
Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases

Abstract
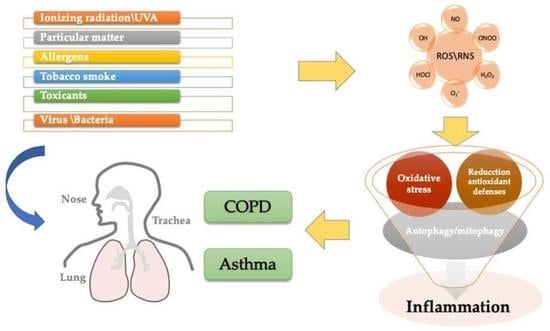
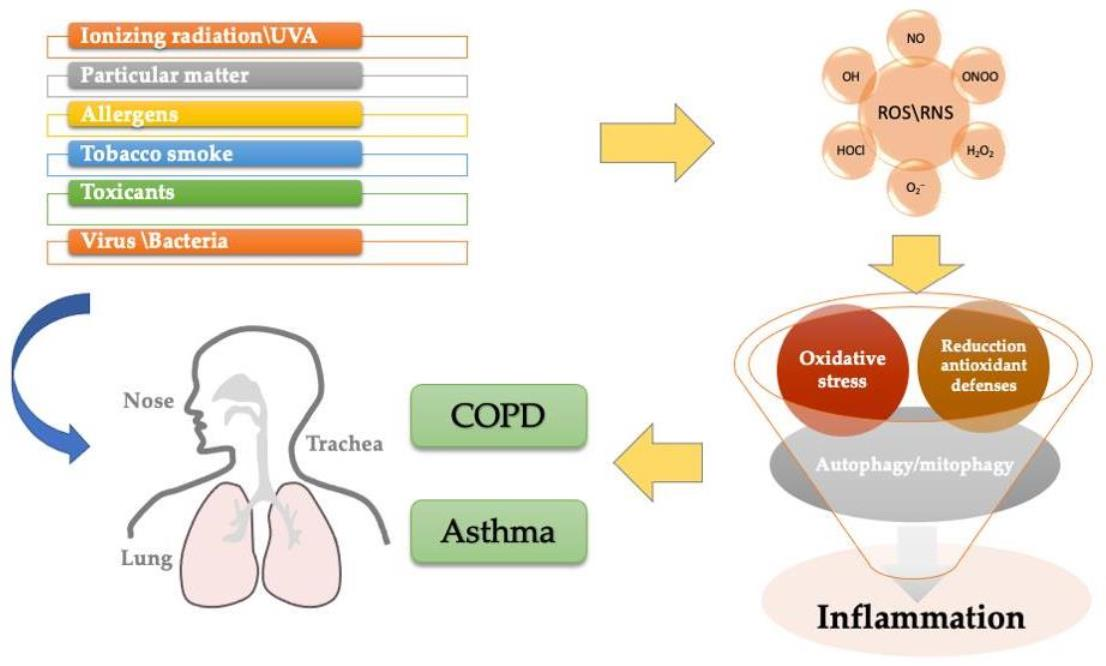
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
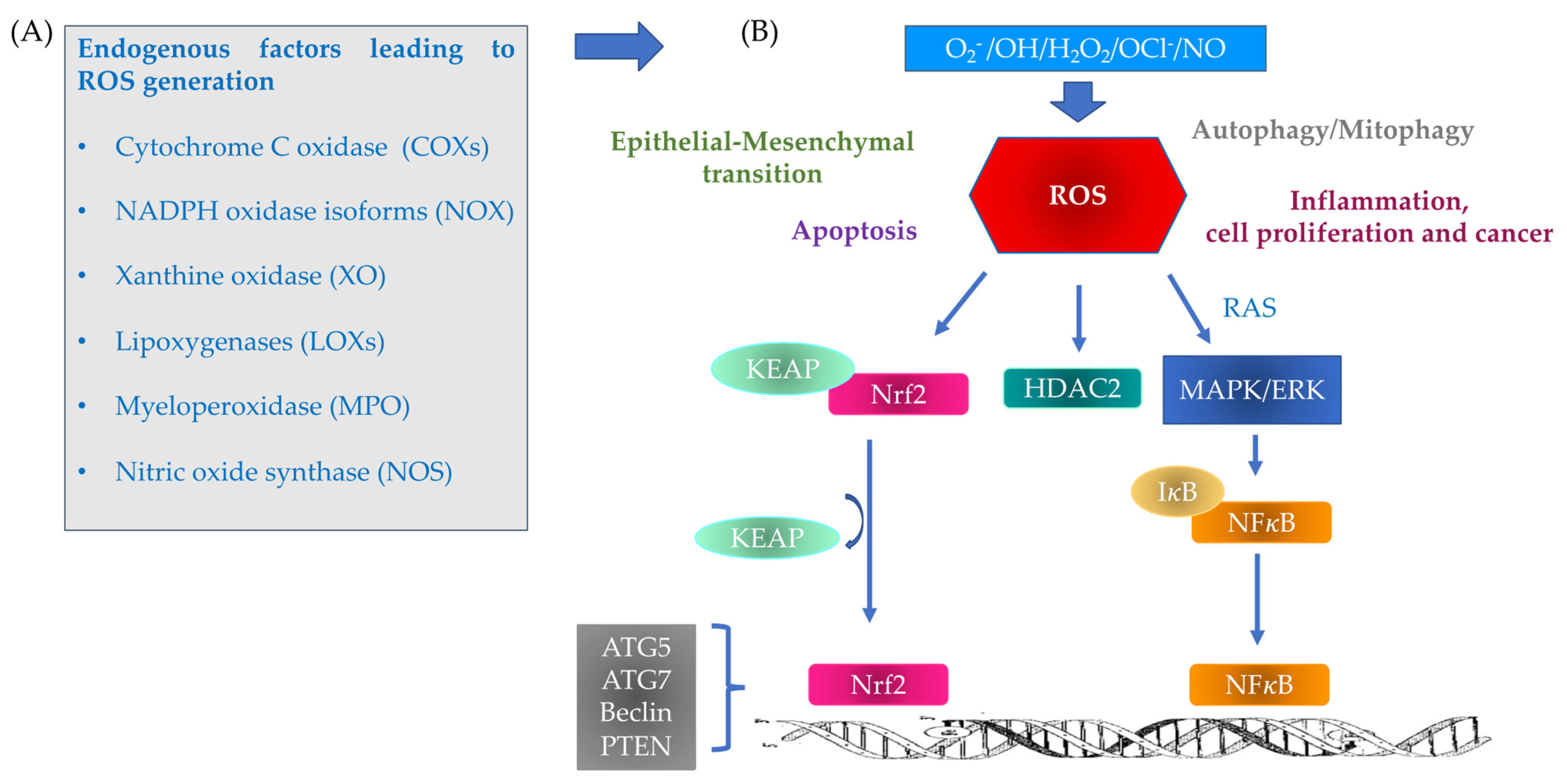
2. Mechanism of Oxidative Stress
3. ROS Inflammation and Autophagy/Mitophagy Processes
4. Oxidative Stress in Asthma
5. Oxidative Stress in COPD
6. Environmental Pollution and OS in the Airways
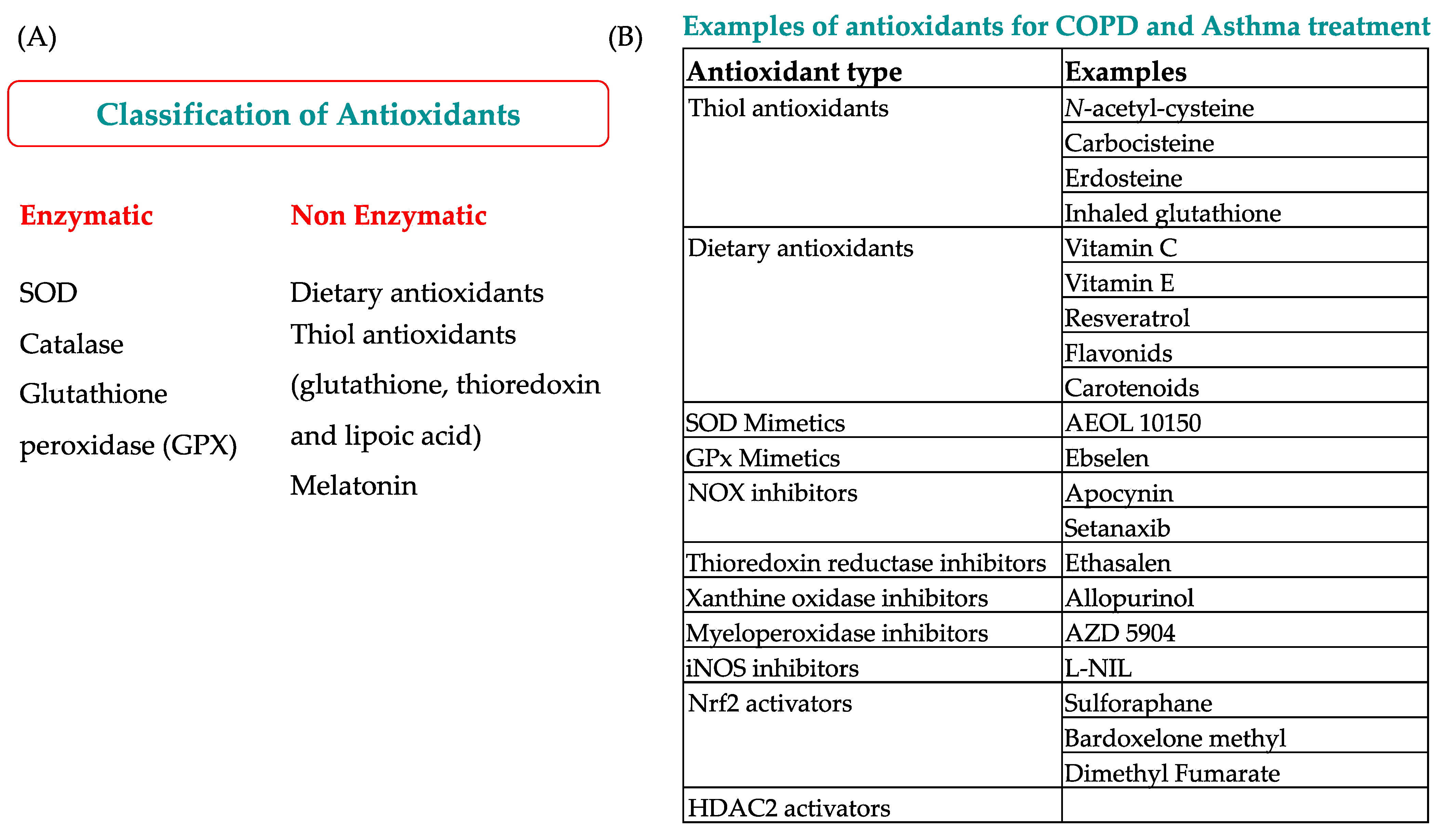
7. New Pharmacological Perspectives in Airway Diseases: Antioxidants
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Oxidative stress | OS |
| Reactive oxygen species | ROS |
| Reactive nitrogen species | RNS |
| Cytochrome c oxidase | COX |
| NADPH oxidase | NOX |
| Myeloperoxidase | MPO |
| Xanthine oxidase | XO |
| Nitric oxidase synthase | NOS |
| Dual oxidases | DUOX |
| Hydroperoxyeicosatetraenoic acids | HPETEs |
| Cyclooxygenases | COXs |
| Arachidonic acid | AA |
| Nitric oxide | NO |
| Tumor microenvironment | TME |
| Cytochrome P | CYP |
| Oxidative phosphorylation system | OXPHOS |
| Mitochondrial ROS | MtROS |
| Hypoxia-inducible factor-1 | HIF-1 |
| Nuclear Factor kappa B | NF-kB |
| Nuclear factor erythroid 2-related factor | Nfr2 |
| Kelch Like ECH Associated Protein 1 | Keap1 |
| PTEN-induced kinase 1 | PINK1 |
| Cellular prion protein | PrPC |
| Cigarette smoke | CS |
| Pathogen-associated molecular patterns | PAMP |
| Toll-Like Receptor | TLR |
References
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cui, H.; Xu, Y. Quantitative Estimation of Oxidative Stress in Cancer Tissue Cells Through Gene Expression Data Analyses. Front. Genet. 2020, 11, 494. