

Microglial Dysfunction in Neurodegenerative Diseases via RIPK1 and ROS

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
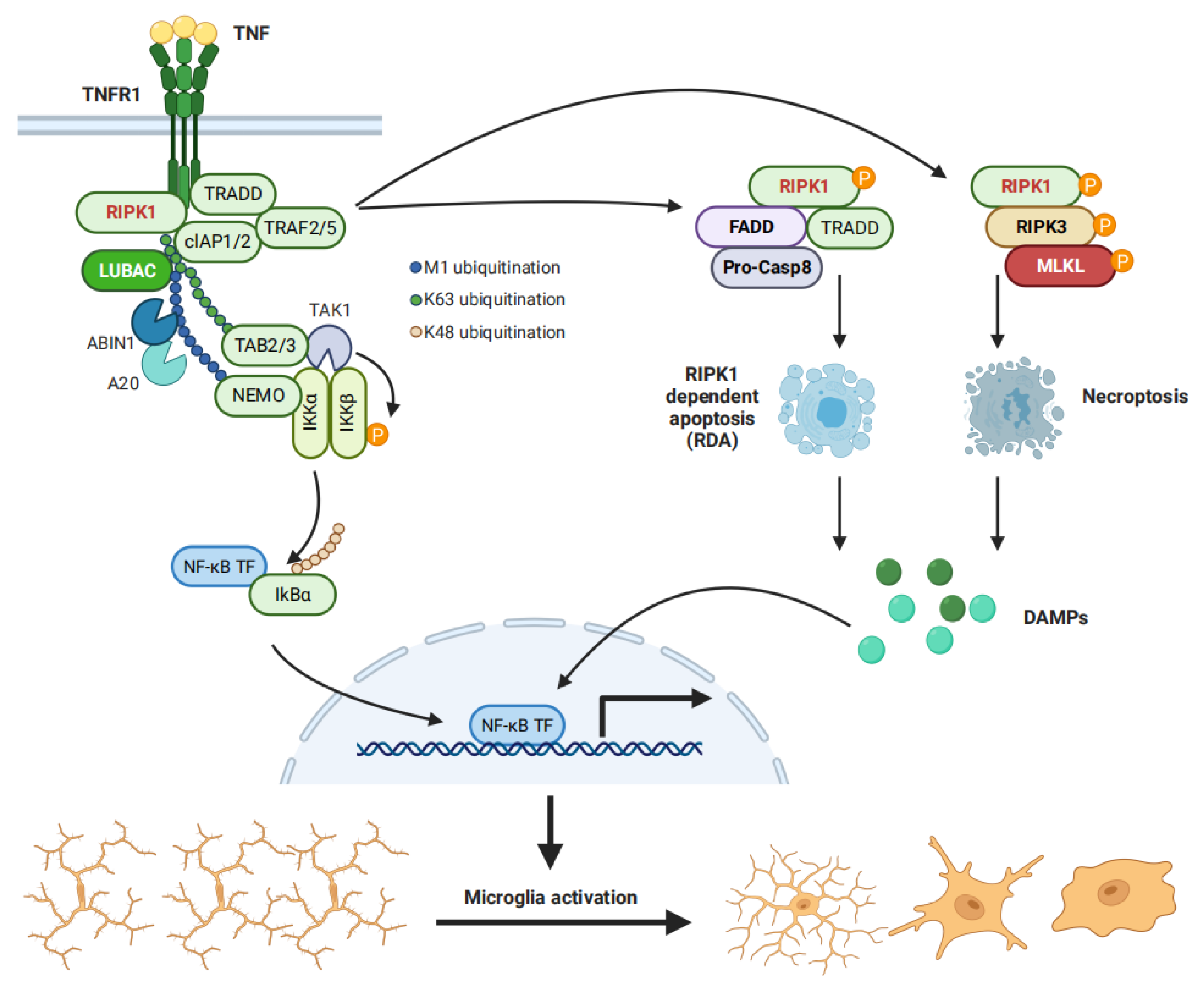
2. NF-κB Pathway Is Essential for Microglial Activation and Subsequent Neurotoxicity
3. RIPK1 Mediates NF-κB Pathway Activation
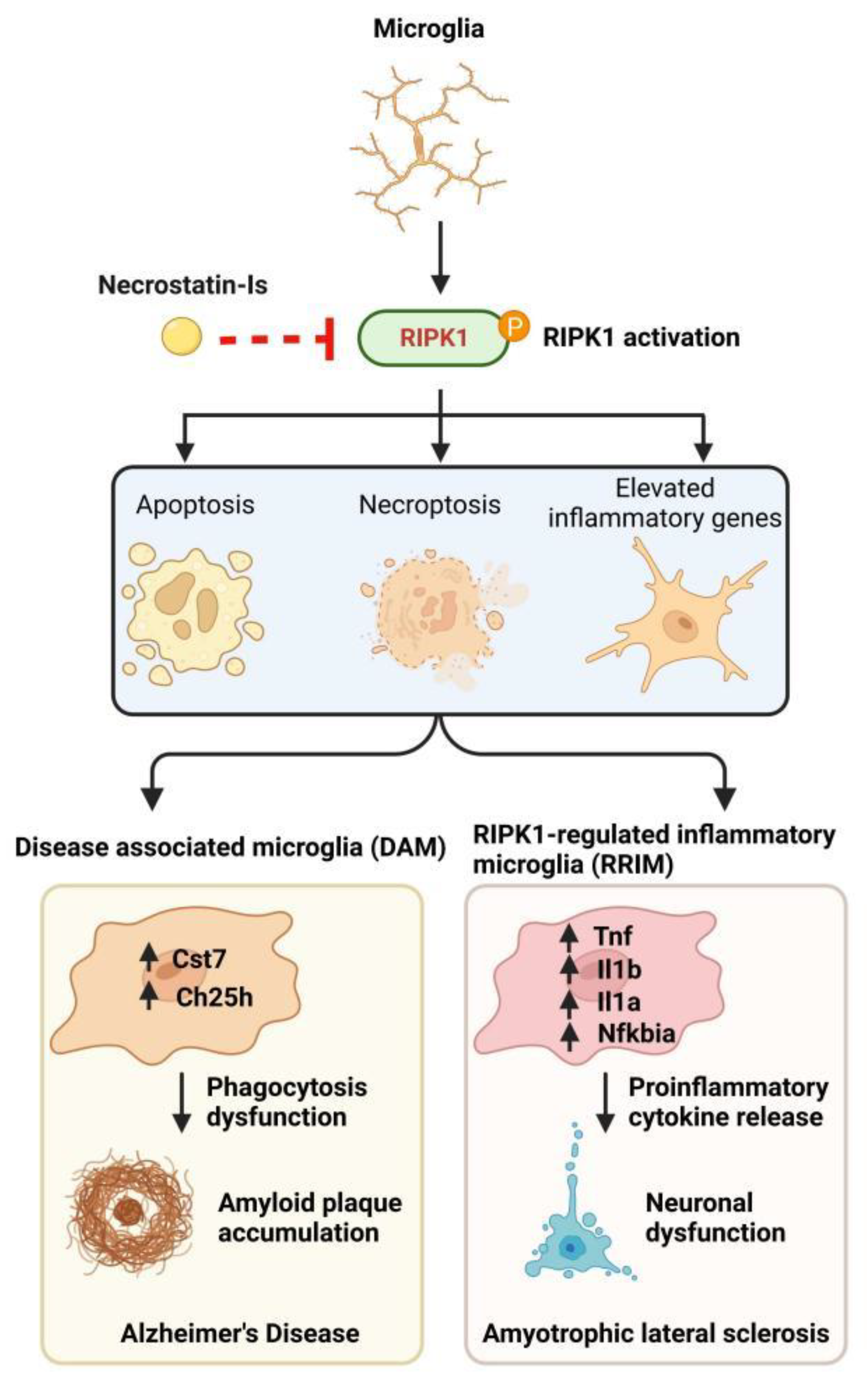
4. RIPK1 Regulates Microglia Activity in Neurodegenerative Diseases
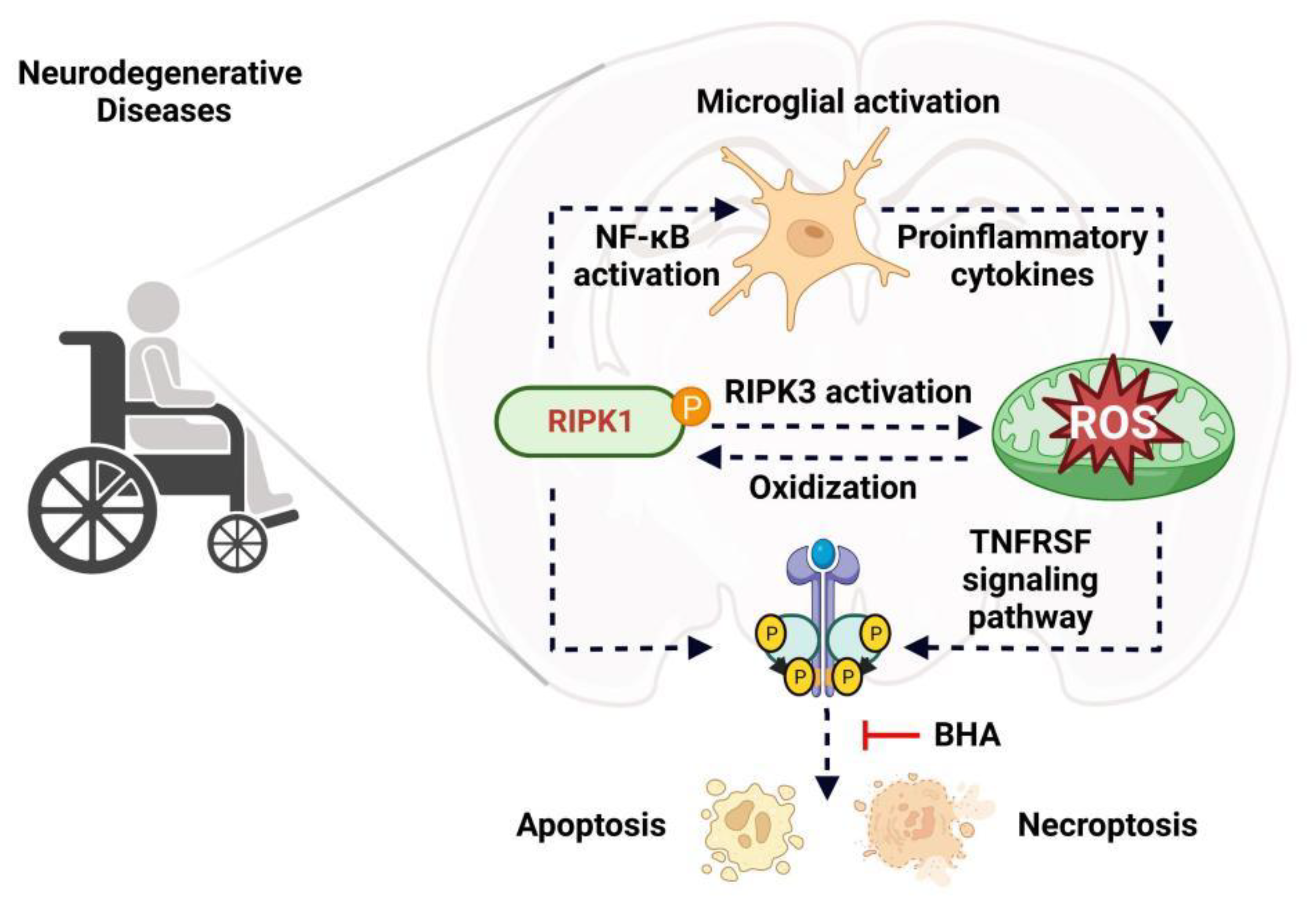
5. RIPK1 and ROS Activate Each Other in a Positive Feedback Loop
6. Conclusions
Funding
Conflicts of Interest
References
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.P.; Song, D.Y.; Sugama, S.; Shin, D.H.; Shimizu, Y.; Kim, S.S.; Kim, Y.S.; Joh, T.H. Pathological dynamics of activated microglia following medial forebrain bundle transection. Glia 2006, 53, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Reith, W. Neurodegenerative diseases. Radiologe 2018, 58, 241–258. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide- and Abeta-mediated microglial inflammation by inhibiting the TLR4/NF-kappaB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-kappaB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Bras, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [Green Version]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambare, D.; Bellini, E.; Ordazzo, G.; et al. Author Correction: Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 7359. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kaltschmidt, C. NF-kappa B: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Kim, B.W.; More, S.V.; Yun, Y.S.; Ko, H.M.; Kwak, J.H.; Lee, H.; Suk, K.; Kim, I.S.; Choi, D.K. A novel synthetic compound MCAP suppresses LPS-induced murine microglial activation in vitro via inhibiting NF-kB and p38 MAPK pathways. Acta Pharmacol. Sin. 2016, 37, 334–343. [Google Scholar] [CrossRef]
