
Targeting Mitochondrial Metabolism to Reverse Radioresistance: An Alternative to Glucose Metabolism


Abstract
:1. Introduction
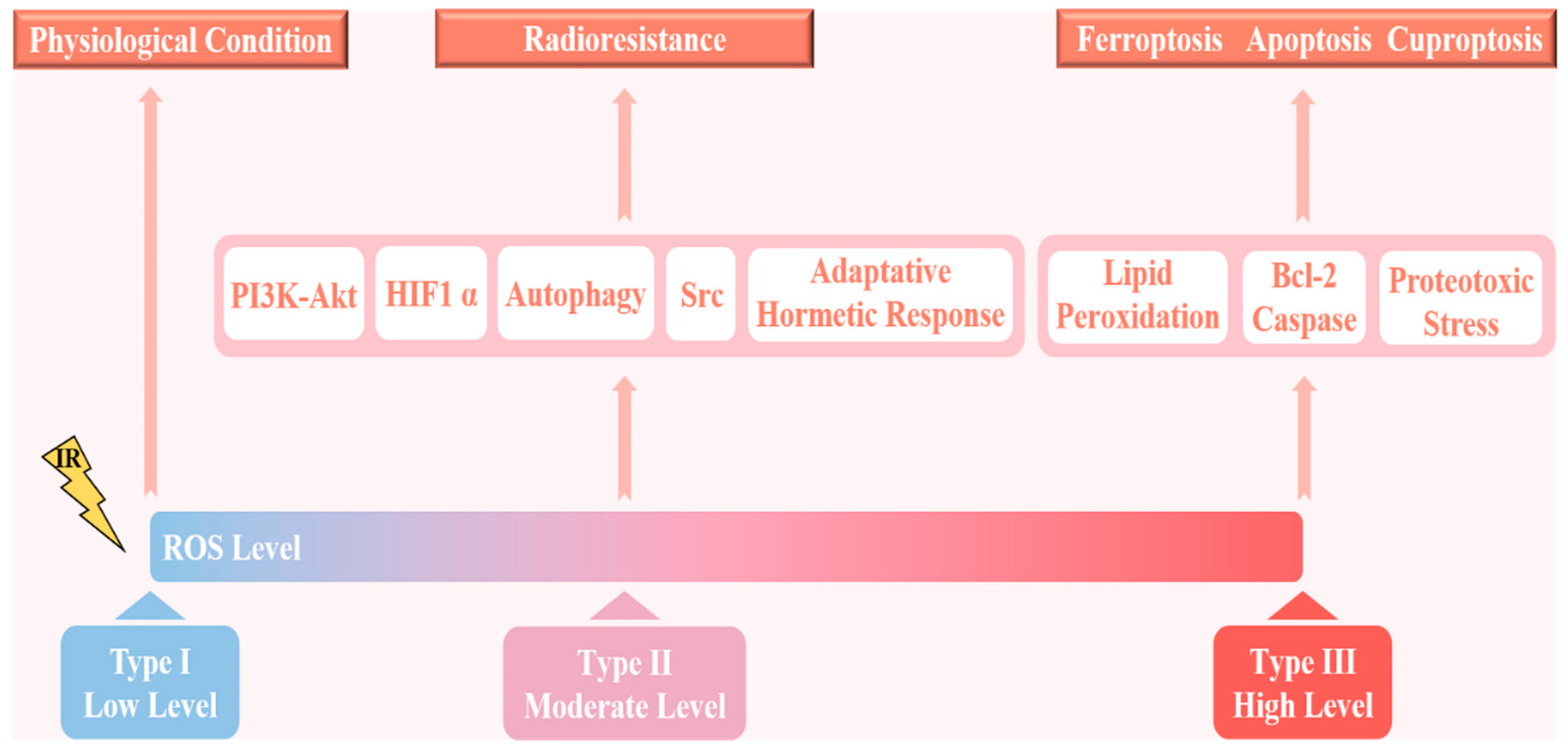
2. ROS and Radioresistance
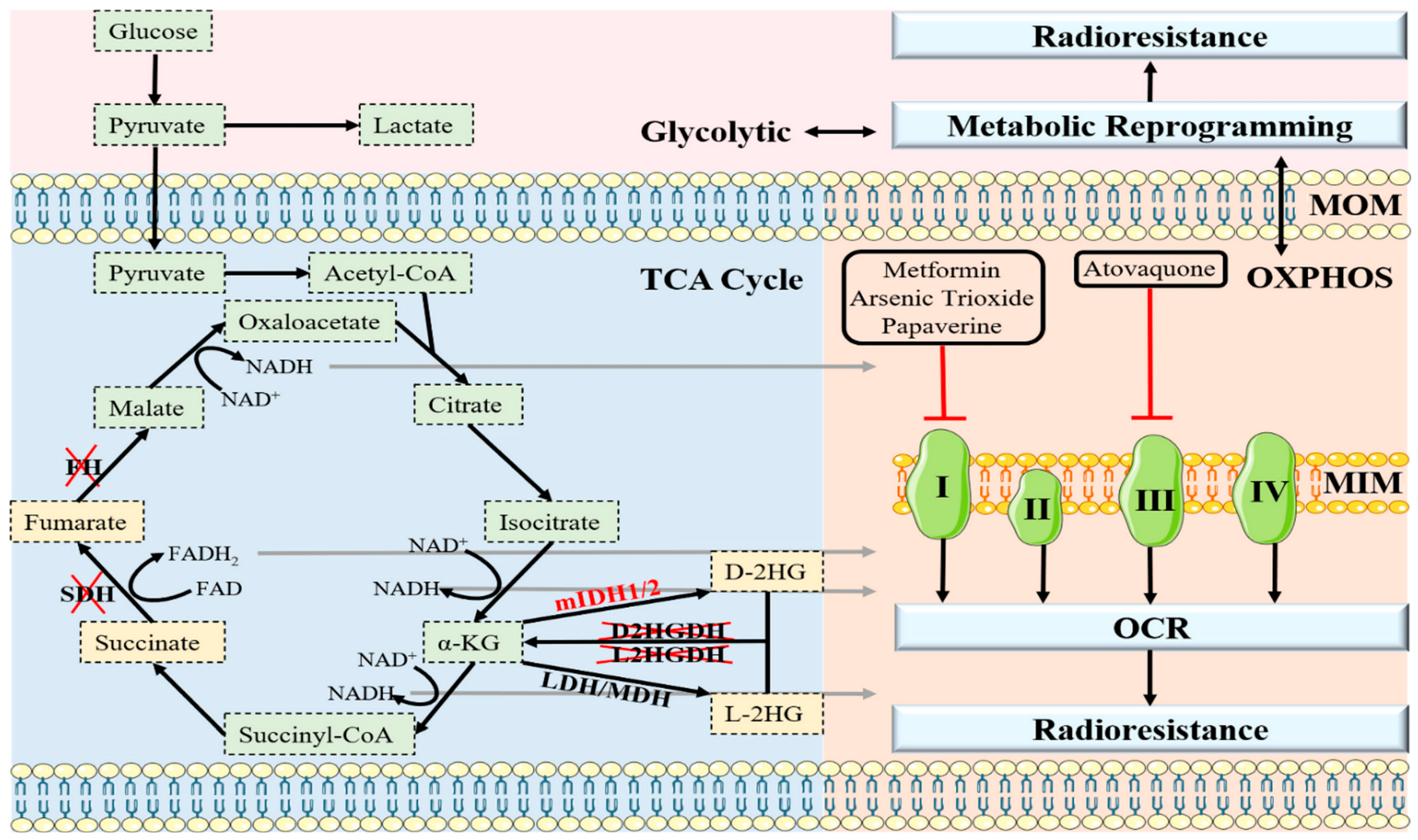
3. OXPHOS and Radioresistance
4. Oncometabolites and Radioresistance
5. Apoptosis and Radioresistance
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Clinton, S.K.; Giovannucci, E.L.; Hursting, S.D. The World Cancer Research Fund/American Institute for Cancer Research Third Expert Report on Diet, Nutrition, Physical Activity, and Cancer: Impact and Future Directions. J. Nutr. 2020, 150, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Luo, J.; Zhou, H. Radiotherapy targeting cancer stem cells “awakens” them to induce tumour relapse and metastasis in oral cancer. Int. J. Oral Sci. 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.Y.C.; Filippi, A.R.; Dabaja, B.S.; Yahalom, J.; Specht, L. Total Body Irradiation: Guidelines from the International Lymphoma Radiation Oncology Group (ILROG). Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, K.D.; Kaplan, A.B.; Brastianos, P.K. Seminoma with Neoplastic Meningitis Treated with Craniospinal Irradiation. Oncologist 2018, 23, 1385–1387. [Google Scholar] [CrossRef] [Green Version]
- Demoor-Goldschmidt, C.; Chiavassa, S.; Josset, S.; Mahé, M.A.; Supiot, S. Respiratory-gated bilateral pulmonary radiotherapy for Ewing’s sarcoma and nephroblastoma in children and young adults: Dosimetric and clinical feasibility studies. Cancer Radiother. 2017, 21, 124–129. [Google Scholar] [CrossRef]
- Lee, J. Mitochondrial drug targets in neurodegenerative diseases. Bioorg. Med. Chem. Lett. 2016, 26, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Kam, W.W.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Averbeck, D.; Rodriguez-Lafrasse, C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. Int. J. Mol. Sci. 2021, 22, 11047. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; De Koning, L.; Court, F.; Pereira, B.; Verrelle, P.; Dutreix, M. Predictive biomarkers of resistance to hypofractionated radiotherapy in high grade glioma. Radiat. Oncol. 2017, 12, 123. [Google Scholar] [CrossRef]
- Huang, R.; Xiang, J.; Zhou, P. Vitamin D, gut microbiota, and radiation-related resistance: A love-hate triangle. J. Exp. Clin. Cancer Res. 2019, 38, 493. [Google Scholar] [CrossRef]
- McBride, W.H.; Schaue, D. Radiation-induced tissue damage and response. J. Pathol. 2020, 250, 647–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śliwińska-Mossoń, M.; Wadowska, K.; Trembecki, Ł.; Bil-Lula, I. Markers Useful in Monitoring Radiation-Induced Lung Injury in Lung Cancer Patients: A Review. J. Pers. Med. 2020, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; He, Y.; Zhang, P.; Wang, J.; Fan, C.; Yang, L.; Xiong, F.; Zhang, S.; Gong, Z.; Nie, S.; et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Mol. Cancer 2018, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.M.; Butz, L.; Stegen, B.; Klumpp, D.; Braun, N.; Ruth, P.; Eckert, F. Ionizing radiation, ion transports, and radioresistance of cancer cells. Front. Physiol. 2013, 4, 212. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef]
- Yu, J.; Wei, Z.; Li, Q.; Wan, F.; Chao, Z.; Zhang, X.; Lin, L.; Meng, H.; Tian, L. Advanced Cancer Starvation Therapy by Simultaneous Deprivation of Lactate and Glucose Using a MOF Nanoplatform. Adv. Sci. 2021, 8, e2101467. [Google Scholar] [CrossRef]