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals, antioxidants, and human disease: Curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Jan Mohammad, M.; Charitra, M.R. A gentle introduction to gasotransmitters with special reference to nitric oxide: Biological and chemical implications. Rev. Inorg. Chem. 2018, 38, 193–220. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Antosova, M.; Mokra, D.; Pepucha, L.; Plevkova, J.; Buday, T.; Sterusky, M.; Bencova, A. Physiology of nitric oxide in the respiratory system. Physiol. Res. 2017, 66 (Suppl. S2), S159–S172. [Google Scholar] [CrossRef]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox. Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef] [Green Version]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Al Ghouleh, I.; Khoo, N.K.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011, 51, 1271–1288. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Beers, M.F.; Morrisey, E.E. The three R’s of lung health and disease: Repair, remodeling, and regeneration. J. Clin. Investig. 2011, 121, 2065–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghio, A.J.; Soukup, J.M.; Madden, M.C. The toxicology of air pollution predicts its epidemiology. Inhal. Toxicol. 2018, 30, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH Oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar] [CrossRef]
- Kolářová, H.; Binó, L.; Pejchalová, K.; Kubala, L. The expression of NADPH oxidases and production of reactive oxygen species by human lung adenocarcinoma epithelial cell line A549. Folia Biol. 2010, 56, 211–217. [Google Scholar]
- Van der Vliet, A. NADPH oxidases in lung biology and pathology: Host defense enzymes, and more. Free Radic. Biol. Med. 2008, 44, 938–955. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK10757/ (accessed on 12 November 2022).
