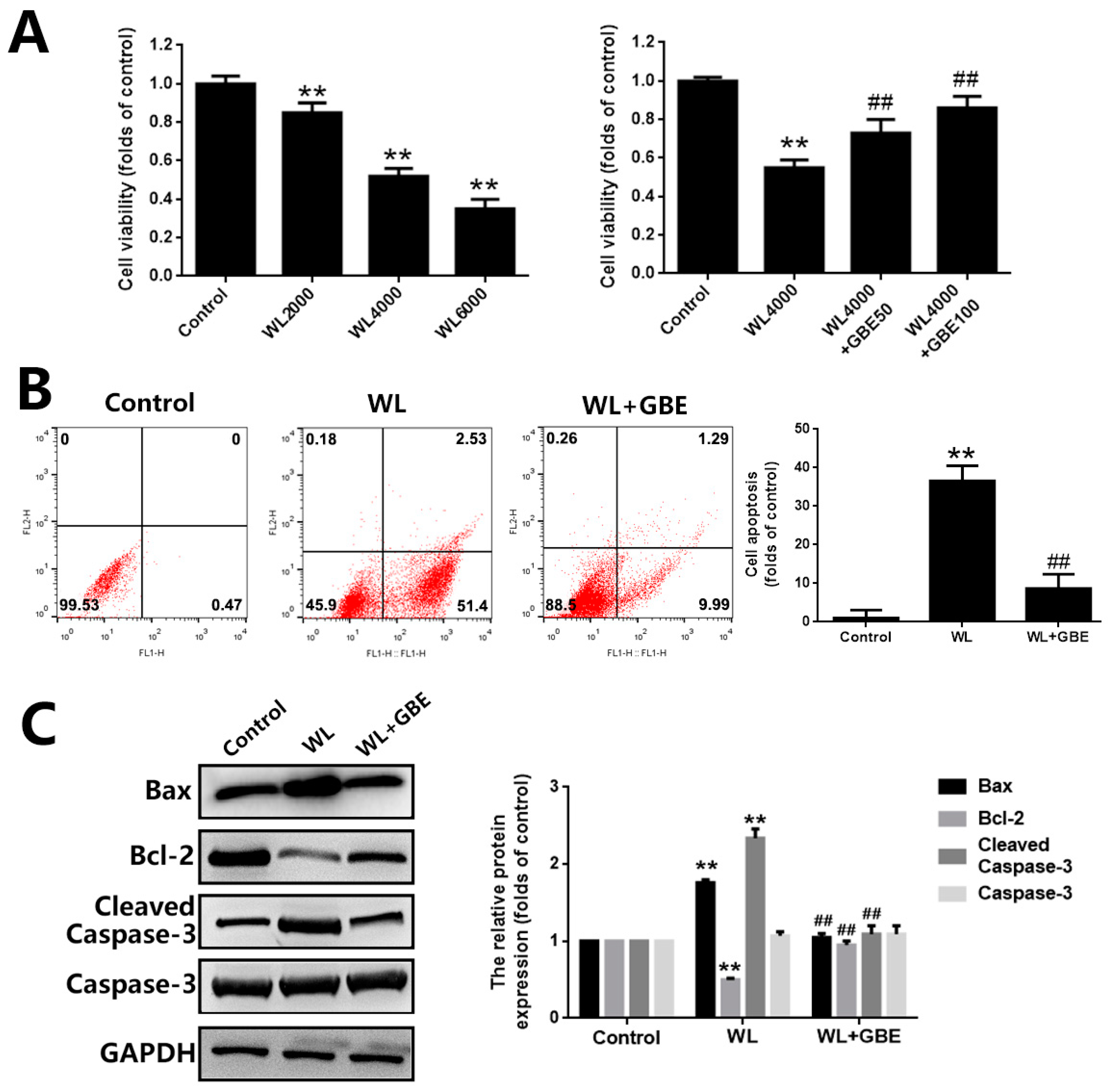
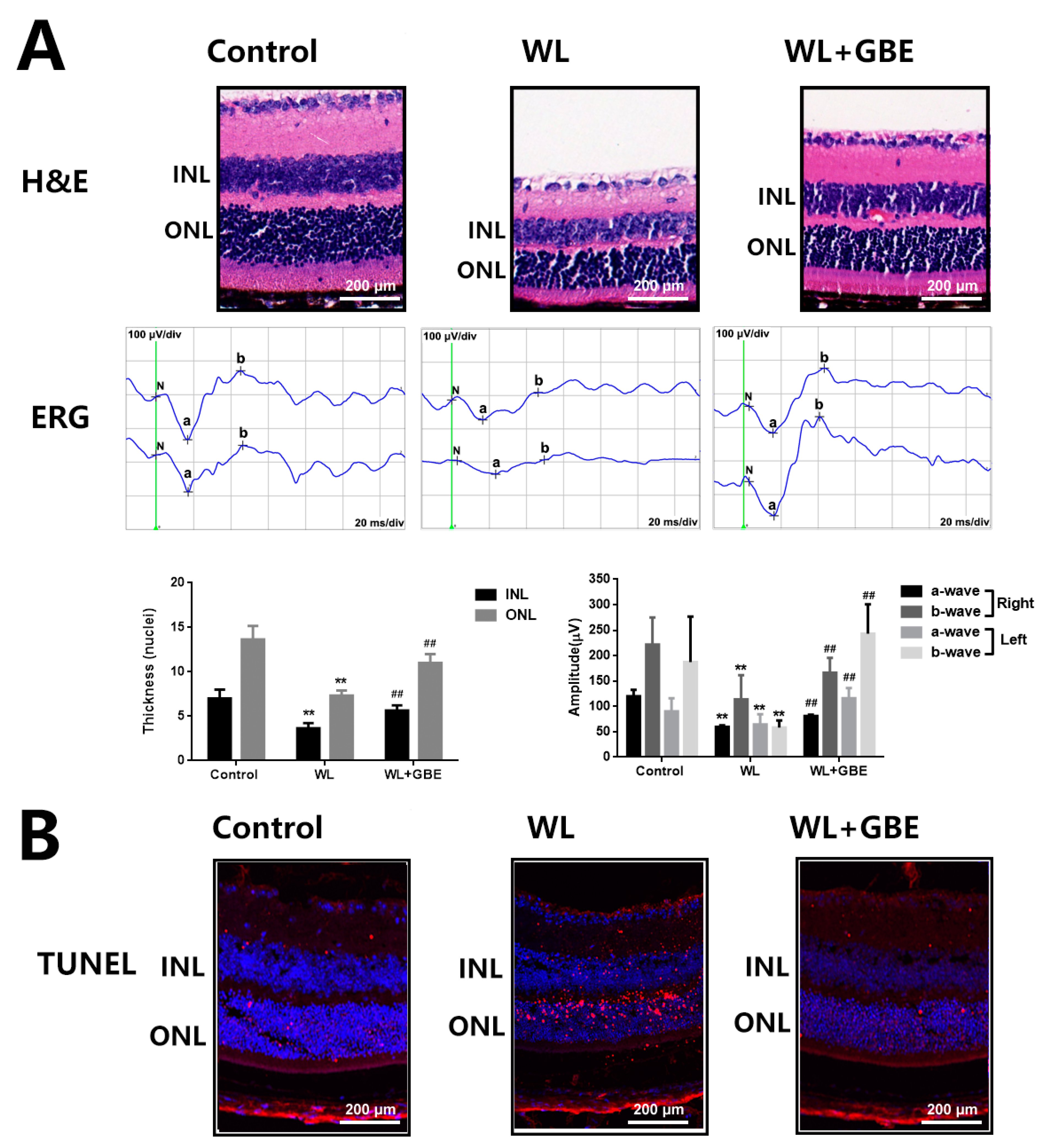
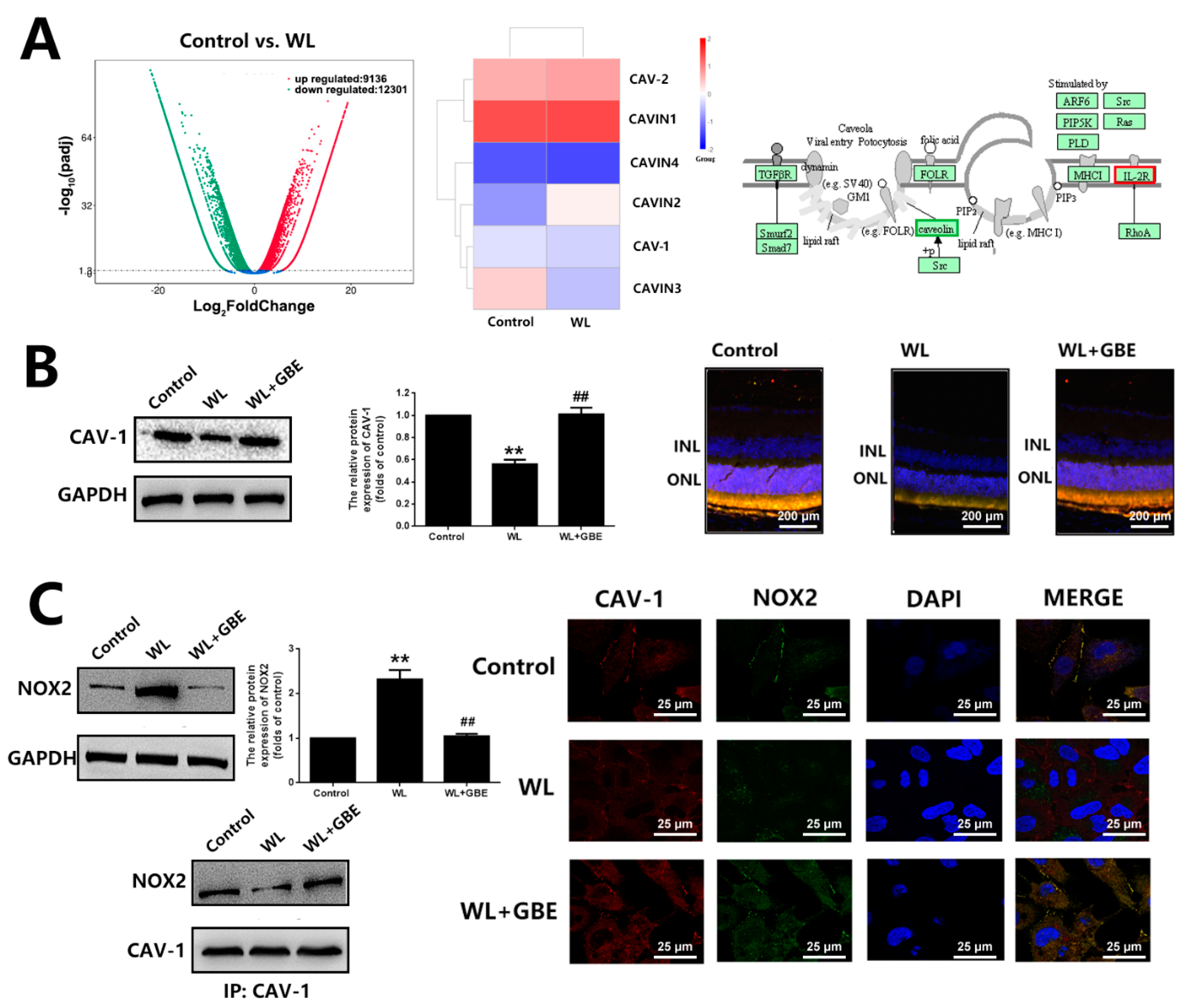
3.1. GBE Attenuates Photoreceptor Degeneration in In Vitro and In Vivo Models Exposed to WL Illumination
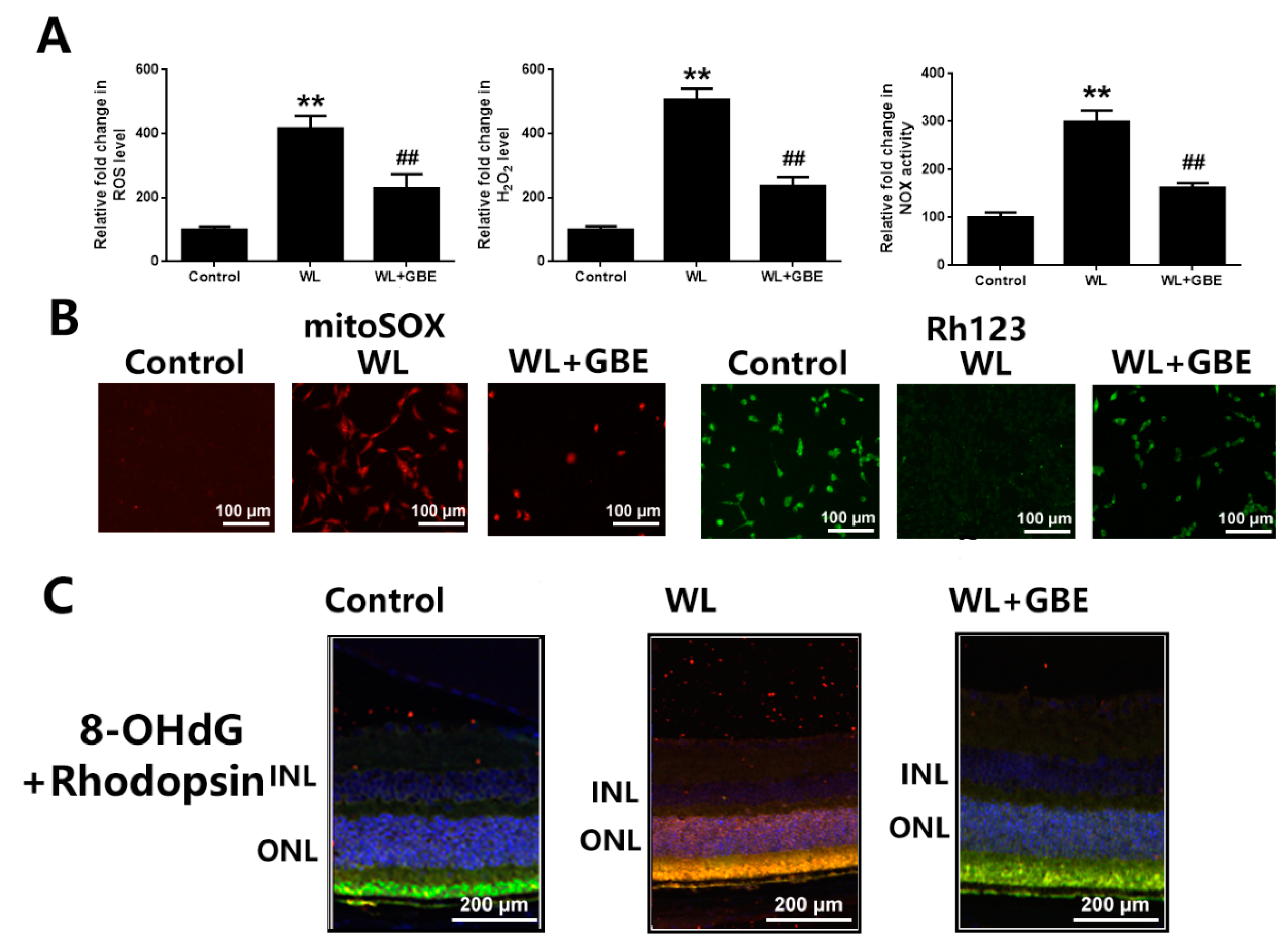
3.2. GBE Alleviated Redoxosome-Dependent Oxidative Stress and Mitochondrial Dysfunction
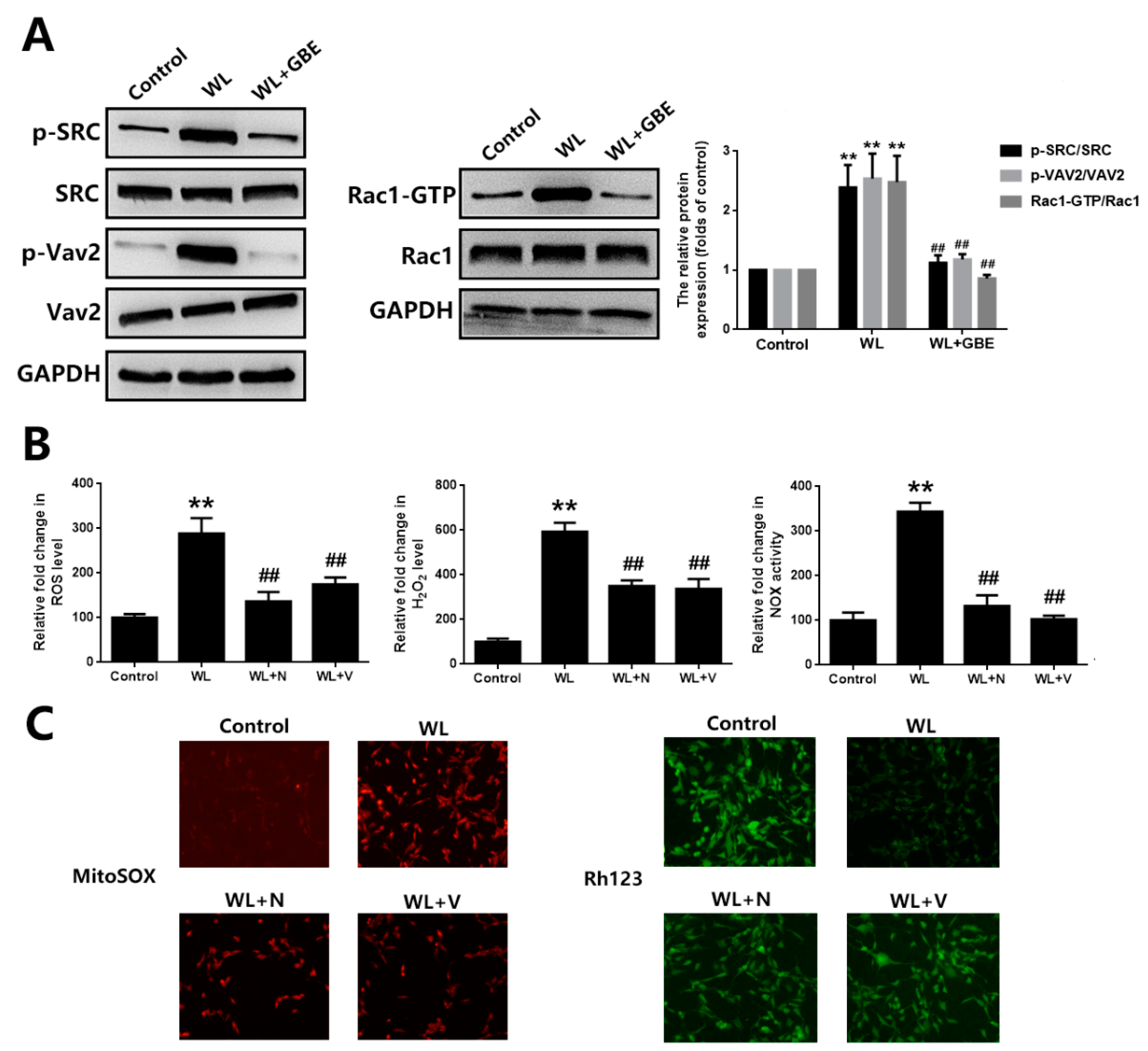
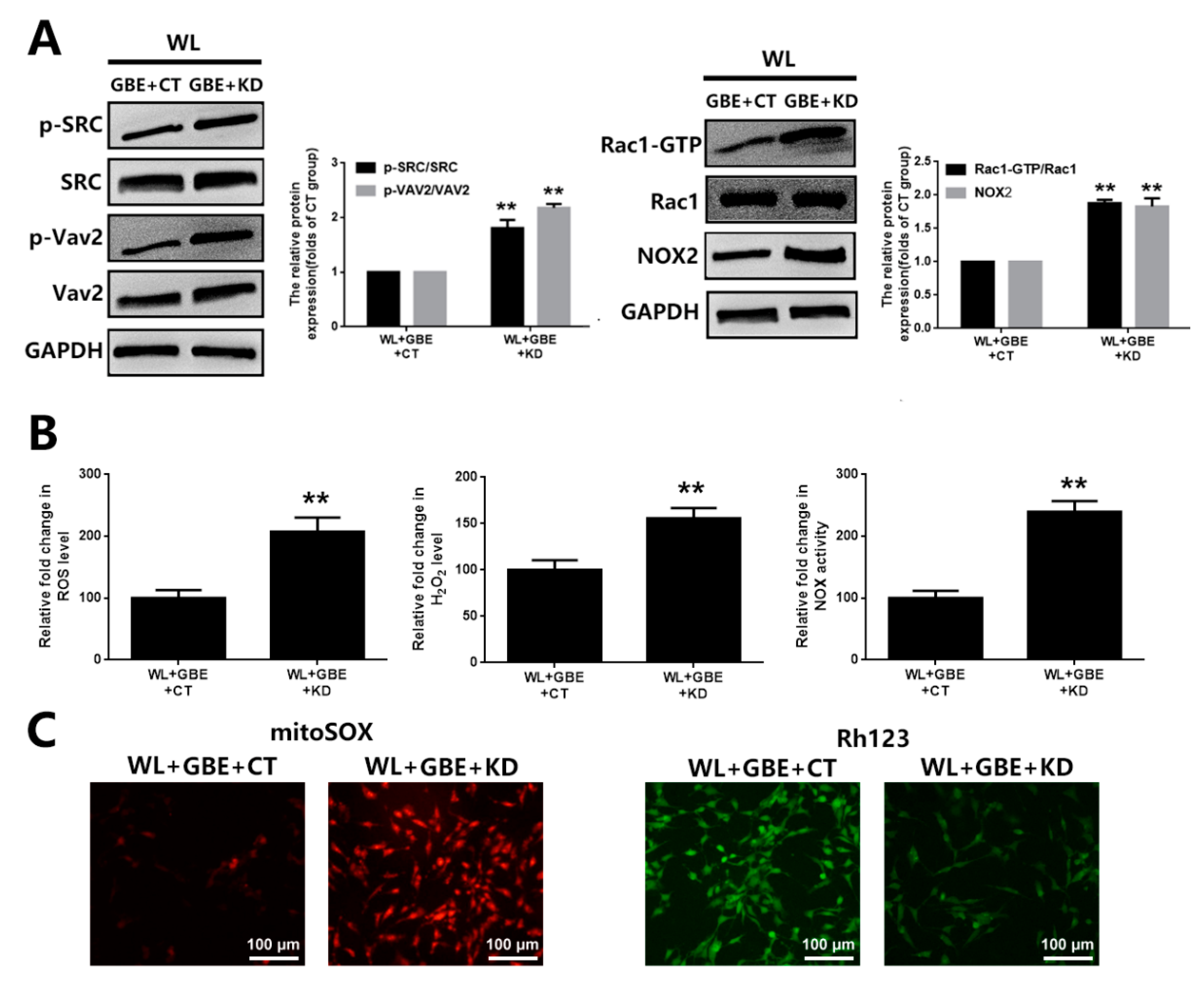
3.3. GBE Reduces Redoxosome Activation by Influencing the Interaction of CAV-1 and NOX2
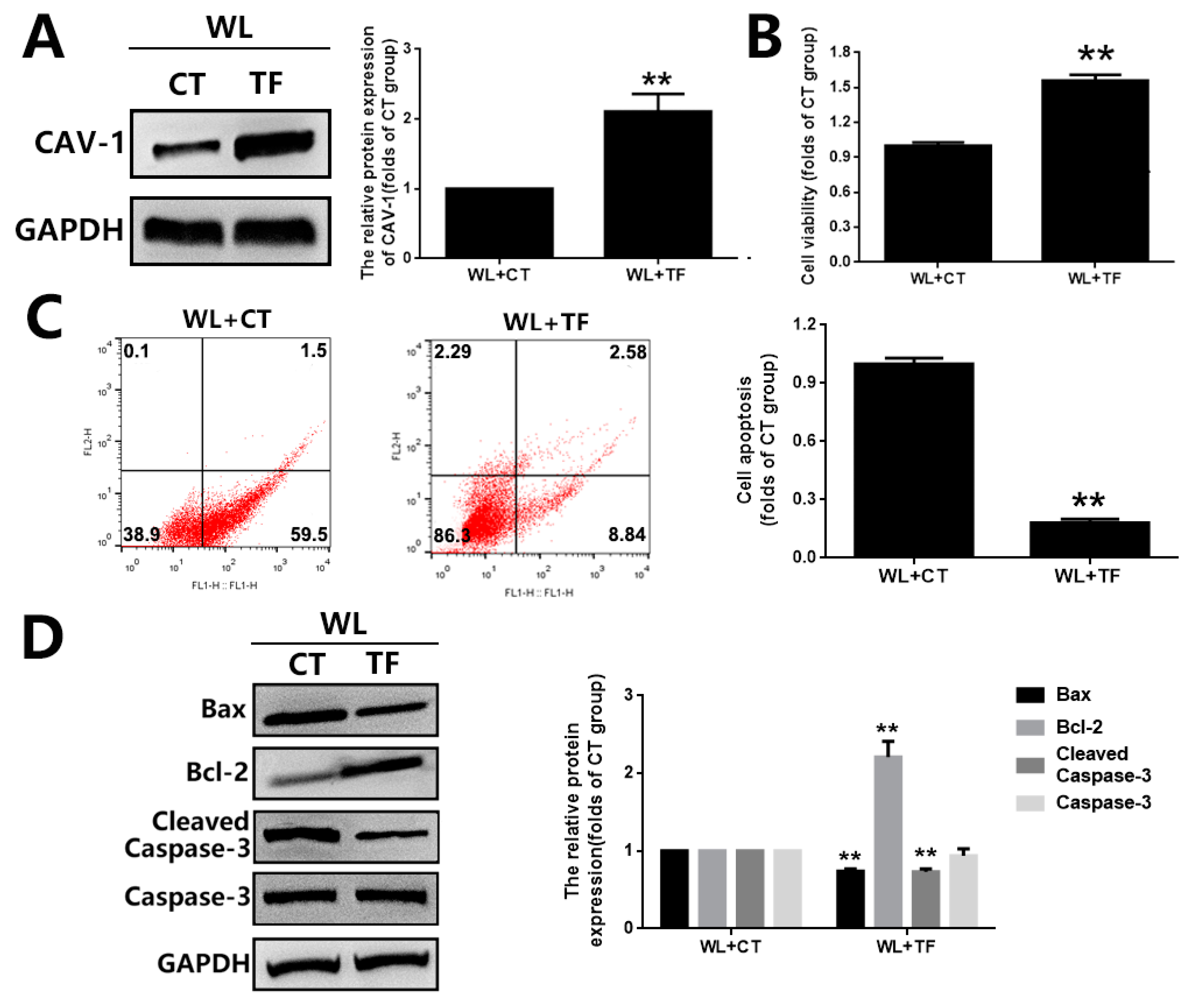
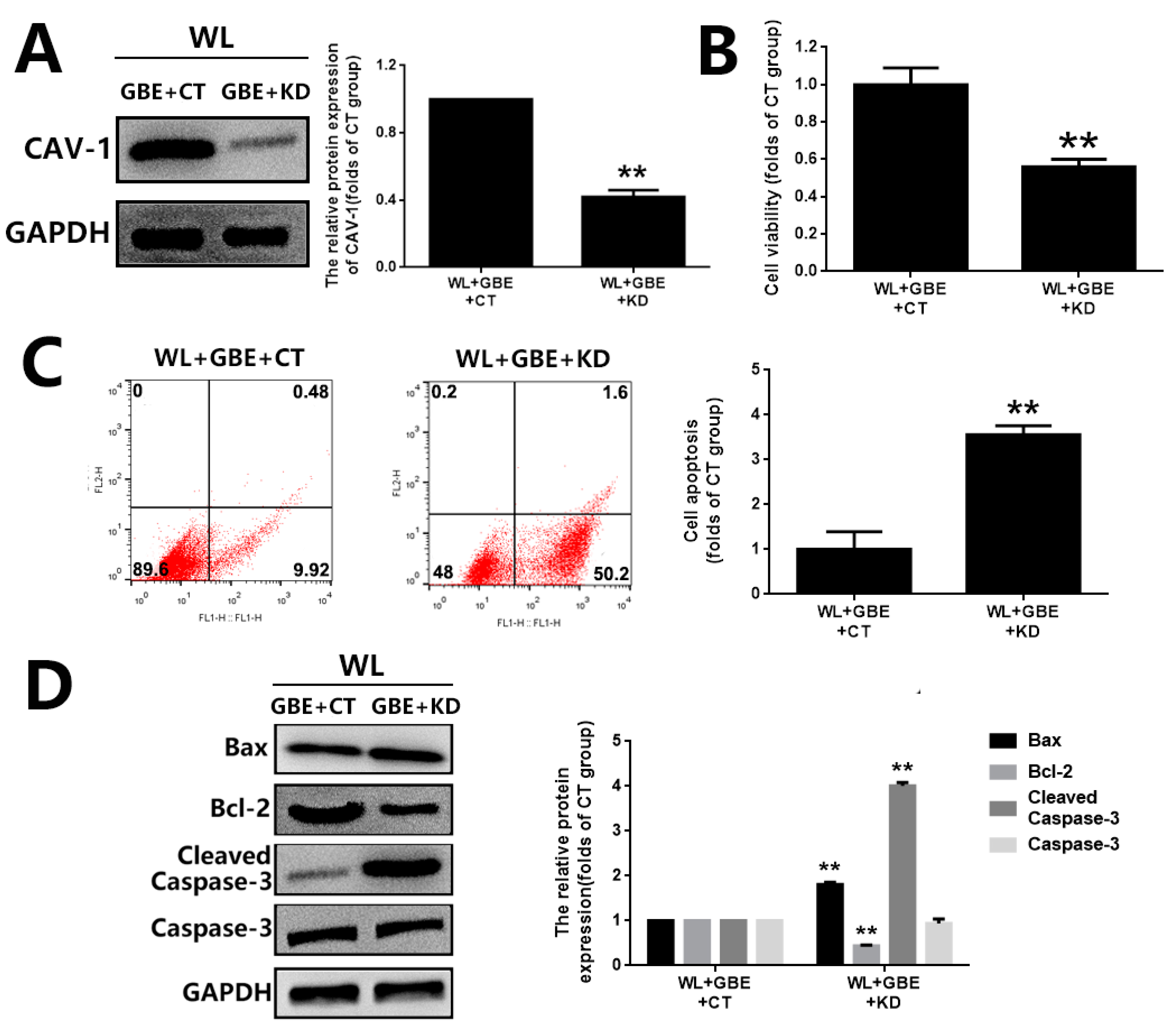
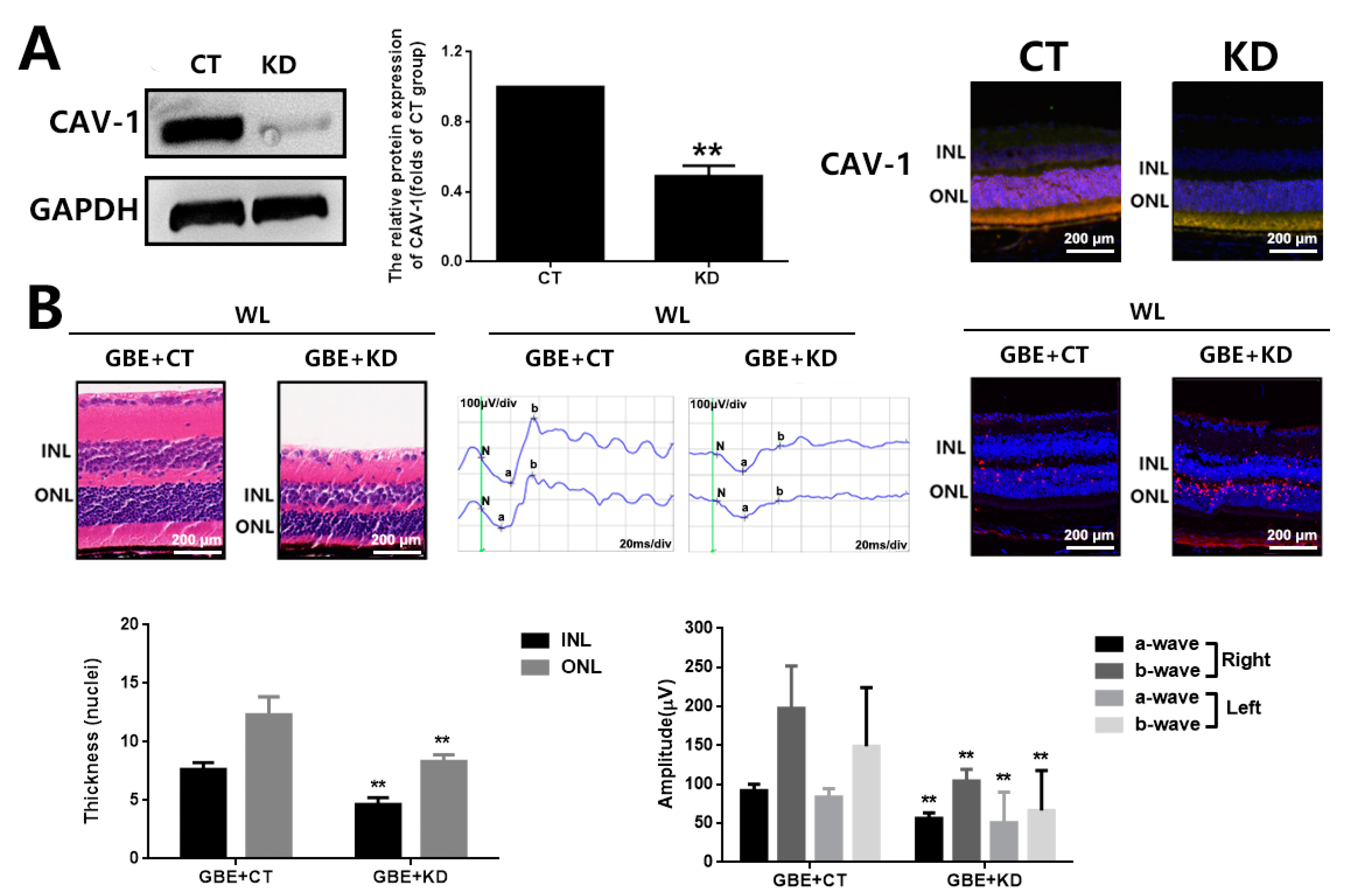
3.4. The Cytoprotective Effect of GBE on Photoreceptor Degeneration Is Exerted in a CAV-1 Dependent Manner
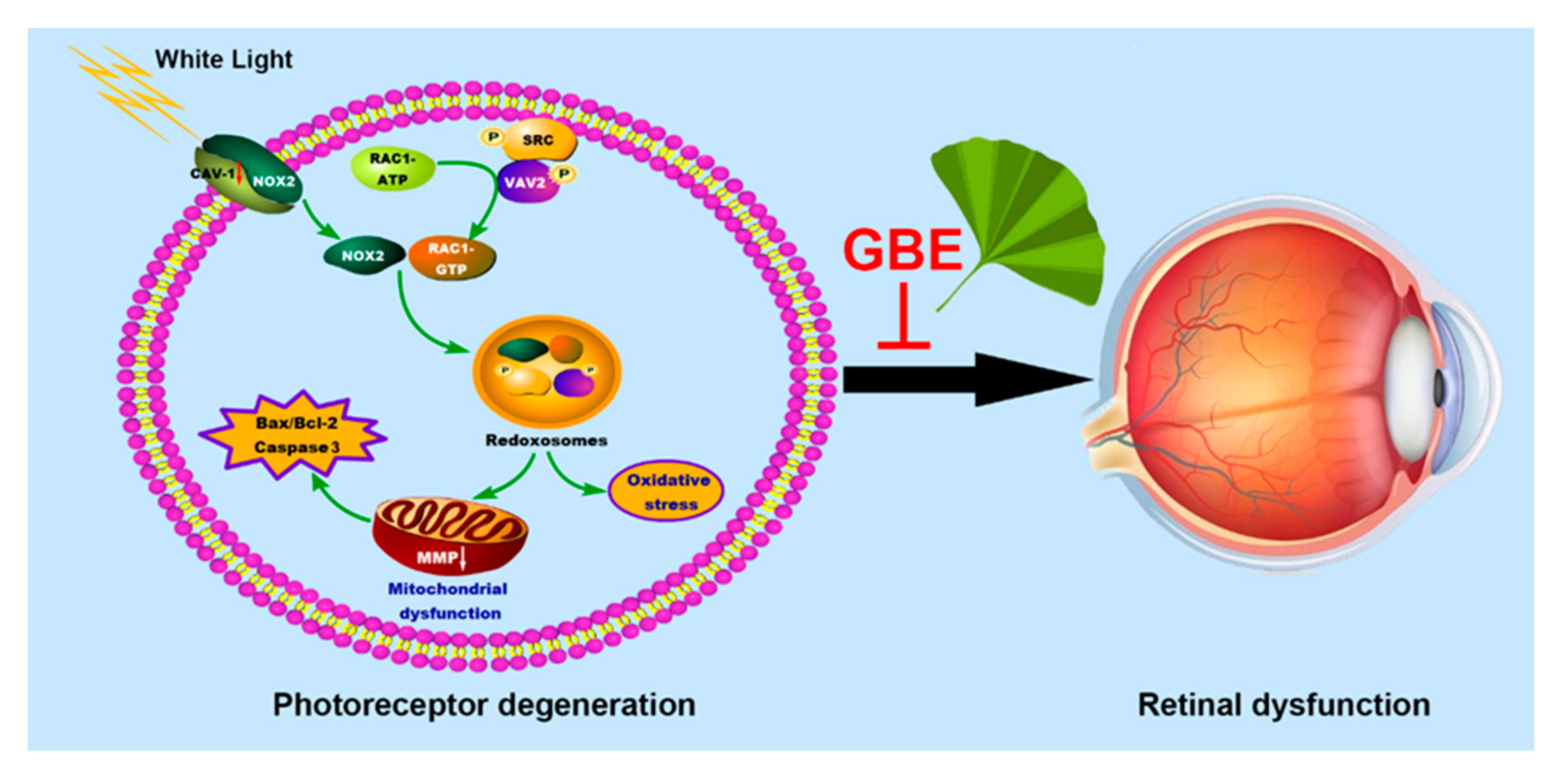
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, C.J.; Ma, Y.; Jin, Z.B. The road to restore vision with photoreceptor regeneration. Exp. Eye Res. 2021, 202, 108283. [Google Scholar] [CrossRef] [PubMed]
- Arshavsky, V.Y.; Burns, M.E. Photoreceptor signaling: Supporting vision across a wide range of light intensities. J. Biol. Chem. 2012, 287, 1620–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef]
- Nowak, J.Z. Age-related macular degeneration (AMD): Pathogenesis and therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar]