- Laussel, C.; Léon, S. Cellular toxicity of the metabolic inhibitor 2-deoxyglucose and associated resistance mechanisms. Biochem. Pharmacol. 2020, 182, 114213. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Sobhakumari, A.; Orcutt, K.P.; Love-Homan, L.; Kowalski, C.E.; Parsons, A.D.; Knudson, C.M.; Simons, A.L. 2-Deoxy-d-glucose Suppresses the In Vivo Antitumor Efficacy of Erlotinib in Head and Neck Squamous Cell Carcinoma Cells. Oncol. Res. 2016, 24, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashmi, R.; Huang, X.; Floberg, J.M.; Elhammali, A.E.; McCormick, M.L.; Patti, G.J.; Spitz, D.R.; Schwarz, J.K. Radioresistant Cervical Cancers Are Sensitive to Inhibition of Glycolysis and Redox Metabolism. Cancer Res. 2018, 78, 1392–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Fang, D.; Li, S.; Zhong, Z.; Jiang, X.; Qi, Q.; Liu, Y.; Zhang, W.; Xu, X.; Liu, Y.; et al. Thioredoxin 1 supports colorectal cancer cell survival and promotes migration and invasion under glucose deprivation through interaction with G6PD. Int. J. Biol. Sci. 2022, 18, 5539–5553. [Google Scholar] [CrossRef] [PubMed]
- Rather, G.M.; Pramono, A.A.; Szekely, Z.; Bertino, J.R.; Tedeschi, P.M. In cancer, all roads lead to NADPH. Pharmacol. Ther. 2021, 226, 107864. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein. Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.H.; Cha, Y.H.; Lee, J.; Lee, S.H.; Yang, J.H.; Yun, J.S.; Cho, E.S.; Zhang, X.; Nam, M.; Kim, N.; et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 2017, 8, 14374. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, J.; Duan, R.; Gu, R.; Wang, W.; Wu, J.; Lian, H.; Hu, Y.; Yuan, A. High-Z-Sensitized Radiotherapy Synergizes with the Intervention of the Pentose Phosphate Pathway for In Situ Tumor Vaccination. Adv. Mater. 2022, 34, e2109726. [Google Scholar] [CrossRef]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef] [Green Version]
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Zhang, X.; Zhang, S.; Zhu, T.; Garg, M.; Lobie, P.E.; Pandey, V. Mitochondria: The metabolic switch of cellular oncogenic transformation. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188534. [Google Scholar] [CrossRef]
- Payen, V.L.; Zampieri, L.X.; Porporato, P.E.; Sonveaux, P. Pro- and antitumor effects of mitochondrial reactive oxygen species. Cancer Metastasis Rev. 2019, 38, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Grasso, D.; Medeiros, H.C.D.; Zampieri, L.X.; Bol, V.; Danhier, P.; van Gisbergen, M.W.; Bouzin, C.; Brusa, D.; Grégoire, V.; Smeets, H.; et al. Fitter Mitochondria Are Associated With Radioresistance in Human Head and Neck SQD9 Cancer Cells. Front. Pharmacol. 2020, 11, 263. [Google Scholar] [CrossRef]
- Gao, X.; Yang, Y.; Wang, J.; Zhang, L.; Sun, C.; Wang, Y.; Zhang, J.; Dong, H.; Zhang, H.; Gao, C.; et al. Inhibition of mitochondria NADH-Ubiquinone oxidoreductase (complex I) sensitizes the radioresistant glioma U87MG cells to radiation. Biomed. Pharmacother. 2020, 129, 110460. [Google Scholar] [CrossRef]
- Tomita, K.; Kuwahara, Y.; Igarashi, K.; Roudkenar, M.H.; Roushandeh, A.M.; Kurimasa, A.; Sato, T. Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy. Genes 2021, 12, 1348. [Google Scholar] [CrossRef]
