
Mutual Regulation between Redox and Hypoxia-Inducible Factors in Cardiovascular and Renal Complications of Diabetes
 , , , and
, , , and 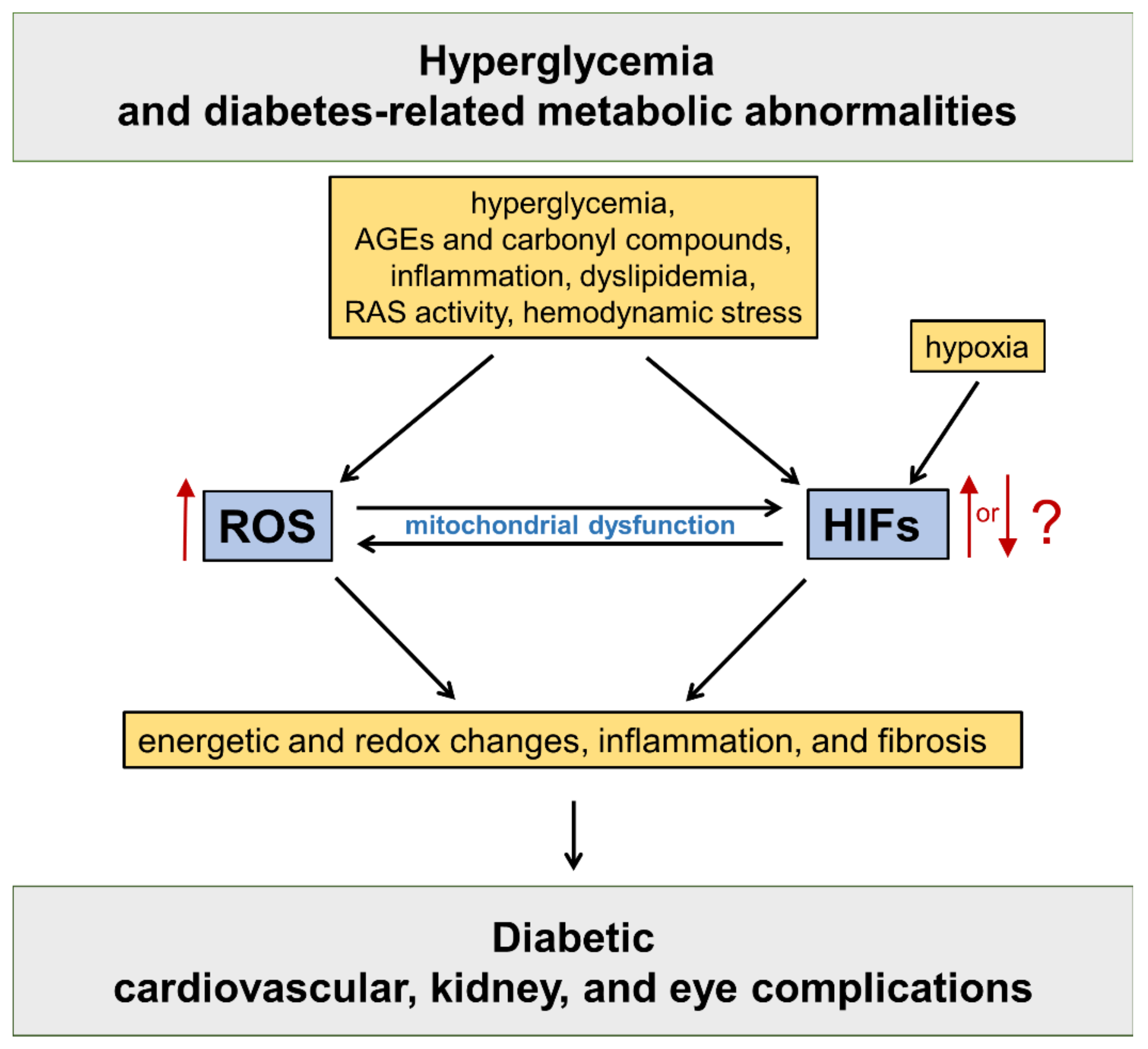