- Jaadane, I.; Boulenguez, P.; Chahory, S.; Carré, S.; Savoldelli, M.; Jonet, L.; Behar-Cohen, F.; Martinsons, C.; Torriglia, A. Retinal damage induced by commercial light emitting diodes (LEDs). Free Radic. Biol. Med. 2015, 84, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef]
- Yu, T.Y.; Acosta, M.L.; Ready, S.; Cheong, Y.L.; Kalloniatis, M. Light exposure causes functional changes in the retina: Increased photoreceptor cation channel permeability, photoreceptor apoptosis, and altered retinal metabolic function. J. Neurochem. 2007, 103, 714–724. [Google Scholar] [CrossRef]
- Kong, H.; Ren, X.; Zhang, H.; Wang, N.; Zhang, C.; Li, L.; Xia, X.; Kong, L.; Zhang, M.; Xu, M. Thioredoxin is a potential therapy for light-induced photoreceptor degeneration in diabetic mice. Neuro Endocrinol. Lett. 2019, 39, 561–566. [Google Scholar]
- Grimm, C.; Remé, C.E. Light Damage Models of Retinal Degeneration. Methods Mol. Biol. 2019, 1834, 167–178. [Google Scholar] [CrossRef]
- Benedetto, M.M.; Contin, M.A. Oxidative Stress in Retinal Degeneration Promoted by Constant LED Light. Front. Cell. Neurosci. 2019, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Baksheeva, V.E.; Tiulina, V.V.; Tikhomirova, N.K.; Gancharova, O.S.; Komarov, S.V.; Philippov, P.P.; Zamyatnin, A.A., Jr.; Senin, I.I.; Zernii, E.Y. Suppression of Light-Induced Oxidative Stress in the Retina by Mitochondria-Targeted Antioxidant. Antioxidants 2018, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.W.; Zhang, H.; Grant, S.J.; Wan, X.; Li, G. Single herbal medicine for diabetic retinopathy. Cochrane Database Syst. Rev. 2018, 12, CD007939. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.H.; Lam, H.I.; Choi, K.Y.; Li, S.Z.; Lakshmanan, Y.; Yu, W.Y.; Chang, R.C.; Lai, J.S.; So, K.F. Delay of cone degeneration in retinitis pigmentosa using a 12-month treatment with Lycium barbarum supplement. J. Ethnopharmacol. 2019, 236, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Jahanshahi, M.; Nikmahzar, E.; Yadollahi, N.; Ramazani, K. Protective effects of Ginkgo biloba extract (EGB 761) on astrocytes of rat hippocampus after exposure with scopolamine. Anat. Cell Biol. 2012, 45, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yallapragada, P.R.; Velaga, M.K. Effect of Ginkgo biloba Extract on Lead-Induced Oxidative Stress in Different Regions of Rat Brain. J. Environ. Pathol. Toxicol. Oncol. 2015, 34, 161–173. [Google Scholar] [CrossRef]
- Kanowski, S.; Herrmann, W.M.; Stephan, K.; Wierich, W.; Hörr, R. Proof of efficacy of the ginkgo biloba special extract EGb 761 in outpatients suffering from mild to moderate primary degenerative dementia of the Alzheimer type or multi-infarct dementia. Pharmacopsychiatry 1996, 29, 47–56. [Google Scholar] [CrossRef]