- Caetano-Silva, M.E.; Rund, L.A.; Vailati-Riboni, M.; Pacheco, M.T.B.; Johnson, R.W. Copper-Binding Peptides Attenuate Microglia Inflammation through Suppression of NF-kB Pathway. Mol. Nutr. Food Res. 2021, 65, e2100153. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, F.; Shi, J. Effect of sevoflurane treatment on microglia activation, NF-kB and MAPK activities. Immunobiology 2019, 224, 638–644. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Xuan, Z.; Yang, M.; Wang, C.; Tao, T.; Wang, Q.; Cui, W. CSB6B prevents beta-amyloid-associated neuroinflammation and cognitive impairments via inhibiting NF-kappaB and NLRP3 in microglia cells. Int. Immunopharmacol. 2020, 81, 106263. [Google Scholar] [CrossRef]
- Xiao, T.; Wan, J.; Qu, H.; Li, Y. Tripartite-motif protein 21 knockdown extenuates LPS-triggered neurotoxicity by inhibiting microglial M1 polarization via suppressing NF-kappaB-mediated NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2021, 706, 108918. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.W.; Egan, L.; Li, Z.W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NF-kappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Mankan, A.K.; Canli, O.; Schwitalla, S.; Ziegler, P.; Tschopp, J.; Korn, T.; Greten, F.R. TNF-alpha-dependent loss of IKKbeta-deficient myeloid progenitors triggers a cytokine loop culminating in granulocytosis. Proc. Natl. Acad. Sci. USA 2011, 108, 6567–6572. [Google Scholar] [CrossRef] [Green Version]
- Jie, Z.; Ko, C.J.; Wang, H.; Xie, X.; Li, Y.; Gu, M.; Zhu, L.; Yang, J.Y.; Gao, T.; Ru, W.; et al. Microglia promote autoimmune inflammation via the noncanonical NF-kappaB pathway. Sci. Adv. 2021, 7, eabh0609. [Google Scholar] [CrossRef]