- Wei, J.; Fang, T.; Wong, C.; Lakey, P.S.J.; Nizkorodov, S.A.; Shiraiwa, M. Superoxide Formation from Aqueous Reactions of Biogenic Secondary Organic Aerosols. Environ. Sci. Technol. 2021, 55, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2021, 23, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.; Huang, L.; Du, Y.; Gan, F.; Li, Y.; Yao, Y. Antioxidative Stress: Inhibiting Reactive Oxygen Species Production as a Cause of Radioresistance and Chemoresistance. Oxid. Med. Cell. Longev. 2021, 2021, 6620306. [Google Scholar] [CrossRef]
- Liu, J.; Liu, M.; Zhang, H.; Guo, W. High-Contrast Fluorescence Diagnosis of Cancer Cells/Tissues Based on β-Lapachone-Triggered ROS Amplification Specific in Cancer Cells. Angew. Chem. Int. Ed. Engl. 2021, 60, 12992–12998. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.L.; Ryu, H.; Son, A.R.; Seo, B.; Kim, J.; Jung, S.Y.; Song, J.Y.; Hwang, S.G.; Ahn, J. TGF-β and Hypoxia/Reoxygenation Promote Radioresistance of A549 Lung Cancer Cells through Activation of Nrf2 and EGFR. Oxid. Med. Cell. Longev. 2016, 2016, 6823471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, Y.; Li, Y.; Zheng, F.; Zou, K.; Li, Z.; Chen, M.; Hu, S.; Tian, C.; Yu, W.; Guo, W.; et al. TRIP4 promotes tumor growth and metastasis and regulates radiosensitivity of cervical cancer by activating MAPK, PI3K/AKT, and hTERT signaling. Cancer Lett. 2019, 452, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, Q.F.; Ye, Y.C.; Tashiro, S.; Onodera, S.; Ikejima, T. Activated O2•− and H2O2 mediated cell survival in SU11274-treated non-small-cell lung cancer A549 cells via c-Met-PI3K-Akt and c-Met-Grb2/SOS-Ras-p38 pathways. J. Pharmacol. Sci. 2012, 119, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox. Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Assi, M.; Rébillard, A. The Janus-Faced Role of Antioxidants in Cancer Cachexia: New Insights on the Established Concepts. Oxid. Med. Cell. Longev. 2016, 2016, 9579868. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. (Lond) 2015, 128, 863–875. [Google Scholar] [CrossRef]
- Kim, T.W.; Hong, D.W.; Kang, C.M.; Hong, S.H. A novel PPARɣ ligand, PPZ023, overcomes radioresistance via ER stress and cell death in human non-small-cell lung cancer cells. Exp. Mol. Med. 2020, 52, 1730–1743. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Minafra, L.; Cammarata, F.P.; Savoca, G.; Calvaruso, M.; Vicario, N.; Maccari, L.; Pérès, E.A.; Özçelik, H.; Bernaudin, M.; et al. SRC Tyrosine Kinase Inhibitor and X-rays Combined Effect on Glioblastoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 13917. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- O’Day, S.; Gonzalez, R.; Lawson, D.; Weber, R.; Hutchins, L.; Anderson, C.; Haddad, J.; Kong, S.; Williams, A.; Jacobson, E. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. J. Clin. Oncol. 2009, 27, 5452–5458. [Google Scholar] [CrossRef]
- Rae, C.; Mairs, R.J. Evaluation of the radiosensitizing potency of chemotherapeutic agents in prostate cancer cells. Int. J. Radiat. Biol. 2017, 93, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Oh, J.; Kim, S.M.; Kim, W.G.; Jeong, H.; Ahn, S.A.; Kim, S.H.; Jang, J.Y.; Yoo, B.C.; Kim, C.W.; et al. SOCS1 counteracts ROS-mediated survival signals and promotes apoptosis by modulating cell cycle to increase radiosensitivity of colorectal cancer cells. BMB Rep. 2022, 55, 198–203. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, R.; Yan, X.; Fan, K. Superoxide dismutase nanozymes: An emerging star for anti-oxidation. J. Mater. Chem. B 2021, 9, 6939–6957. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Patwardhan, R.S.; Jayakumar, S.; Sharma, D.; Sandur, S.K. Oxidative stress associated metabolic adaptations regulate radioresistance in human lung cancer cells. J. Photochem. Photobiol. B 2020, 213, 112080. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Ben-Sahra, I.; Lockwood, S.E.; Timson, R.C.; Byles, V.; Henning, G.T.; Gao, P.; Selfors, L.M.; Asara, J.M.; Manning, B.D. Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science 2019, 363, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Pirpour Tazehkand, A.; Salehi, R.; Velaei, K.; Samadi, N. The potential impact of trigonelline loaded micelles on Nrf2 suppression to overcome oxaliplatin resistance in colon cancer cells. Mol. Biol. Rep. 2020, 47, 5817–5829. [Google Scholar] [CrossRef]
- Yao, W.; Lin, Z.; Shi, P.; Chen, B.; Wang, G.; Huang, J.; Sui, Y.; Liu, Q.; Li, S.; Lin, X.; et al. Delicaflavone induces ROS-mediated apoptosis and inhibits PI3K/AKT/mTOR and Ras/MEK/Erk signaling pathways in colorectal cancer cells. Biochem. Pharmacol. 2020, 171, 113680. [Google Scholar] [CrossRef]
- Fisher, C.J.; Goswami, P.C. Mitochondria-targeted antioxidant enzyme activity regulates radioresistance in human pancreatic cancer cells. Cancer Biol. Ther. 2008, 7, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Sarsour, E.H.; Kalen, A.L.; Li, L.; Kumar, M.G.; Goswami, P.C. Late ROS accumulation and radiosensitivity in SOD1-overexpressing human glioma cells. Free Radic. Biol. Med. 2008, 45, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O(2)(⋅-) and H(2)O(2)-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e488. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updat. 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Weinhouse, S. Hepatomas. Science 1967, 158, 542–543. [Google Scholar] [CrossRef]
- Wu, W.; Zhao, S. Metabolic changes in cancer: Beyond the Warburg effect. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, T.; Luo, P.; Gao, L.; Liao, X.; Ma, L.; Jiang, Z.; Liu, D.; Yang, Z.; Jiang, Q.; et al. Near-infrared oxidative phosphorylation inhibitor integrates acute myeloid leukemia-targeted imaging and therapy. Sci. Adv 2021, 7, eabb6104. [Google Scholar] [CrossRef]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: Opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22, 323–339. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Tan, S.; Liu, L.; Hu, S.; Huo, H.; Li, M.; Cui, Q.; Yu, M. Nutrient deprivation-related OXPHOS/glycolysis interconversion via HIF-1α/C-MYC pathway in U251 cells. Tumour. Biol. 2016, 37, 6661–6671. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Nile, D.L.; Rae, C.; Walker, D.J.; Waddington, J.C.; Vincent, I.; Burgess, K.; Gaze, M.N.; Mairs, R.J.; Chalmers, A.J. Inhibition of glycolysis and mitochondrial respiration promotes radiosensitisation of neuroblastoma and glioma cells. Cancer Metab. 2021, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; You, M.; Wang, F.; Wang, Z.; Gao, X.; Jing, C.; Liu, J.; Guo, M.; Li, J.; Luo, A.; et al. Multifunctional Graphdiyne-Cerium Oxide Nanozymes Facilitate MicroRNA Delivery and Attenuate Tumor Hypoxia for Highly Efficient Radiotherapy of Esophageal Cancer. Adv. Mater. 2021, 33, e2100556. [Google Scholar] [CrossRef]
- Harrison, D.K.; Vaupel, P. Heterogeneity in tissue oxygenation: From physiological variability in normal tissues to pathophysiological chaos in malignant tumours. Adv. Exp. Med. Biol. 2014, 812, 25–31. [Google Scholar] [CrossRef]