- Mix, J.A.; Crews, W.D., Jr. A double-blind, placebo-controlled, randomized trial of Ginkgo biloba extract EGb 761 in a sample of cognitively intact older adults: Neuropsychological findings. Hum. Psychopharmacol. 2002, 17, 267–277. [Google Scholar] [CrossRef]
- Mohammed, N.A.; Abdou, H.M.; Tass, M.A.; Alfwuaires, M.; Abdel-Moneim, A.M.; Essawy, A.E. Oral Supplements of Ginkgo biloba Extract Alleviate Neuroinflammation, Oxidative Impairments and Neurotoxicity in Rotenone-Induced Parkinsonian Rats. Curr. Pharm. Biotechnol. 2020, 21, 1259–1268. [Google Scholar] [CrossRef]
- Johnson, S.K.; Diamond, B.J.; Rausch, S.; Kaufman, M.; Shiflett, S.C.; Graves, L. The effect of Ginkgo biloba on functional measures in multiple sclerosis: A pilot randomized controlled trial. Explore 2006, 2, 19–24. [Google Scholar] [CrossRef]
- Decker, L.; Basta, D.; Burkart, M.; Ernst, A. Balance Training With Vibrotactile Neurofeedback and Ginkgo Biloba Extract in Age-Related Vertigo. Front. Neurol. 2021, 12, 691917. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Srivastav, S.; Castellani, R.J.; Plascencia-Villa, G.; Perry, G. Neuroprotective and Antioxidant Effect of Ginkgo biloba Extract Against AD and Other Neurological Disorders. Neurotherapeutics 2019, 16, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Coley, N.; Ousset, P.J.; Berrut, G.; Dartigues, J.F.; Dubois, B.; Grandjean, H.; Pasquier, F.; Piette, F.; Robert, P.; et al. Long-term use of standardised Ginkgo biloba extract for the prevention of Alzheimer’s disease (GuidAge): A randomised placebo-controlled trial. Lancet Neurol. 