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [Green Version]
- Gujral, J.S.; Hinson, J.A.; Jaeschke, H. Chlorotyrosine protein adducts are reliable biomarkers of neutrophil-induced cytotoxicity in vivo. Comp. Hepatol. 2004, 3 (Suppl. S1), S48. [Google Scholar] [CrossRef]
- Van Der Vliet, A.; Nguyen, M.N.; Shigenaga, M.K.; Eiserich, J.P.; Marelich, G.P.; Cross, C.E. Myeloperoxidase and protein oxidation in cystic fibrosis. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2000, 279, L537–L546. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef] [Green Version]
- Poss, W.B.; Huecksteadt, T.P.; Panus, P.C.; Freeman, B.A.; Hoidal, J.R. Regulation of xanthine dehydrogenase and xanthine oxidase activity by hypoxia. Am. J. Physiol. 1996, 270, L941–L946. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayarri, M.A.; Milara, J.; Estornut, C.; Cortijo, J. Nitric Oxide System and Bronchial Epithelium: More Than a Barrier. Front. Physiol. 2021, 12, 687381. [Google Scholar] [CrossRef] [PubMed]
- Aminuddin, F.; Hackett, T.L.; Stefanowicz, D.; Saferali, A.; Paré, P.D.; Gulsvik, A.; Bakke, P.; Cho, M.H.; Litonjua, A.; Lomas, D.A.; et al. Nitric oxide synthase polymorphisms, gene expression and lung function in chronic obstructive pulmonary disease. BMC Pulm. Med. 2013, 13, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesslinger, C.; Strub, A.; Boer, R.; Ulrich, W.R.; Lehner, M.D.; Braun, C. Inhibition of inducible nitric oxide synthase in respiratory diseases. Biochem. Soc. Trans. 2009, 37, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, V.; Reiß, K.; Kollau, A.; Mayer, B.; Gorren, A.C.F. Site and mechanism of uncoupling of nitric-oxide synthase: Uncoupling by monomerization and othr misconceptions. Nitric Oxide 2019, 89, 14–21. [Google Scholar] [CrossRef] [PubMed]
- García-Sánchez, A.; Miranda-Díaz, A.G.; Cardona-Muñoz, E.G. The Role of Oxidative Stress in Physiopathology and Pharmacological Treatment with Pro- and Antioxidant Properties in Chronic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 2082145. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Rojanasakul, Y.; Vallyathan, V. Inflammation and lung cancer: Roles of reactive oxygen/nitrogen species. J. Toxicol. Environ. Health B Crit. Rev. 2008, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Granot, Z.; Fridlender, Z.G. The diversity of circulating neutrophils in cancer. Immunobiology 2017, 222, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef] [PubMed]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, D.J.; Islam, M.N.; Das, S.R.; Huertas, A.; Quadri, S.K.; Horiuchi, K.; Inamdar, N.; Emin, M.T.; Lindert, J.; Ten, V.S. Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J. Clin. Investig. 2011, 121, 1986–1999. [Google Scholar] [CrossRef] [Green Version]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Li, N.L.; Li, J.T.; Yu, F.; Zhao, Y.L.; Wang, L.; Yi, J.; Wang, L.; Bian, J.F.; et al. Subanesthetic isoflurane reduces zymosan-induced inflammation in murine Kupffer cells by inhibiting ROS-activated p38 MAPK/NF-κB signaling. Oxid. Med. Cell. Longev. 2014, 2014, 851692. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Nakajima, A.; Sakon-Komazawa, S.; Piao, J.H.; Xue, X.; Okumura, K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell. Death. Differ. 2006, 13, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Wu, T.; Zhao, F.; Lau, A.; Birch, C.M.; Zhang, D.D. KPNA6 (Importin α7)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol. Cell. Biol. 2011, 31, 1800–1811. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA. 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Slocum, S.L.; Kensler, T.W. Nrf2: Control of sensitivity to carcinogens. Arch. Toxicol. 2011, 85, 273–284. [Google Scholar] [CrossRef]
- Chavda, V.; Chaurasia, B.; Garg, K.; Deora, H.; Umana, G.E.; Palmisciano, P.; Scalia, G.; Lu, B. Molecular mechanisms of oxidative stress in stroke and cancer. Brain Disord. 2022, 5, 100029. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 18. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell. Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Montuschi, P.; Corradi, M.; Ciabattoni, G.; Nightingale, J.; Kharitonov, S.A.; Barnes, P.J. Increased 8-isoprostane, a marker of oxidative stress, in exhaled condensate of asthma patients. Am. J. Respir. Crit. Care Med. 1999, 160, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredi, P.; Kharitonov, S.A.; Barnes, P.J. Elevation of exhaled ethane concentration in asthma. Am. J. Respir. Crit. Care Med 2000, 162 (4 Pt 1), 1450–1454. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Moon, H.B. The role of oxidative stress in the pathogenesis of asthma. Allergy Asthma Immunol. Res. 2010, 2, 183–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, T.A. Human exhaled breath analysis. Ann. Allergy Asthma Immunol. 2011, 106, 451–456. [Google Scholar] [CrossRef]
- Teng, Y.; Sun, P.; Zhang, J.; Yu, R.; Bai, J.; Yao, X.; Huang, M.; Adcock, I.M.; Barnes, P.J. Hydrogen peroxide in exhaled breath condensate in patients with asthma: A promising biomarker? Chest 2011, 140, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Borrill, Z.L.; Roy, K.; Singh, D. Exhaled breath condensate biomarkers in COPD. Eur. Respir. J. 2008, 32, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Conrad, D.H.; Chow, S.; Tran, V.H.; Yates, D.H.; Thomas, P.S. Collection devices influence the constituents of exhaled breath condensate. Eur. Respir. J. 2007, 30, 807–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, V.; Banga, J.; Silveyra, P. Oxidative stress and cellular pathways of asthma and inflammation: Therapeutic strategies and pharmacological targets. Pharmacol. Ther. 2018, 181, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Horváth, I.; Donnelly, L.E.; Kiss, A.; Kharitonov, S.A.; Lim, S.; Fan Chung, K.; Barnes, P.J. Combined use of exhaled hydrogen peroxide and nitric oxide in monitoring asthma. Am. J. Respir. Crit. Care Med. 1998, 158, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Loukides, S.; Bouros, D.; Papatheodorou, G.; Panagou, P.; Siafakas, N.M. The relationships among hydrogen peroxide in expired breath condensate, airway inflammation, and asthma severity. Chest 2002, 121, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.; Erzurum, S.C. Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 93–124, Erratum in Antioxid. Redox. Signal. 2010, 12, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dut, R.; Dizdar, E.A.; Birben, E.; Sackesen, C.; Soyer, O.U.; Besler, T.; Kalayci, O. Oxidative stress and its determinants in the airways of children with asthma. Allergy 2008, 63, 1605–1609. [Google Scholar] [CrossRef]
- Profita, M.; Montuschi, P.; Bonanno, A.; Riccobono, L.; Montalbano, A.M.; Ciabattoni, G.; Albano, G.D.; Liotta, G.; Bousquet, J.; Gjomarkaj, M.; et al. Novel perspectives in the detection of oral and nasal oxidative stress and inflammation in pediatric united airway diseases. Int. J. Immunopathol. Pharmacol. 2010, 23, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, A.; Chhabra, S.K.; Masood, A.; Raj, H.G. Increased oxidative stress and altered levels of antioxidants in asthma. J Allergy. Clin. Immunol. 2003, 111, 72–78. [Google Scholar] [CrossRef]
- Ozaras, R.; Tahan, V.; Turkmen, S.; Talay, F.; Besirli, K.; Aydin, S.; Uzun, H.; Cetinkaya, A. Changes in malondialdehyde levels in bronchoalveolar fluid and serum by the treatment of asthma with inhaled steroid and beta2-agonist. Respirology 2000, 5, 289–292. [Google Scholar] [CrossRef]