- Kim, B.; Lee, J.J.; Shin, J.S.; Suh, J.W.; Jung, S.; Hwang, G.S.; Lee, H.Y.; Lee, K.J. Nm23-H1 activator phenylbutenoid dimer exerts cytotoxic effects on metastatic breast cancer cells by inducing mitochondrial dysfunction only under glucose starvation. Sci. Rep. 2021, 11, 23549. [Google Scholar] [CrossRef]
- Boreel, D.F.; Span, P.N.; Heskamp, S.; Adema, G.J.; Bussink, J. Targeting Oxidative Phosphorylation to Increase the Efficacy of Radio- and Immune-Combination Therapy. Clin. Cancer Res. 2021, 27, 2970–2978. [Google Scholar] [CrossRef] [PubMed]
- de Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides Radiosensitize Hypoxic Colorectal Cancer Cells Through a Decrease in Oxygen Consumption. Front. Pharmacol. 2018, 9, 1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zeng, Y.C. A candidate for lung cancer treatment: Arsenic trioxide. Clin. Transl. Oncol. 2019, 21, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Grégoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors. Cancer Res. 2012, 72, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benej, M.; Hong, X.; Vibhute, S.; Scott, S.; Wu, J.; Graves, E.; Le, Q.T.; Koong, A.C.; Giaccia, A.J.; Yu, B.; et al. Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 10756–10761. [Google Scholar] [CrossRef] [Green Version]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Harada, Y.; Ishii, I.; Hatake, K.; Kasahara, T. Pyrvinium pamoate inhibits proliferation of myeloma/erythroleukemia cells by suppressing mitochondrial respiratory complex I and STAT3. Cancer Lett. 2012, 319, 83–88. [Google Scholar] [CrossRef]
- Yan, B.; Stantic, M.; Zobalova, R.; Bezawork-Geleta, A.; Stapelberg, M.; Stursa, J.; Prokopova, K.; Dong, L.; Neuzil, J. Mitochondrially targeted vitamin E succinate efficiently kills breast tumour-initiating cells in a complex II-dependent manner. BMC Cancer 2015, 15, 401. [Google Scholar] [CrossRef] [Green Version]
- Pruss, M.; Dwucet, A.; Tanriover, M.; Hlavac, M.; Kast, R.E.; Debatin, K.M.; Wirtz, C.R.; Halatsch, M.E.; Siegelin, M.D.; Westhoff, M.A.; et al. Dual metabolic reprogramming by ONC201/TIC10 and 2-Deoxyglucose induces energy depletion and synergistic anti-cancer activity in glioblastoma. Br. J. Cancer 2020, 122, 1146–1157. [Google Scholar] [CrossRef]
- Rossato, L.G.; Costa, V.M.; Dallegrave, E.; Arbo, M.; Silva, R.; Ferreira, R.; Amado, F.; Dinis-Oliveira, R.J.; Duarte, J.A.; de Lourdes Bastos, M.; et al. Mitochondrial cumulative damage induced by mitoxantrone: Late onset cardiac energetic impairment. Cardiovasc. Toxicol. 2014, 14, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, M.; Schcolnik-Cabrera, A.; Dueñas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar] [PubMed]
- Yiallouris, A.; Patrikios, I.; Johnson, E.O.; Sereti, E.; Dimas, K.; De Ford, C.; Fedosova, N.U.; Graier, W.F.; Sokratous, K.; Kyriakou, K.; et al. Annonacin promotes selective cancer cell death via NKA-dependent and SERCA-dependent pathways. Cell Death Dis. 2018, 9, 764. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Lan, W.; Fraunhoffer, N.; Meilerman, A.; Iovanna, J.; Santofimia-Castaño, P. Dissecting the Anticancer Mechanism of Trifluoperazine on Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 1869. [Google Scholar] [CrossRef] [Green Version]
- Rottscholl, R.; Haegele, M.; Jainsch, B.; Xu, H.; Respondek, G.; Höllerhage, M.; Rösler, T.W.; Bony, E.; Le Ven, J.; Guérineau, V.; et al. Chronic consumption of Annona muricata juice triggers and aggravates cerebral tau phosphorylation in wild-type and MAPT transgenic mice. J. Neurochem. 2016, 139, 624–639. [Google Scholar] [CrossRef]