- Xu, D.; Zou, C.; Yuan, J. Genetic Regulation of RIPK1 and Necroptosis. Annu. Rev. Genet. 2021, 55, 235–263. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.A.; Michallet, M.C.; Papadopoulou, N.; Utermohlen, O.; Kranidioti, K.; Kollias, G.; Tschopp, J.; Pasparakis, M. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 2008, 9, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Pobezinskaya, Y.L.; Kim, Y.S.; Choksi, S.; Morgan, M.J.; Li, T.; Liu, C.; Liu, Z. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 2008, 9, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shan, B.; Liang, Y.; Wei, H.; Yuan, J. Parkin regulates NF-kappaB by mediating site-specific ubiquitination of RIPK1. Cell Death Dis. 2018, 9, 732. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fuhrer, M.; Bahrami, E.; Socha, P.; Klaudel-Dreszler, M.; Bouzidi, A.; Liu, Y.; Lehle, A.S.; Magg, T.; Hollizeck, S.; et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 970–975. [Google Scholar] [CrossRef] [Green Version]
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 2017, 277, 113–127. [Google Scholar] [CrossRef]
- Karunakaran, D.; Nguyen, M.A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates With Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-kappaB Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177. [Google Scholar] [CrossRef]
- Wong, W.W.; Gentle, I.E.; Nachbur, U.; Anderton, H.; Vaux, D.L.; Silke, J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010, 17, 482–487. [Google Scholar] [CrossRef]
- Li, W.; Shan, B.; Zou, C.; Wang, H.; Zhang, M.M.; Zhu, H.; Naito, M.G.; Xu, D.; Manuel, V.J.; Mifflin, L.; et al. Nuclear RIPK1 promotes chromatin remodeling to mediate inflammatory response. Cell Res. 2022, 32, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Burns, K.; Hofmann, K.; Blancheteau, V.; Martinon, F.; Kelliher, M.; Tschopp, J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 2004, 5, 503–507. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.A.; Ting, A.T. NFkappaB and ubiquitination: Partners in disarming RIPK1-mediated cell death. Immunol. Res. 2012, 54, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Wang, C.; Telpoukhovskaia, M.A.; Bahr, B.A.; Chen, X.; Gan, L. Endo-lysosomal dysfunction: A converging mechanism in neurodegenerative diseases. Curr. Opin. Neurobiol. 