- Karadogan, B.; Beyaz, S.; Gelincik, A.; Buyukozturk, S.; Arda, N. Evaluation of oxidative stress biomarkers and antioxidant parameters in allergic asthma patients with different level of asthma control. J. Asthma 2021, 59, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, O. Oxidative stress in asthma: Part of the puzzle. Pediatr. Allergy Immunol. 2018, 29, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.; Rahman, I. Oxidative stress in asthma and COPD: Antioxidants as a therapeutic strategy. Pharmacol. Ther. 2006, 111, 476–494. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Bacsi, A.; Choudhury, B.K.; Dharajiya, N.; Alam, R.; Hazra, T.K.; Mitra, S.; Goldblum, R.M.; Sur, S. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J. Clin. Investig. 2005, 115, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387. [Google Scholar] [CrossRef] [Green Version]
- Barreto, M.; Villa, M.P.; Olita, C.; Martella, S.; Ciabattoni, G.; Montuschi, P. 8-Isoprostane in exhaled breath condensate and exercise- induced bronchoconstriction in asthmatic children and adolescents. Chest 2009, 135, 66–73. [Google Scholar] [CrossRef]
- Barreto, M.; Villa, M.P.; Olita, C.; Martella, S.; Ciabattoni, G.; Montuschi, P. NADPH oxidase-4 overexpression is associated with epithelial ciliary dysfunction in neutrophilic asthma. Chest 2016, 149, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Sutcliffe, A.; Hollins, F.; Gomez, E.; Saunders, R.; Doe, C.; Cooke, M.; Challiss, R.A.J.; Brightling, C.E. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Samoszuk, M.K.; Comhair, S.A.; Thomassen, M.J.; Farver, C.F.; Dweik, R.A.; Kavuru, M.S.; Erzurum, S.C.; Hazen, S.L. Eosinophils generate brominating oxidants in allergen-induced asthma. J. Clin. Investig. 2000, 105, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Aruoma, O.I.; Halliwell, B. Action of hypochlorous acid on the antioxidant protective enzymes superoxide dismutase, catalase and glutathione peroxidase. Biochem. J. 1987, 248, 973–976. [Google Scholar] [CrossRef] [Green Version]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef]
- Cattani-Cavalieri, I.; Valença, H.D.M.; Moraes, J.A.; Brito-Gitirana, L.; Romana-Souza, B.; Schmidt, M.; Valença, S.S. Dimethyl fumarate attenuates lung inflammation and oxidative stress induced by chronic exposure to diesel exhaust particles in mice. Int. J. Mol. Sci. 2020, 21, 9658. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, J.C.; Comhair, S.A.; Erzurum, S.C.; Klein, D.F.; Lipscomb, M.F.; Kavuru, M.S.; Samoszuk, M.K.; Hazen, S.L. Eosinophils are a major source of nitric oxide-derived oxidants in severe asthma: Characterization of pathways available to eosinophils for generating reactive nitrogen species. J. Immunol. 2001, 166, 5763–5772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, D.; Ernst, P.; Lim, S.; Barnes, P.J.; Giaid, A. Increased formation of the potent oxidant peroxynitrite in the airways of asthmatic patients is associated with induction of nitric oxide synthase: Effect of inhaled glucocorticoid. FASEB J. 1998, 12, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osoata, G.O.; Hanazawa, T.; Brindicci, C.; Ito, M.; Barnes, P.J.; Kharitonov, S.; Ito, K. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest 2009, 135, 1513–1520. [Google Scholar] [CrossRef]
- Dickinson, J.D.; Sweeter, J.M.; Warren, K.J.; Ahmad, I.M.; De Deken, X.; Zimmerman, M.C.; Brody, S.L. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 2018, 14, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Koff, J.L.; Shao, M.X.; Ueki, I.F.; Nadel, J.A. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am. J. Physiol. Lung. Cell Mol. Physiol. 2008, 294, L1068–L1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhou, X.; Wen, S.; Xiao, Q. Flagellin/TLR5 responses induce mucus hypersecretion by activating EGFR via an epithelial cell signaling cascades. Exp. Cell. Res. 2012, 318, 723–731. [Google Scholar] [CrossRef]
- Sanders, P.N.; Koval, O.M.; Jaffer, O.A.; Prasad, A.M.; Businga, T.R.; Scott, J.A.; Hayden, P.J.; Luczak, E.D.; Dickey, D.D.; Allamargot, C.; et al. CaMKII is essential for the proasthmatic effects of oxidation. Sci. Transl. Med. 2013, 5, 195ra97. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Do, D.C.; Zhou, Y.; Luczak, E.; Mitzner, W.; Anderson, M.E.; Gao, P. Oxidized CaMKII promotes asthma through the activation of mast cells. JCI. Insight 2017, 2, e90139. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Ro, M.; Cho, K.J.; Kim, J.H. Lipopolysaccharide/TLR4 stimulates IL-13 production through a MyD88-BLT2-linked cascade in mast cells, potentially contributing to the allergic response. J. Immunol. 2017, 199, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, A.; Budai, M.M.; Milesz, S.; Bácsi, A.; Tőzsér, J.; Benkő, S. Ragweed pollen extract intensifies lipopolysaccharide-induced priming of NLRP3 inflammasome in human macrophages. Immunology 2013, 138, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.M.; Milton, M.J.; Schreiner, S.; Profozich, J.L.; Trucco, M.; Piganelli, J.D. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J. Immunol. 2007, 178, 908–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef]
- Hubert, M.T.; Thayer, T.C.; Steele, C.; Cuda, C.M.; Morel, L.; Piganelli, J.D.; Mathews, C.E. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J. Immunol. 2010, 185, 5247–5258. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.-I.; Kim, T.W.; Shin, K.; Kim, Y.H.; Yuk, C.M.; Yuk, J.-M.; Shin, D.-M.; Jo, E.-K.; Lee, C.-H.; Lee, S.-H. Enhanced Th2 cell differentiation and function in the absence of Nox2. Allergy 2017, 72, 252–265. [Google Scholar] [CrossRef]
- Barnes, P.J. Chronic obstructive pulmonary disease: Effects beyond the lungs. PLoS Med. 2010, 7, e1000220. [Google Scholar] [CrossRef]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic aspects of chronic obstructive pulmonary disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef]
- Kodgule, R.; Salvi, S. Exposure to biomass smoke as a cause for airway disease in women and children. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 82–90. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Louhelainen, N.; Rytilä, P.; Haahtela, T.; Kinnula, V.L.; Djukanović, R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm. Med. 2009, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haegens, A.; Vernooy, J.H.; Heeringa, P.; Mossman, B.T.; Wouters, E.F. Myeloperoxidase modulates lung epithelial responses to pro-inflammatory agents. Eur. Respir. J. 2008, 31, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, S.; Leitch, A.E.; Duffin, R.; Haslett, C.; Rossi, A.G. Neutrophil apoptosis: Relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2010, 2, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Rutgers, S.R.; Postma, D.S.; ten Hacken, N.H.; Kauffman, H.F.; van Der Mark, T.W.; Koëter, G.H.; Timens, W. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000, 55, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Taraseviciene-Stewart, L.; Douglas, I.S.; Nana-Sinkam, P.S.; Lee, J.D.; Tuder, R.M.; Nicolls, M.R.; Voelkel, N.F. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc. Am. Thorac. Soc. 2006, 3, 687–690. [Google Scholar] [CrossRef]