- Schultz, C.W.; McCarthy, G.A.; Nerwal, T.; Nevler, A.; DuHadaway, J.B.; McCoy, M.D.; Jiang, W.; Brown, S.Z.; Goetz, A.; Jain, A.; et al. The FDA-Approved Anthelmintic Pyrvinium Pamoate Inhibits Pancreatic Cancer Cells in Nutrient-Depleted Conditions by Targeting the Mitochondria. Mol. Cancer Ther. 2021, 20, 2166–2176. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Schafer, X.; Fu, D.; Nehrke, K.; Munger, J.; Brookes, P.S. Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J. Biol. Chem. 2016, 291, 20188–20197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.; Neijman, K.; Timmers, H. MITOCHONDRIA: Succinate dehydrogenase subunit B-associated phaeochromocytoma and paraganglioma. Int. J. Biochem. Cell Biol. 2021, 134, 105949. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.J.; Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef]
- Yong, C.; Stewart, G.D.; Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020, 16, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Terra, X.; Ceperuelo-Mallafré, V.; Merma, C.; Benaiges, E.; Bosch, R.; Castillo, P.; Flores, J.C.; León, X.; Valduvieco, I.; Basté, N.; et al. Succinate Pathway in Head and Neck Squamous Cell Carcinoma: Potential as a Diagnostic and Prognostic Marker. Cancers 2021, 13, 1653. [Google Scholar] [CrossRef]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- Núñez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11, eaaq1427. [Google Scholar] [CrossRef]
- Yu, H.E.; Wang, F.; Yu, F.; Zeng, Z.L.; Wang, Y.; Lu, Y.X.; Jin, Y.; Wang, D.S.; Qiu, M.Z.; Pu, H.Y.; et al. Suppression of fumarate hydratase activity increases the efficacy of cisplatin-mediated chemotherapy in gastric cancer. Cell Death Dis. 2019, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Forde, C.; Lim, D.H.K.; Alwan, Y.; Burghel, G.; Butland, L.; Cleaver, R.; Dixit, A.; Evans, D.G.; Hanson, H.; Lalloo, F.; et al. Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur. Urol. Oncol. 2020, 3, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerins, M.J.; Milligan, J.; Wohlschlegel, J.A.; Ooi, A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018, 109, 2757–2766. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Li, Q.; Zhang, F.; Xian, X.; Wang, S.; Xia, J.; Li, J.; Tuo, Z.; Xiao, G.; Liu, L.; et al. SDH5 Depletion Enhances Radiosensitivity by Regulating p53: A New Method for Noninvasive Prediction of Radiotherapy Response. Theranostics 2019, 9, 6380–6395. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Xiong, Y. Tumour metabolites hinder DNA repair. Nature 2020, 582, 492–494. [Google Scholar] [CrossRef]
- Jie, X.; Fong, W.P.; Zhou, R.; Zhao, Y.; Zhao, Y.; Meng, R.; Zhang, S.; Dong, X.; Zhang, T.; Yang, K.; et al. USP9X-mediated KDM4C deubiquitination promotes lung cancer radioresistance by epigenetically inducing TGF-β2 transcription. Cell Death Differ. 2021, 28, 2095–2111. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D.; Naik, A.A. Cellular demolition: Proteins as molecular players of programmed cell death. Int. J. Biol. Macromol. 2019, 138, 492–503. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.S.; Cheung, A.N.; Ngan, H.Y. Differential gene expression in cervical cancer cell lines before and after ionizing radiation. Int. J. Oncol. 2003, 22, 1091–1099. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Baine, M.J.; Souchek, J.J.; Menning, M.; Kaur, S.; Yan, Y.; Ouellette, M.M.; Jain, M.; Lin, C.; Batra, S.K. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 69–92. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal. Transduct. Target Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Rasmi, R.R.; Sakthivel, K.M.; Guruvayoorappan, C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed. Pharmacother. 2020, 130, 110569. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Jardin, S.E.; Dahl, H.; Nawas, A.F.; Bautista, M.; Delk, N.A. NF-κB signaling promotes castration-resistant prostate cancer initiation and progression. Pharmacol. Ther. 2020, 211, 107538. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, L.; Yang, M.; Wu, X.; Wang, X.; Huang, W.; Yuan, L.; Pan, H.; Wang, Y.; Wang, Z.; et al. Cancer-associated fibroblasts promote the survival of irradiated nasopharyngeal carcinoma cells via the NF-κB pathway. J. Exp. Clin. Cancer Res. 2021, 40, 87. [Google Scholar] [CrossRef] [PubMed]
- Haselager, M.; Thijssen, R.; West, C.; Young, L.; Van Kampen, R.; Willmore, E.; Mackay, S.; Kater, A.; Eldering, E. Regulation of Bcl-XL by non-canonical NF-κB in the context of CD40-induced drug resistance in CLL. Cell Death Differ. 2021, 28, 1658–1668. [Google Scholar] [CrossRef]
- Mortezaee, K.; Salehi, E.; Mirtavoos-Mahyari, H.; Motevaseli, E.; Najafi, M.; Farhood, B.; Rosengren, R.J.; Sahebkar, A. Mechanisms of apoptosis modulation by curcumin: Implications for cancer therapy. J. Cell. Physiol. 2019, 234, 12537–12550. [Google Scholar] [CrossRef]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef]
- McCann, E.; O’Sullivan, J.; Marcone, S. Targeting cancer-cell mitochondria and metabolism to improve radiotherapy response. Transl. Oncol. 2021, 14, 100905. [Google Scholar] [CrossRef]
- Wu, C.; Guo, E.; Ming, J.; Sun, W.; Nie, X.; Sun, L.; Peng, S.; Luo, M.; Liu, D.; Zhang, L.; et al. Radiation-Induced DNMT3B Promotes Radioresistance in Nasopharyngeal Carcinoma through Methylation of p53 and p21. Mol. Ther. Oncolytics 2020, 17, 306–319. [Google Scholar] [CrossRef]
- Maimon, N.; Zamir, Z.Z.; Kalkar, P.; Zeytuni-Timor, O.; Schif-Zuck, S.; Larisch, S.; Ariel, A. The pro-apoptotic ARTS protein induces neutrophil apoptosis, efferocytosis, and macrophage reprogramming to promote resolution of inflammation. Apoptosis 2020, 25, 558–573. [Google Scholar] [CrossRef]
- Bian, C.; Su, J.; Zheng, Z.; Wei, J.; Wang, H.; Meng, L.; Xin, Y.; Jiang, X. ARTS, an unusual septin, regulates tumorigenesis by promoting apoptosis. Biomed. Pharmacother. 2022, 152, 113281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Dong, J.; Wang, L.; Xia, Q.; Zhang, D.; Kim, H.; Yin, T.; Fan, S.; Shen, Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene 2018, 37, 5292–5304. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, H.; Wen, H.; Ding, Q.; Feng, C. Knockdown of tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ) enhances tumorigenesis both in vivo and in vitro in bladder cancer. Oncol. Rep. 2018, 39, 2127–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.C.; Li, C.F.; Chen, I.H.; Lai, M.T.; Lin, Z.J.; Korla, P.K.; Chai, C.Y.; Ko, G.; Chen, C.M.; Hwang, T.; et al. YWHAZ amplification/overexpression defines aggressive bladder cancer and contributes to chemo-/radio-resistance by suppressing caspase-mediated apoptosis. J. Pathol. 2019, 248, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Macedo-Silva, C.; Miranda-Gonçalves, V.; Lameirinhas, A.; Lencart, J.; Pereira, A.; Lobo, J.; Guimarães, R.; Martins, A.T.; Henrique, R.; Bravo, I.; et al. JmjC-KDMs KDM3A and KDM6B modulate radioresistance under hypoxic conditions in esophageal squamous cell carcinoma. Cell Death Dis. 2020, 11, 1068. [Google Scholar] [CrossRef]