{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
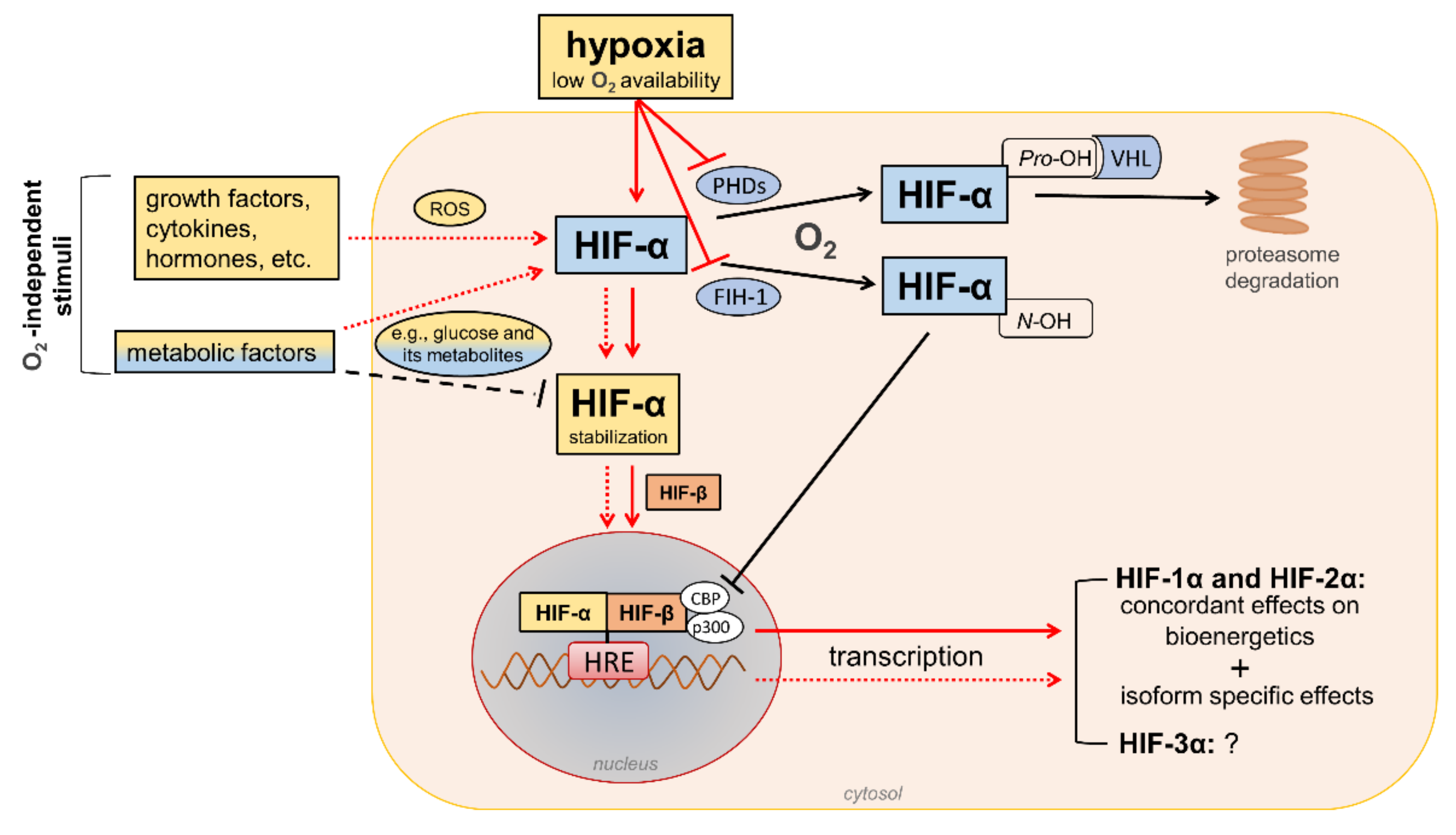
2. The HIFs Family
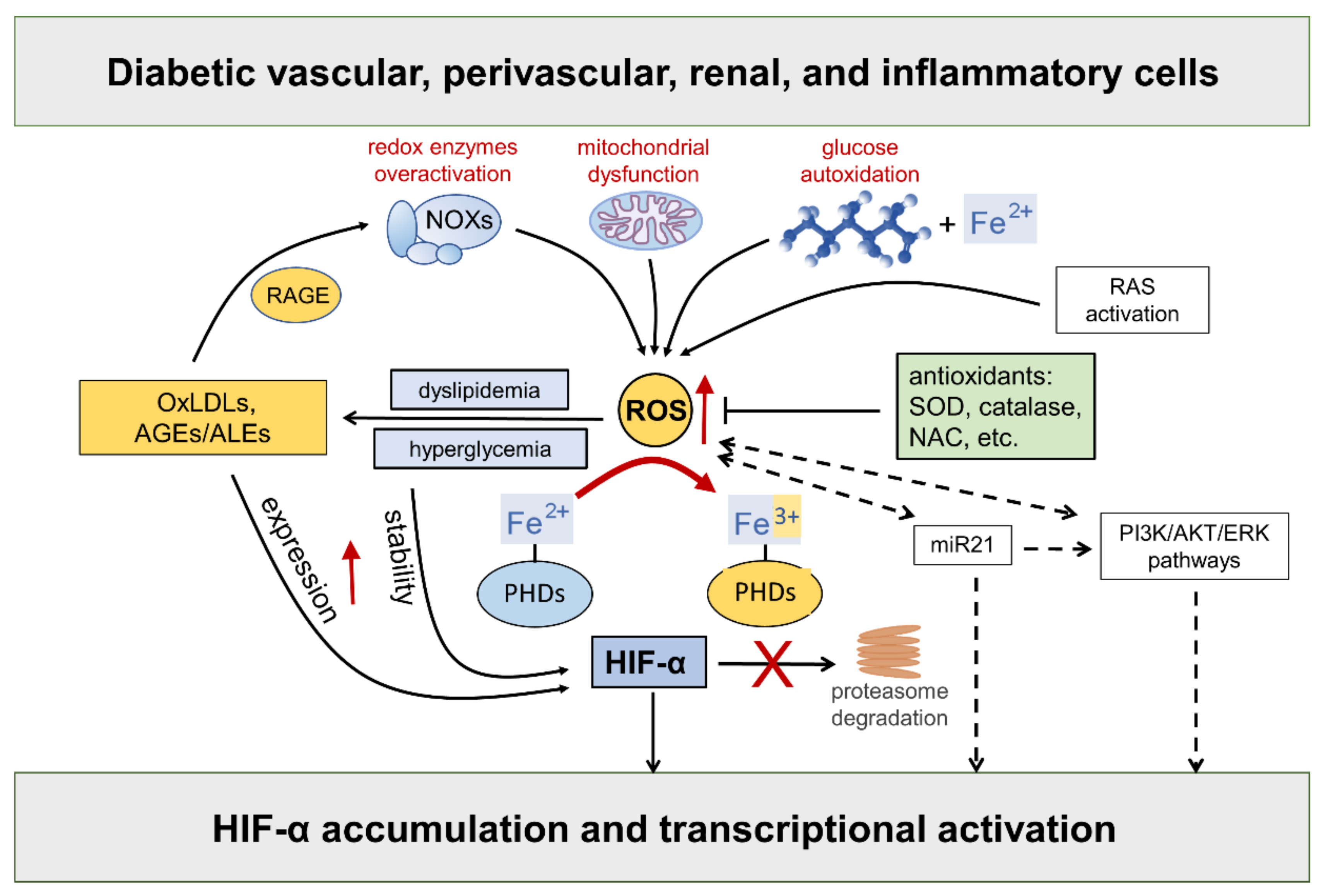