- Stone, H.; McNab, G.; Wood, A.M.; Stockley, R.A.; Sapey, E. Variability of sputum inflammatory mediators in COPD and α1-antitrypsin deficiency. Eur. Respir. J. 2012, 40, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.E.; Thorley, A.; Culpitt, S.V.; Dodd, S.; Donnelly, L.E.; Demattos, C.; Fitzgerald, M.; Barnes, P.J. Alveolar macrophage-mediated elastolysis: Roles of matrix metalloproteinases, cysteine, and serine proteases. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L867–L873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voynow, J.A.; Young, L.R.; Wang, Y.; Horger, T.; Rose, M.C.; Fischer, B.M. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am. J. Physiol. 1999, 276, L835–L843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Horváth, I.; Hunt, J.; Barnes, P.J.; Alving, K.; Antczak, A.; Baraldi, E.; Becher, G.; van Beurden, W.J.; Corradi, M.; Dekhuijzen, R.; et al. Exhaled breath condensate: Methodological recommendations and unresolved questions. Eur. Respir. J. 2005, 26, 523–548. [Google Scholar] [CrossRef]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154, 813–816. [Google Scholar] [CrossRef] [Green Version]
- Nowak, D.; Kasielski, M.; Antczak, A.; Pietras, T.; Bialasiewicz, P. Increased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: No significant effect of cigarette smoking. Respir. Med. 1999, 93, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., II. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, M.L.; Novelli, F.; Costa, F.; Malagrinò, L.; Melosini, L.; Bacci, E.; Cianchetti, S.; Dente, F.L.; Di Franco, A.; Vagaggini, B.; et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediators Inflamm. 2011, 2011, 891752. [Google Scholar] [CrossRef] [PubMed]
- Kluchová, Z.; Petrášová, D.; Joppa, P.; Dorková, Z.; Tkáčová, R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol. Res. 2007, 56, 51–56. [Google Scholar] [CrossRef]
- Ichinose, M.; Sugiura, H.; Yamagata, S.; Koarai, A.; Shirato, K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 2000, 162, 701–706. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.; Caramori, G.; Ito, K.; Capelli, A.; Brun, P.; Abatangelo, G.; Papi, A.; Chung, K.F.; Adcock, I.; Barnes, P.J.; et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J. Allergy. Clin. Immunol. 2005, 116, 1028–1035. [Google Scholar] [CrossRef]
- Pesci, A.; Majori, M.; Cuomo, A.; Borciani, N.; Bertacco, S.; Cacciani, G.; Gabrielli, M. Neutrophils infiltrating bronchial epithelium in chronic obstructive pulmonary disease. Respir. Med. 1998, 92, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Stockley, J.A.; Walton, G.M.; Lord, J.M.; Sapey, E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: The neutrophil as an immunotherapeutic target. Int. Immunopharmacol. 2013, 17, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Stockley, J.A.; Greenwood, H.; Ahmad, A.; Bayley, D.; Lord, J.M.; Insall, R.H.; Stockley, R.A. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2011, 183, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agustí, A.G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef]
- Rahman, I.; Morrison, D.; Donaldson, K.; MacNee, W. Systemic oxidative stress in asthma, COPD, and smokers. Am. J. Respir. Crit. Care. Med. 1996, 154, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Renkema, T.E.; Postma, D.S.; Noordhoek, J.A.; Sluiter, H.J.; Kauffman, H.F. Influence of in vivo prednisolone on increased in vitro O2- generation by neutrophils in emphysema. Eur. Respir. J. 1993, 6, 90–95. [Google Scholar] [PubMed]
- Van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, C.; Newbold, P.; White, P.; Thong, B.; Stone, H.; Stockley, R.A. 3-Chlorotyrosine in sputum of COPD patients: Relationship with airway inflammation. COPD 2010, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkham, P.A.; Caramori, G.; Casolari, P.; Papi, A.A.; Edwards, M.; Shamji, B.; Triantaphyllopoulos, K.; Hussain, F.; Pinart, M.; Khan, Y.; et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 796–802. [Google Scholar] [CrossRef] [Green Version]
- Gagliardo, R.; Chanez, P.; Profita, M.; Bonanno, A.; Albano, G.D.; Montalbano, A.M.; Pompeo, F.; Gagliardo, C.; Merendino, A.M.; Gjomarkaj, M. IκB kinase-driven nuclear factor-κB activation in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2011, 128, e1–e2. [Google Scholar] [CrossRef]
- Gorowiec, M.R.; Borthwick, L.A.; Parker, S.M.; Kirby, J.A.; Saretzki, G.C.; Fisher, A.J. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 1024–1032. [Google Scholar] [CrossRef]
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L295–L304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osoata, G.O.; Yamamura, S.; Ito, M.; Vuppusetty, C.; Adcock, I.M.; Barnes, P.J.; Ito, K. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem. Biophys. Res. Commun. 2009, 384, 366–371. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Hwang, J.W.; Kirkham, P.A.; Rahman, I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr. Med. Chem. 2013, 20, 1496–1530. [Google Scholar] [CrossRef]
- Thomson, N.C. Targeting oxidant-dependent mechanisms for the treatment of respiratory diseases and their comorbidities. Curr. Opin. Pharmacol. 2018, 40, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Zheng, X.Y.; Chung, K.F.; Zhong, N.S. Impact of air pollution on the burden of chronic respiratory diseases in China: Time for urgent action. Lancet 2016, 388, 1939–1951. [Google Scholar] [CrossRef]
- Szyszkowicz, M.; Kousha, T.; Valacchi, G. Ambient air pollution and emergency department visits for skin conditions. Glob. Dermatol. 2016, 3, 323–329. [Google Scholar] [CrossRef]
- Kampa, M.; Castanas, E. Human health effects of air pollution. Environ. Pollut. 2008, 151, 362–367. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994, Erratum in Lancet 2019, 393, 132. [Google Scholar] [CrossRef] [Green Version]
- Buiarelli, F.; Di Filippo, P.; Massimi, L.; Pomata, D.; Riccardi, C.; Simonetti, G.; Sonego, E. Ultrafine, fine and coarse airborne particle mass concentration in workplaces. Atmos. Pollut. Res. 2019, 10, 1685–1690. [Google Scholar] [CrossRef]
- Kumar, P.; Morawska, L.; Birmili, W.; Paasonen, P.; Hu, M.; Kulmala, M.; Harrison, R.M.; Norford, L.; Britter, R. Ultrafine particles in cities. Environ. Int. 2014, 66, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Schraufnagel, D.E. The health effects of ultrafine particles. Exp. Mol. Med. 2020, 52, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.K.; Cai, S.; Chen, Y.; Xiao, B.; Chen, P.; Xiang, X.D. The effect of pollutional haze on pulmonary function. J. Thorac. Dis. 2016, 8, E41–E56. [Google Scholar] [CrossRef]
- Van Donkelaar, A.; Martin, R.V.; Brauer, M.; Hsu, N.C.; Kahn, R.A.; Levy, R.C.; Lyapustin, A.; Sayer, A.M.; Winker, D.M. Global Estimates of Fine Particulate Matter using a Combined Geophysical-Statistical Method with Information from Satellites, Models, and Monitors. Environ. Sci. Technol. 2016, 50, 3762–3772. [Google Scholar] [CrossRef]
- Park, J.; Kim, H.J.; Lee, C.H.; Lee, C.H.; Lee, H.W. Impact of long-term exposure to ambient air pollution on the incidence of chronic obstructive pulmonary disease: A systematic review and meta-analysis. Environ. Res. 2021, 194, 110703. [Google Scholar] [CrossRef]
- Yang, X.Y.; Wen, B.; Han, F.; Wang, C.; Zhang, S.P.; Wang, J.; Xu, D.Q.; Wang, Q. Acute Effects of Individual Exposure to Fine Particulate Matter on Pulmonary Function in Schoolchildren. Biomed. Environ. Sci. 2020, 33, 647–659. [Google Scholar] [CrossRef]
- Genc, S.; Zadeoglulari, Z.; Fuss, S.H.; Genc, K. The adverse effects of air pollution on the nervous system. J. Toxicol. 2012, 2012, 782462. [Google Scholar] [CrossRef] [PubMed]