- Mao, A.; Tang, J.; Tang, D.; Wang, F.; Liao, S.; Yuan, H.; Tian, C.; Sun, C.; Si, J.; Zhang, H.; et al. MicroRNA-29b-3p enhances radiosensitivity through modulating WISP1-mediated mitochondrial apoptosis in prostate cancer cells. J. Cancer 2020, 11, 6356–6364. [Google Scholar] [CrossRef]
- Codenotti, S.; Marampon, F.; Triggiani, L.; Bonù, M.L.; Magrini, S.M.; Ceccaroli, P.; Guescini, M.; Gastaldello, S.; Tombolini, V.; Poliani, P.L.; et al. Caveolin-1 promotes radioresistance in rhabdomyosarcoma through increased oxidative stress protection and DNA repair. Cancer Lett. 2021, 505, 1–12. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OXPHOS Inhibitor | Identifier | Phase | Cancer Type | ECT Target | Ref. |
|---|---|---|---|---|---|
| Metformin | NCT04275713, NCT04414540, NCT04945148, NCT04387630 | II, II, II, II | Cervical cancer, head and neck squamous cell carcinoma, glioblastoma (IDH-wildtype), breast cancer | Complex I | [92] |
| Phenformin | NCT03026517 | I | Melanoma | Complex I | [92] |
| Arsenic Trioxide | NCT02066870, NCT03503864 | I, II | Non-small-cell lung cancer, neuroblastoma | Complex I | [93] |
| Papaverine | NCT05136846, NCT03824327 | I, I | Locally advanced or unresectable non-small-cell lung cancer | Complex I | [95] |
| Atovaquone | NCT04648033, NCT02628080 | I, I | Locally advanced non-small-cell lung cancer, non-small-cell lung cancer | Complex III | [96] |
| Proguanil | N.A. | N.A. | Acts synergistically with atovaquone | Complex I | [97] |
| Pyrvinium Pamoate | NCT05055323 | I | Resectable pancreatic ductal adenocarcinoma | Complex I | [98] |
| Vitamin E | NCT01871454 | II | Non-small-cell lung cancer | Complex II | [99] |
| ONC201 | NCT04055649 | II | Platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, diffuse midline gliomas | Complex I, II | [100] |
| Mitoxantrone | NCT04927481, NCT03839446, NCT03258320, NCT04718402 | II, II, I, I | Breast cancer, acute myeloid leukemia, prostate cancer patients, advanced gastric carcinoma | Complex V | [101] |
| Ivermectin | N.A. | N.A. | Induces the death of renal cancer cells, chronic myeloid leukemia cells, and glioblastoma cells * | Complex I | [102] |
| Anonacin | N.A. | N.A. | Delays the growth of pancreatic cancer cells * | Complex I | [103] |
| Trifluoperazine | N.A. | N.A. | Induces pancreatic ductal adenocarcinoma cell death in combination with bortezomib * | Mitochondrial Stress | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, C.; Zheng, Z.; Su, J.; Wang, H.; Chang, S.; Xin, Y.; Jiang, X. Targeting Mitochondrial Metabolism to Reverse Radioresistance: An Alternative to Glucose Metabolism. Antioxidants 2022, 11, 2202. https://doi.org/10.3390/antiox11112202
Bian C, Zheng Z, Su J, Wang H, Chang S, Xin Y, Jiang X. Targeting Mitochondrial Metabolism to Reverse Radioresistance: An Alternative to Glucose Metabolism. Antioxidants. 2022; 11(11):2202. https://doi.org/10.3390/antiox11112202
Chicago/Turabian StyleBian, Chenbin, Zhuangzhuang Zheng, Jing Su, Huanhuan Wang, Sitong Chang, Ying Xin, and Xin Jiang. 2022. "Targeting Mitochondrial Metabolism to Reverse Radioresistance: An Alternative to Glucose Metabolism" Antioxidants 11, no. 11: 2202. https://doi.org/10.3390/antiox11112202