2018, 48, 52–58. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Schaffert, L.N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Nagano, S.; Araki, T. Axonal Transport and Local Translation of mRNA in Neurodegenerative Diseases. Front. Mol. Neurosci. 2021, 14, 697973. [Google Scholar] [CrossRef] [PubMed]
- Zelic, M.; Pontarelli, F.; Woodworth, L.; Zhu, C.; Mahan, A.; Ren, Y.; LaMorte, M.; Gruber, R.; Keane, A.; Loring, P.; et al. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 2021, 35, 109112. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.N.; Shiao, Y.J.; Shie, F.S.; Guo, B.S.; Chen, P.H.; Cho, C.Y.; Chen, Y.J.; Huang, F.L.; Tsay, H.J. Mechanism mediating oligomeric Abeta clearance by naive primary microglia. Neurobiol. Dis. 2011, 42, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Liu, Y.; Cooper, C.; Liu, B.; Wilson, B.; Hong, J.S. Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 2002, 83, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Walker, D.G.; Rogers, J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol. Aging 2001, 22, 945–956. [Google Scholar] [CrossRef]
- Dheen, S.T.; Jun, Y.; Yan, Z.; Tay, S.S.; Ling, E.A. Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia 2005, 50, 21–31. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Kos, J.; Nanut, M.P.; Prunk, M.; Sabotic, J.; Dautovic, E.; Jewett, A. Cystatin F as a regulator of immune cell cytotoxicity. Cancer Immunol. Immunother. 2018, 67, 1931–1938. [Google Scholar] [CrossRef]