2012, 11, 851–859. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, Z.; Wang, K.; Zhu, X.; Ali, Y.; Shu, W.; Bao, X.; Zhu, L.; Fan, X.; Murray, M.; et al. Procyanidin B2 and rutin in Ginkgo biloba extracts protect human retinal pigment epithelial (RPE) cells from oxidative stress by modulating Nrf2 and Erk1/2 signalling. Exp. Eye Res. 2021, 207, 108586. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.R. Ginkgo biloba extract for age-related macular degeneration. Cochrane Database Syst. Rev. 2013, 2013, CD001775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Wu, X.; Gong, Y.; Song, Y.; Qiu, Q.; Li, C. Intraperitoneal injection of Ginkgo biloba extract enhances antioxidation ability of retina and protects photoreceptors after light-induced retinal damage in rats. Curr. Eye Res. 2007, 32, 471–479. [Google Scholar] [CrossRef]
- Fies, P.; Dienel, A. [Ginkgo extract in impaired vision--treatment with special extract EGb 761 of impaired vision due to dry senile macular degeneration]. Wien. Med. Wochenschr. 2002, 152, 423–426. [Google Scholar] [CrossRef]
- Ranchon, I.; Gorrand, J.M.; Cluzel, J.; Droy-Lefaix, M.T.; Doly, M. Functional protection of photoreceptors from light-induced damage by dimethylthiourea and Ginkgo biloba extract. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1191–1199. [Google Scholar]