- Kelly, F.J.; Fussell, J.C. Air pollution and airway disease. Clin. Exp. Allergy 2011, 41, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Faustini, A.; Stafoggia, M.; Colais, P.; Berti, G.; Bisanti, L.; Cadum, E.; Cernigliaro, A.; Mallone, S.; Scarnato, C.; Forastiere, F.; et al. Air pollution and multiple acute respiratory outcomes. Eur. Respir. J. 2013, 42, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.E. Air pollution and heart disease. Rev. Cardiovasc. Med. 2006, 7, 44. [Google Scholar]
- Pan, G.; Zhang, S.; Feng, Y.; Takahashi, K.; Kagawa, J.; Yu, L.; Wang, P.; Liu, M.; Liu, Q.; Hou, S.; et al. Air pollution and children’s respiratory symptoms in six cities of Northern China. Respir. Med. 2010, 104, 1903–1911. [Google Scholar] [CrossRef] [Green Version]
- Millman, A.; Tang, D.; Perera, F.P. Air pollution threatens the health of children in China. Pediatrics 2008, 122, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Chaloulakou, A.; Mavroidis, I.; Gavriil, I. Compliance with the annual NO2 air quality standard in Athens. Required NOx levels and expected health implications. Atmos. Environ. 2008, 42, 454–465. [Google Scholar] [CrossRef]
- Albano, G.D.; Montalbano, A.M.; Gagliardo, R.; Anzalone, G.; Profita, M. Impact of Air Pollution in Airway Diseases: Role of the Epithelial Cells (Cell Models and Biomarkers). Int. J. Mol. Sci. 2022, 23, 2799. [Google Scholar] [CrossRef]
- Olivieri, D.; Scoditti, E. Impact of environmental factors on lung defences. Eur. Respir. Rev. 2005, 14, 51–56. [Google Scholar] [CrossRef]
- Salazar, L.M.; Herrera, A.M. Fibrotic response of tissue remodeling in COPD. Lung 2011, 189, 101–109. [Google Scholar] [CrossRef]
- Ko, F.W.S.; Hui, D.S.C. Effects of Air Pollution on Lung Health. Clin. Pulm. Med. 2010, 17, 300–304. [Google Scholar] [CrossRef]
- Lee, Y.G.; Lee, P.H.; Choi, S.M.; An, M.H.; Jang, A.S. Effects of Air Pollutants on Airway Diseases. Int. J. Environ. Res. Public Health 2021, 18, 9905. [Google Scholar] [CrossRef]
- Lodovici, M.; Bigagli, E. Oxidative stress and air pollution exposure. J. Toxicol. 2011, 2011, 487074. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Hayashi, S.; Hogg, J.C.; Fujii, T.; Goto, Y.; Sakamoto, N.; Mukae, H.; Vincent, R.; van Eeden, S.F. Alveolar macrophage-epithelial cell interaction following exposure to atmospheric particles induces the release of mediators involved in monocyte mobilization and recruitment. Respir. Res. 2005, 6, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, P.; Loft, S. Oxidative damage to DNA and lipids as biomarkers of exposure to air pollution. Environ. Health Perspect. 2010, 118, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2008, 26, 339–362. [Google Scholar] [CrossRef]
- Jeng, H.A. Chemical composition of ambient particulate matter and redox activity. Environ. Monit. Assess. 2010, 169, 597–606. [Google Scholar] [CrossRef]
- Andersson, H.; Piras, E.; Demma, J.; Hellman, B.; Brittebo, E. Low levels of the air pollutant 1-nitropyrene induce DNA damage, increased levels of reactive oxygen species and endoplasmic reticulum stress in human endothelial cells. Toxicology 2009, 262, 57–64. [Google Scholar] [CrossRef]
- Park, J.H.; Troxel, A.B.; Harvey, R.G.; Penning, T.M. Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by the aldo-keto-reductases (AKRs) generate abasic sites, oxidized pyrimidines, and 8-oxo-dGuo via reactive oxygen species. Chem. Res. Toxicol. 2006, 19, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Stojić, S.S.; Stanišić, N.; Stojić, A.; Šoštarić, A. Single and combined effects of air pollutants on circulatory and respiratory system-related mortality in Belgrade, Serbia. J. Toxicol. Environ. Health A 2016, 79, 17–27. [Google Scholar] [CrossRef]
- Todokoro, M.; Mochizuki, H.; Tokuyama, K.; Utsugi, M.; Dobashi, K.; Mori, M.; Morikawa, A. Effect of ozone exposure on intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation 2004, 28, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Schnelle-Kreis, J.; Küpper, U.; Sklorz, M.; Cyrys, J.; Briedé, J.J.; Peters, A.; Zimmermann, R. Daily measurement of organic compounds in ambient particulate matter in Augsburg, Germany: New aspects on aerosol sources and aerosol related health effects. Biomarkers 2009, 14 (Suppl. S1), 39–44. [Google Scholar] [CrossRef] [PubMed]
- Chirino, Y.I.; Sánchez-Pérez, Y.; Osornio-Vargas, A.R.; Morales-Bárcenas, R.; Gutiérrez-Ruíz, M.C.; Segura-García, Y.; Rosas, I.; Pedraza-Chaverri, J.; García-Cuellar, C.M. PM(10) impairs the antioxidant defense system and exacerbates oxidative stress driven cell death. Toxicol. Lett. 2010, 193, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ferecatu, I.; Borot, M.C.; Bossard, C.; Leroux, M.; Boggetto, N.; Marano, F.; Baeza-Squiban, A.; Andreau, K. Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor. Part Fibre Toxicol. 2010, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delfino, R.J.; Staimer, N.; Tjoa, T.; Arhami, M.; Polidori, A.; Gillen, D.L.; George, S.C.; Shafer, M.M.; Schauer, J.J.; Sioutas, C. Associations of primary and secondary organic aerosols with airway and systemic inflammation in an elderly panel cohort. Epidemiology 2010, 21, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, M.; Schins, R.P.; Hertel, O.; Loft, S. Transition metals in personal samples of PM2.5 and oxidative stress in human volunteers. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1340–1343. [Google Scholar] [CrossRef] [Green Version]
- Gerde, P.; Muggenburg, B.A.; Lundborg, M.; Tesfaigzi, Y.; Dahl, A.R. Respiratory epithelial penetration and clearance of particle-borne benzo[a]pyrene. Res. Rep. 2001, 101, 5–27. [Google Scholar]
- Kim, K.B.; Lee, B.M. Oxidative stress to DNA, protein, and antioxidant enzymes (superoxide dismutase and catalase) in rats treated with benzo(a)pyrene. Cancer Lett. 1997, 113, 205–212. [Google Scholar] [CrossRef]
- Churg, A.; Xie, C.; Wang, X.; Vincent, R.; Wang, R.D. Air pollution particles activate NF-kappaB on contact with airway epithelial cell surfaces. Toxicol. Appl. Pharmacol. 2005, 208, 37–45. [Google Scholar] [CrossRef]
- Becker, S.; Dailey, L.A.; Soukup, J.M.; Grambow, S.C.; Devlin, R.B.; Huang, Y.C. Seasonal variations in air pollution particle-induced inflammatory mediator release and oxidative stress. Environ. Health Perspect. 2005, 113, 1032–1038. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Mundandhara, S.; Devlin, R.B.; Madden, M. Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: Further mechanistic studies. Toxicol. Appl. Pharmacol. 2005, 207 (Suppl. S2), 269–275. [Google Scholar] [CrossRef]
- Karoly, E.D.; Li, Z.; Dailey, L.A.; Hyseni, X.; Huang, Y.C. Up-regulation of tissue factor in human pulmonary artery endothelial cells after ultrafine particle exposure. Environ. Health Perspect. 2007, 115, 535–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hegelan, M.; Tighe, R.M.; Castillo, C.; Hollingsworth, J.W. Ambient ozone and pulmonary innate immunity. Immunol. Res. 2011, 49, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Michaudel, C.; Mackowiak, C.; Maillet, I.; Fauconnier, L.; Akdis, C.A.; Sokolowska, M.; Dreher, A.; Tan, H.T.; Quesniaux, V.F.; Ryffel, B.; et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J. Allergy Clin. Immunol. 2018, 142, 942–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunekreef, B.; Holgate, S.T. Air pollution and health. Lancet 2002, 360, 1233–1242. [Google Scholar] [CrossRef]
- Lin, Y.K.; Chang, S.C.; Lin, C.; Chen, Y.C.; Wang, Y.C. Comparing ozone metrics on associations with outpatient visits for respiratory diseases in Taipei Metropolitan area. Environ. Pollut. 2013, 177, 177–184. [Google Scholar] [CrossRef]