- Sebastian Monasor, L.; Muller, S.A.; Colombo, A.V.; Tanrioever, G.; Konig, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Abeta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 2020, 9, e54083. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Park, S.; Jin Hur, H.; Cho, H.J.; Hwang, I.; Pyo Kang, Y.; Im, I.; Lee, H.; Lee, E.; Yang, W.; et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun. 2016, 7, 13129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mifflin, L.; Hu, Z.; Dufort, C.; Hession, C.C.; Walker, A.J.; Niu, K.; Zhu, H.; Liu, N.; Liu, J.S.; Levin, J.Z.; et al. A RIPK1-regulated inflammatory microglial state in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2025102118. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Ferre-Gonzalez, L.; Pena-Bautista, C.; Baquero, M.; Chafer-Pericas, C. Assessment of Lipid Peroxidation in Alzheimer’s Disease Differential Diagnosis and Prognosis. Antioxidants 2022, 11, 551. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Picciano, M.; La Francois, J.; Duff, K. Fibrillar beta-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer’s disease. Neuroscience 2001, 104, 609–613. [Google Scholar] [CrossRef]
- Arslan, J.; Jamshed, H.; Qureshi, H. Early Detection and Prevention of Alzheimer’s Disease: Role of Oxidative Markers and Natural Antioxidants. Front. Aging Neurosci. 2020, 12, 231. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Goossens, V.; Grooten, J.; De Vos, K.; Fiers, W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 8115–8119. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Choksi, S.; Shen, H.M.; Yang, Q.F.; Hur, G.M.; Kim, Y.S.; Tran, J.H.; Nedospasov, S.A.; Liu, Z.G. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 2004, 279, 10822–10828. [Google Scholar] [CrossRef] [Green Version]
- Shindo, R.; Kakehashi, H.; Okumura, K.; Kumagai, Y.; Nakano, H. Critical contribution of oxidative stress to TNFalpha-induced necroptosis downstream of RIPK1 activation. Biochem. Biophys. Res. Commun. 