- Natoli, R.; Jiao, H.; Barnett, N.L.; Fernando, N.; Valter, K.; Provis, J.M.; Rutar, M. A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp. Eye Res. 2016, 147, 114–127. [Google Scholar] [CrossRef] [Green Version]
- Yoo, D.Y.; Nam, Y.; Kim, W.; Yoo, K.Y.; Park, J.; Lee, C.H.; Choi, J.H.; Yoon, Y.S.; Kim, D.W.; Won, M.H.; et al. Effects of Ginkgo biloba extract on promotion of neurogenesis in the hippocampal dentate gyrus in C57BL/6 mice. J. Vet. Med. Sci. 2011, 73, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Bai, Y.; Wang, B.; Cui, H.; Wu, H.; Lv, J.R.; Mei, Y.; Zhang, J.S.; Liu, S.; Qi, L.W.; et al. Suppression of experimental abdominal aortic aneurysms in the mice by treatment with Ginkgo biloba extract (EGb 761). J. Ethnopharmacol. 2013, 150, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.T. In situ detection of apoptosis by the TUNEL assay: An overview of techniques. Methods Mol. Biol. 2011, 682, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, C.; Yu, M.; Feng, S.; Ma, Y.; Liu, Z.; Zhang, J.; Ding, T.; Li, B.; Wang, X. The Effect of Ginkgo Biloba Dropping Pills on Hemorheology and Blood Lipid: A Systematic Review of Randomized Trials. Evid. Based Complement. Altern. Med. 2019, 2019, 2609625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Zhang, P.; Li, J.; Liu, T.; Zhang, Y.; Wang, Q.; Zhang, J.; Lu, X.; Fan, X. Neuroprotective effects