- Song, H.; Tan, W.; Zhang, X. Ozone induces inflammation in bronchial epithelial cells. J. Asthma. 2011, 48, 79–83. [Google Scholar] [CrossRef]
- Mudway, I.S.; Kelly, F.J. Ozone and the lung: A sensitive issue. Mol. Asp. Med. 2000, 21, 1–48. [Google Scholar] [CrossRef]
- Kosmider, B.; Loader, J.E.; Murphy, R.C.; Mason, R.J. Apoptosis induced by ozone and oxysterols in human alveolar epithelial cells. Free Radic. Biol. Med. 2010, 48, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Triantaphyllopoulos, K.; Hussain, F.; Pinart, M.; Zhang, M.; Li, F.; Adcock, I.; Kirkham, P.; Zhu, J.; Chung, K.F. A model of chronic inflammation and pulmonary emphysema after multiple ozone exposures in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L691–L700. [Google Scholar] [CrossRef] [Green Version]
- Wiegman, C.H.; Li, F.; Ryffel, B.; Togbe, D.; Chung, K.F. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11, 1957. [Google Scholar] [CrossRef] [PubMed]
- Vardhan, K.H.; Kumar, P.S.; Panda, R.C. A review on heavy metal pollution, toxicity and remedial measures: Current trends and future perspectives. J. Mol. Liq. 2019, 290, 111197. [Google Scholar] [CrossRef]
- Albano, G.D.; Bonanno, A.; Montalbano, A.M.; Di Sano, C.; Anzalone, G.; Gagliardo, R.; Ruggieri, S.; Profita, M. Cadmium and Cadmium/BDE (47 or 209) Exposure Affect Mitochondrial Function, DNA Damage/Repair Mechanisms and Barrier Integrity in Airway Epithelial Cells. Atmosphere 2022, 13, 201. [Google Scholar] [CrossRef]
- Buha, A.; Baralić, K.; Djukic-Cosic, D.; Bulat, Z.; Tinkov, A.; Panieri, E.; Saso, L. The Role of Toxic Metals and Metalloids in Nrf2 Signaling. Antioxidants 2021, 10, 630. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.R.; Taalab, Y.M.; Heinze, S.; Tariba Lovaković, B.; Pizent, A.; Renieri, E.; Tsatsakis, A.; Farooqi, A.A.; Javorac, D.; Andjelkovic, M.; et al. Toxic-Metal-Induced Alteration in miRNA Expression Profile as a Proposed Mechanism for Disease Development. Cells 2020, 9, 901. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Kim, Y.S.; Kumar, V. Heavy metal toxicity: An update of chelating therapeutic strategies. J. Trace Elem. Med. Biol. 2019, 54, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Sabolić, I. Common mechanisms in nephropathy induced by toxic metals. Nephron Physiol. 2006, 104, 107–114. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Lazzarino, G.; Listorti, I.; Bilotta, G.; Capozzolo, T.; Amorini, A.M.; Longo, S.; Caruso, G.; Lazzarino, G.; Tavazzi, B.; Bilotta, P. Water- and Fat-Soluble Antioxidants in Human Seminal Plasma and Serum of Fertile Males. Antioxidants 2019, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Bouayed, J.; Bohn, T. Exogenous antioxidants—Double-edged swords in cellular redox state: Health beneficial effects at physiologic doses versus deleterious effects at high doses. Oxid. Med. Cell. Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, A.; Siddiqui, N.; Alharbi, N.O.; Alharbi, M.M. Airway and systemic oxidant-antioxidant dysregulation in asthma: A possible scenario of oxidants spill over from lung into blood. Pulm. Pharmacol. Ther. 2014, 29, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Guarneri, F.; Bertino, L.; Pioggia, G.; Casciaro, M.; Gangemi, S. Therapies with Antioxidant Potential in Psoriasis, Vitiligo, and Lichen Planus. Antioxidants 2021, 10, 1087, PMCID:PMC8301010. [Google Scholar] [CrossRef] [PubMed]
- Moussa, Z.; Judeh, Z.M.A.; Ahmed, S.A. Nonenzymatic Exogenous and Endogenous Antioxidants. In Free Radical Medicine and Biology; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unsal, V.; Cicek, M.; Sabancilar, İ. Toxicity of carbon tetrachloride, free radicals and role of antioxidants. Rev. Environ. Health. 2020, 36, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Avdalovic, M. Pulmonary vasculature and critical asthma syndromes: A comprehensive review. Clin. Rev. Allergy Immunol. 2015, 48, 97–103. [Google Scholar] [CrossRef]
- Karamalakova, Y.; Stefanov, I.; Georgieva, E.; Nikolova, G. Pulmonary Protein Oxidation and Oxidative Stress Modulation by Lemna minor L. in Progressive Bleomycin-Induced Idiopathic Pulmonary Fibrosis. Antioxidants 2022, 11, 523. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, J.; Wang, J.; Li, S.; Fukunaga, A.; Yodoi, J.; Tian, H. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct. Target Ther. 2020, 5, 248. [Google Scholar] [CrossRef]
- Nadeem, A.; Masood, A.; Siddiqui, N. Oxidant—Antioxidant imbalance in asthma: Scientific evidence, epidemiological data and possible therapeutic options. Ther. Adv. Respir. Dis. 2008, 2, 215–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 207. [Google Scholar] [CrossRef] [Green Version]
- Victoni, T.; Barreto, E.; Lagente, V.; Carvalho, V.F. Oxidative Imbalance as a Crucial Factor in Inflammatory Lung Diseases: Could Antioxidant Treatment Constitute a New Therapeutic Strategy? Oxid. Med. Cell. Longev. 2021, 2021, 6646923. [Google Scholar] [CrossRef]
- De Flora, S.; Balansky, R.; La Maestra, S. Rationale for the use of N-acetylcysteine in both prevention and adjuvant therapy of COVID-19. FASEB J. 2020, 34, 13185–13193. [Google Scholar] [CrossRef]
- Biswas, S.K.; Rahman, I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Mol. Aspects. Med. 2009, 30, 60–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Tan, Y.; Gu, M.; Kang, T.; Zhang, H.; Guo, L. N-acetyl cysteine inhibits lipopolysaccharide-mediated synthesis of interleukin-1β and tumor necrosis factor-α in human periodontal ligament fibroblast cells through nuclear factor-kappa B signaling. Medicine 2019, 98, e17126. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; MacNee, W. Antioxidant pharmacological therapies for COPD. Curr. Opin. Pharmacol. 2012, 12, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.P.; Wen, F.Q.; Bai, C.X.; Wan, H.Y.; Kang, J.; Chen, P.; Yao, W.Z.; Ma, L.J.; Li, X.; Raiteri, L.; et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2014, 2, 187–194, Erratum in Lancet Respir Med. 2014, 2, e4. [Google Scholar] [CrossRef]
- Grandjean, E.M.; Berthet, P.; Ruffmann, R.; Leuenberger, P. Efficacy of oral long-term N-acetylcysteine in chronic bronchopulmonary disease: A meta-analysis of published double-blind, placebo-controlled clinical trials. Clin. Ther. 2000, 22, 209–221. [Google Scholar] [CrossRef]
- Papi, A.; Zheng, J.; Criner, G.J.; Fabbri, L.M.; Calverley, P.M.A. Impact of smoking status and concomitant medications on the effect of high-dose N-acetylcysteine on chronic obstructive pulmonary disease exacerbations: A post-hoc analysis of the PANTHEON study. Respir. Med. 2019, 147, 37–43. [Google Scholar] [CrossRef]
- Oostwoud, L.C.; Gunasinghe, P.; Seow, H.J.; Ye, J.M.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Apocynin and ebselen reduce influenza A virus-induced lung inflammation in cigarette smoke-exposed mice. Sci. Rep. 2016, 6, 20983. [Google Scholar] [CrossRef] [Green Version]