3. Redox Regulation of HIFs: Role of Diabetes-Related Stimuli
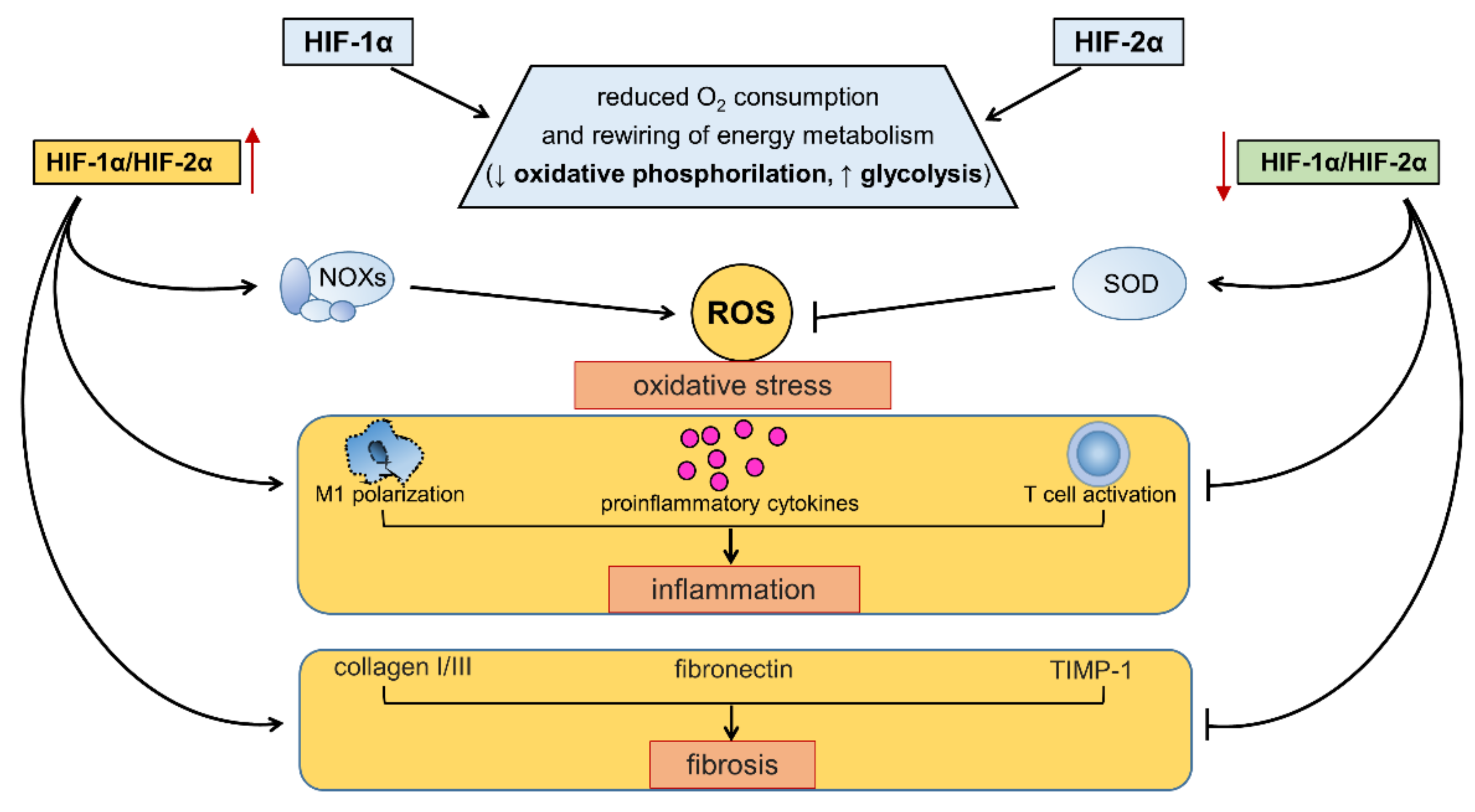
4. Modulation of Redox Homeostasis by HIFs in Response to Changes