of Ginkgo biloba dropping pills in Parkinson’s disease. J. Pharm. Anal. 2021, 11, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhong, W.; Hu, Z.; Tang, X. A review of the role of CAV-1 in neuropathology and neural recovery after ischemic stroke. J. Neuroinflamm. 2018, 15, 348. [Google Scholar] [CrossRef]
- de Almeida, C.J.G. Caveolin-1 and Caveolin-2 Can Be Antagonistic Partners in Inflammation and Beyond. Front. Immunol. 2017, 8, 1530. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Fliesler, S.J.; Zhao, Y.Y.; Stallcup, W.B.; Cohen, A.W.; Elliott, M.H. Loss of caveolin-1 causes blood-retinal barrier breakdown, venous enlargement, and mural cell alteration. Am. J. Pathol. 2014, 184, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; McClellan, M.E.; Tanito, M.; Garteiser, P.; Towner, R.; Bissig, D.; Berkowitz, B.A.; Fliesler, S.J.; Woodruff, M.L.; Fain, G.L.; et al. Loss of caveolin-1 impairs retinal function due to disturbance of subretinal microenvironment. J. Biol. Chem. 2012, 287, 16424–16434. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.H.; Fliesler, S.J.; Ghalayini, A.J. Cholesterol-dependent association of caveolin-1 with the transducin alpha subunit in bovine photoreceptor rod outer segments: Disruption by cyclodextrin and guanosine 5′-O-(3-thiotriphosphate). Biochemistry 2003, 42, 7892–7903. [Google Scholar] [CrossRef]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Black, S.M.; Rafikov, R.; Dou, H.; Bagi, Z.; Han, W.; et al. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Anagnostopoulou, A.; Camargo, L.L.; Rodrigues, D.; Montezano, A.C.; Touyz, R.M. Importance of cholesterol-rich microdomains in the regulation of Nox isoforms and redox signaling in human vascular smooth muscle cells. Sci. Rep. 2020, 10, 17818. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ni, J.; Zhu, X.; Zhu, L.; Li, Y.; Zhou, F. Ginkgo biloba extract protects human neuroblastoma SH-SY5Y cells against oxidative glutamate toxicity by activating redoxosome-p66Shc. Exp. Ther. Med. 2021, 22, 951. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Chen, Y.; Zhu, X.; Zou, W.; Zhou, F. Ginkgo biloba Extract Attenuates Light-Induced Photoreceptor Degeneration by Modulating CAV-1—Redoxosome Signaling. Antioxidants 2022, 11, 1268. https://doi.org/10.3390/antiox11071268
Wang K, Chen Y, Zhu X, Zou W, Zhou F. Ginkgo biloba Extract Attenuates Light-Induced Photoreceptor Degeneration by Modulating CAV-1—Redoxosome Signaling. Antioxidants. 2022; 11(7):1268. https://doi.org/10.3390/antiox11071268
Chicago/Turabian StyleWang, Ke, Yuan Chen, Xue Zhu, Wenjun Zou, and Fanfan Zhou. 2022. "Ginkgo biloba Extract Attenuates Light-Induced Photoreceptor Degeneration by Modulating CAV-1—Redoxosome Signaling" Antioxidants 11, no. 7: 1268. https://doi.org/10.3390/antiox11071268