2013, 436, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lv, X.; Hu, B.; Zhao, L.; Li, S.; Li, Z.; Qing, X.; Liu, H.; Xu, J.; Shao, Z. Critical contribution of RIPK1 mediated mitochondrial dysfunction and oxidative stress to compression-induced rat nucleus pulposus cells necroptosis and apoptosis. Apoptosis 2018, 23, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Wang, J.; Zhang, H.; Cao, Y.; Qu, Y.; Huang, S.; Kong, X.; Song, C.; Li, J.; Li, Q.; et al. Inhibition of microglial receptor-interacting protein kinase 1 ameliorates neuroinflammation following cerebral ischaemic stroke. J. Cell. Mol. Med. 2020, 24, 12585–12598. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wang, Y.; Zhang, Y.; He, X.; Zhong, C.Q.; Ni, H.; Chen, X.; Liang, Y.; Wu, J.; Zhao, S.; et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 2018, 20, 186–197. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koper, M.J.; Van Schoor, E.; Ospitalieri, S.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Tousseyn, T.; Balusu, S.; De Strooper, B.; Thal, D.R. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 2020, 139, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Berger, S.B.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Cook, M.; Finger, J.; Hughes-Earle, A.; et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 2014, 192, 5476–5480. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, X.; Ou, Y.; Liu, M.; Yu, D.; Song, Z.; Niu, L.; Zhang, L.; Shi, J. Advances in RIPK1 kinase inhibitors. Front. Pharmacol. 2022, 13, 976435. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Zou, C. Microglial Dysfunction in Neurodegenerative Diseases via RIPK1 and ROS. Antioxidants 2022, 11, 2201. https://doi.org/10.3390/antiox11112201
Wu Q, Zou C. Microglial Dysfunction in Neurodegenerative Diseases via RIPK1 and ROS. Antioxidants. 2022; 11(11):2201. https://doi.org/10.3390/antiox11112201
Chicago/Turabian StyleWu, Qiaoyan, and Chengyu Zou. 2022. "Microglial Dysfunction in Neurodegenerative Diseases via RIPK1 and ROS" Antioxidants 11, no. 11: 2201. https://doi.org/10.3390/antiox11112201