- Stefanska, J.; Sarniak, A.; Wlodarczyk, A.; Sokolowska, M.; Doniec, Z.; Bialasiewicz, P.; Nowak, D.; Pawliczak, R. Hydrogen peroxide and nitrite reduction in exhaled breath condensate of COPD patients. Pulm. Pharmacol. Ther. 2012, 25, 343–348. [Google Scholar] [CrossRef]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiligianni, I.G.; Van der Molen, T. A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir. Res. 2010, 11, 171. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell. Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comhair, S.A.; Xu, W.; Ghosh, S.; Thunnissen, F.B.; Almasan, A.; Calhoun, W.J.; Janocha, A.J.; Zheng, L.; Hazen, S.L.; Erzurum, S.C. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am. J. Pathol. 2005, 166, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Verma, S.K.; Kumar, S.; Ahmad, M.K.; Nischal, A.; Singh, S.K.; Dixit, R.K. Evaluation of Oxidative Stress and Antioxidant Status in Chronic Obstructive Pulmonary Disease. Scand. J. Immunol. 2017, 85, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myerburg, M.M.; King JDJr Oyster, N.M.; Fitch, A.C.; Magill, A.; Baty, C.J.; Watkins, S.C.; Kolls, J.K.; Pilewski, J.M.; Hallows, K.R. AMPK agonists ameliorate sodium and fluid transport and inflammation in cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Xu, Q.; Wang, Y.; Li, G.; Sun, W.; Ma, D.; Zhou, S.; Liu, Y.; Han, L.; Ni, C. Metformin attenuates silica-induced pulmonary fibrosis via AMPK signaling. J. Transl. Med. 2021, 19, 349. [Google Scholar] [CrossRef]
- Salt, I.P.; Hardie, D.G. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System. Circ. Res. 2017, 120, 1825–1841. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Zhang, Z.; Zhang, P.; Qu, J.; Zheng, C.; Mo, X.; Zhou, W.; Xu, L.; Yao, H.; Gao, J. Nrf2 attenuates inflammatory response in COPD/emphysema: Crosstalk with Wnt3a/β-catenin and AMPK pathways. J. Cell. Mol. Med. 2018, 22, 3514–3525. [Google Scholar] [CrossRef] [Green Version]
- Andrade, I.G.A.; Suano-Souza, F.I.; Fonseca, F.L.A.; Lago, C.S.A.; Sarni, R.O.S. Selenium levels and glutathione peroxidase activity in patients with ataxia-telangiectasia: Association with oxidative stress and lipid status biomarkers. Orphanet J. Rare Dis. 2021, 16, 83. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S. Glutathione peroxidase-1 as a novel therapeutic target for COPD. Redox Rep. 2013, 18, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Brassington, K.; Chan, S.M.H.; De Luca, S.N.; Dobric, A.; Almerdasi, S.A.; Mou, K.; Seow, H.J.; Oseghale, O.; Bozinovski, S.; Selemidis, S.; et al. Ebselen abolishes vascular dysfunction in influenza A virus-induced exacerbations of cigarette smoke-induced lung inflammation in mice. Clin. Sci. 2022, 136, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Brassington, K.; Chan, S.M.H.; Seow, H.J.; Dobric, A.; Bozinovski, S.; Selemidis, S.; Vlahos, R. Ebselen reduces cigarette smoke-induced endothelial dysfunction in mice. Br. J. Pharmacol. 2021, 178, 1805–1818. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Kleeberger, S.R. Nrf2 defends the lung from oxidative stress. Antioxid. Redox Signal. 2006, 8, 76–87. [Google Scholar] [CrossRef]
- Lee, J.; Jang, J.; Park, S.M.; Yang, S.R. An Update on the Role of Nrf2 in Respiratory Disease: Molecular Mechanisms and Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 8406. [Google Scholar] [CrossRef]
- Jiao, Z.; Chang, J.; Li, J.; Nie, D.; Cui, H.; Guo, D. Sulforaphane increases Nrf2 expression and protects alveolar epithelial cells against injury caused by cigarette smoke extract. Mol. Med. Rep. 2017, 16, 1241–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audousset, C.; McGovern, T.; Martin, J.G. Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches—Pulmonary Disease/Asthma. Front. Physiol. 2021, 12, 727806. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Y.; Pan, P.; Huang, Y.; Chen, T.; Yuan, T.; Ma, Y.; Han, G.; Li, J.; Jin, Y.; et al. Pulmonary delivery of resveratrol-β-cyclodextrin inclusion complexes for the prevention of zinc chloride smoke-induced acute lung injury. Drug Deliv. 2022, 29, 1122–1131. [Google Scholar] [CrossRef]
- Nyunoya, T.; March, T.H.; Tesfaigzi, Y.; Seagrave, J. Antioxidant diet protects against emphysema, but increases mortality in cigarette smoke-exposed mice. COPD J. Chronic Obstr. Pulm. Dis. 2011, 8, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Scoditti, E.; Massaro, M.; Garbarino, S.; Toraldo, D.M. Role of Diet in Chronic Obstructive Pulmonary Disease Prevention and Treatment. Nutrients 2019, 11, 1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Wu, T.C.; Chen, P.Y.; Hsieh, L.Y.; Yeh, S.L. Comparison of plasma and intake levels of antioxidant nutrients in patients with chronic obstructive pulmonary disease and healthy people in Taiwan: A case-control study. Asia Pac. J. Clin. Nutr. 2010, 19, 393–401. [Google Scholar]
- Sener, G.; Topaloğlu, N.; Sehirli, A.O.; Ercan, F.; Gedik, N. Resveratrol alleviates bleomycin-induced lung injury in rats. Pulm. Pharmacol. Ther. 2007, 20, 642–649. [Google Scholar] [CrossRef]
- Roberts, L.A.; Suzuki, K. Anti-Inflammatory and Antioxidant Effects of Dietary Supplementation and Lifestyle Factors. Antioxidants 2021, 10, 371. [Google Scholar] [CrossRef]
- Dotan, Y.; Lichtenberg, D.; Pinchuk, I. No evidence supports vitamin E indiscriminate supplementation. Biofactors 2009, 35, 469–473. [Google Scholar] [CrossRef]
- Zhang, L.; Valizadeh, H.; Alipourfard, I.; Bidares, R.; Aghebati-Maleki, L.; Ahmadi, M. Epigenetic Modifications and Therapy in Chronic Obstructive Pulmonary Disease (COPD): An Update Review. COPD J. Chronic Obstr. Pulm. Dis. 2020, 17, 333–342. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Chattopadhyay, I.; Rajasekaran, S. Oxidative Stress Mechanisms in the Pathogenesis of Environmental Lung Diseases. In Oxidative Stress in Lung Diseases; Chakraborti, S., Parinandi, N., Ghosh, R., Ganguly, N., Chakraborti, T., Eds.; Springer: Singapore, 2020. [Google Scholar] [CrossRef]
- Rahman, I. Oxidative stress and gene transcription in asthma and chronic obstructive pulmonary disease: Antioxidant therapeutic targets. Curr. Drug Targets Inflamm. Allergy 2002, 1, 291–315. [Google Scholar] [CrossRef]
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lim, A.Y.H.; Tan, W.S.D.; Abisheganaden, J.; Wong, W.S.F. Restoration of HDAC2 and Nrf2 by andrographolide overcomes corticosteroid resistance in chronic obstructive pulmonary disease. Br. J. Pharmacol. 2020, 177, 3662–3673. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.W.; Ford, M.L.; Rogers, L.K.; Britt, R.D., Jr. Oxidative Stress Promotes Corticosteroid Insensitivity in Asthma and COPD. Antioxidants 2021, 10, 1335. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Rao, N.A.; McCalman, M.T.; Moulos, P.; Francoijs, K.J.; Chatziioannou, A.; Kolisis, F.N.; Alexis, M.N.; Mitsiou, D.J.; Stunnenberg, H.G. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011, 21, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC Complexes in Asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.; Xuan, L.; Cao, S.; Yu, G.; Hou, Q.; Wang, H. Decreased Histone Deacetylase 2 (HDAC2) in Peripheral Blood Monocytes (PBMCs) of COPD Patients. PLoS ONE 2016, 11, e0147380. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.; Caiazzo, E.; McSharry, C.; Guzik, T.J.; Maffia, P. Why do some asthma patients respond poorly to glucocorticoid therapy? Pharmacol. Res. 2020, 160, 105189. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albano, G.D.; Gagliardo, R.P.; Montalbano, A.M.; Profita, M. Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases. Antioxidants 2022, 11, 2237. https://doi.org/10.3390/antiox11112237
Albano GD, Gagliardo RP, Montalbano AM, Profita M. Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases. Antioxidants. 2022; 11(11):2237. https://doi.org/10.3390/antiox11112237
Chicago/Turabian StyleAlbano, Giusy Daniela, Rosalia Paola Gagliardo, Angela Marina Montalbano, and Mirella Profita. 2022. "Overview of the Mechanisms of Oxidative Stress: Impact in Inflammation of the Airway Diseases" Antioxidants 11, no. 11: 2237. https://doi.org/10.3390/antiox11112237