in Oxygen or Energy Substrate Availability
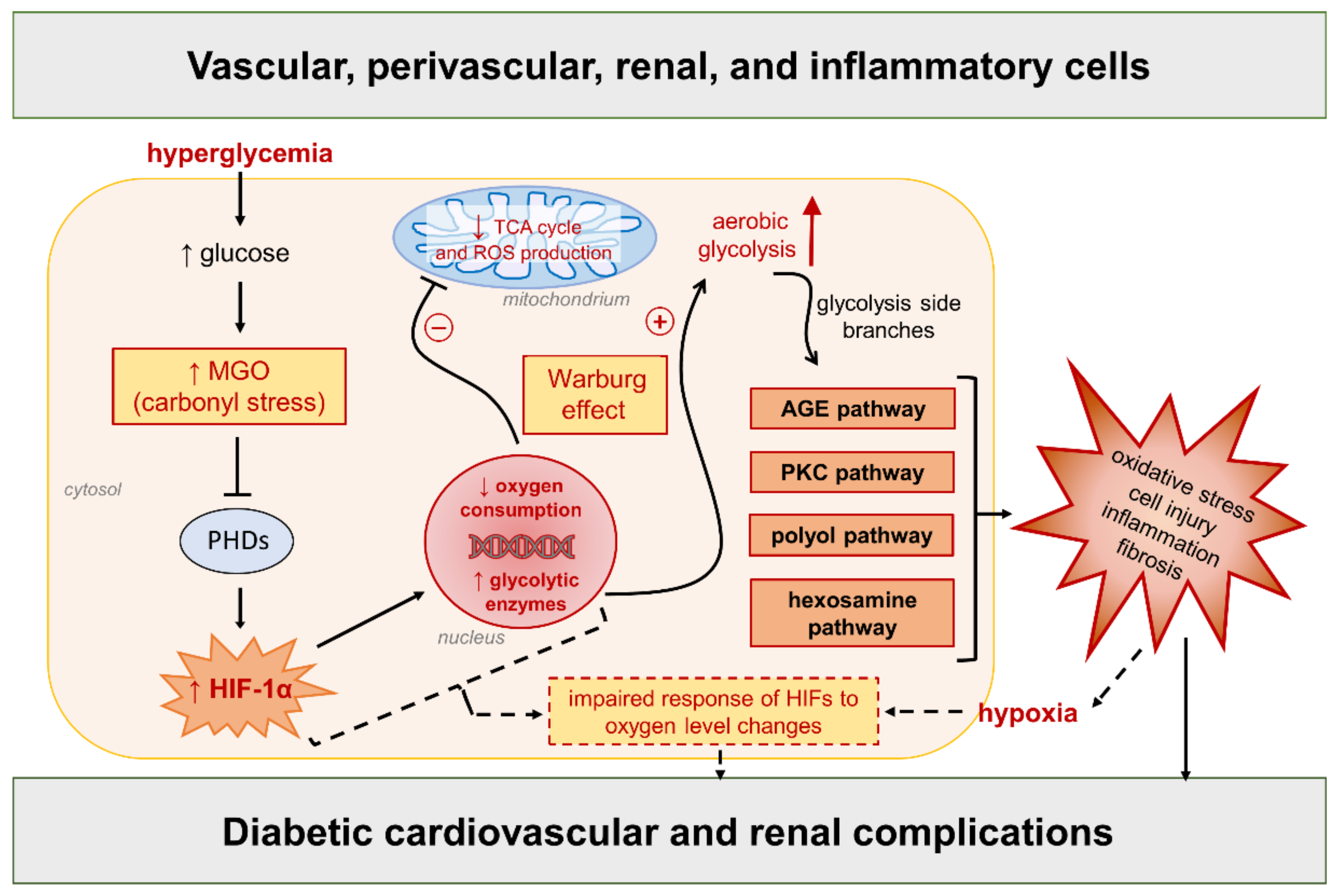
5. Dysregulated HIF Signaling and Redox Homeostasis in Cardiovascular and Renal Complications of Diabetes: Insights from Pharmacological HIF Modifiers and Perspectives for Future Research
5.1. PHD Inhibitors
5.2. Sirtuin-1 Activators
5.2.1. Resveratrol
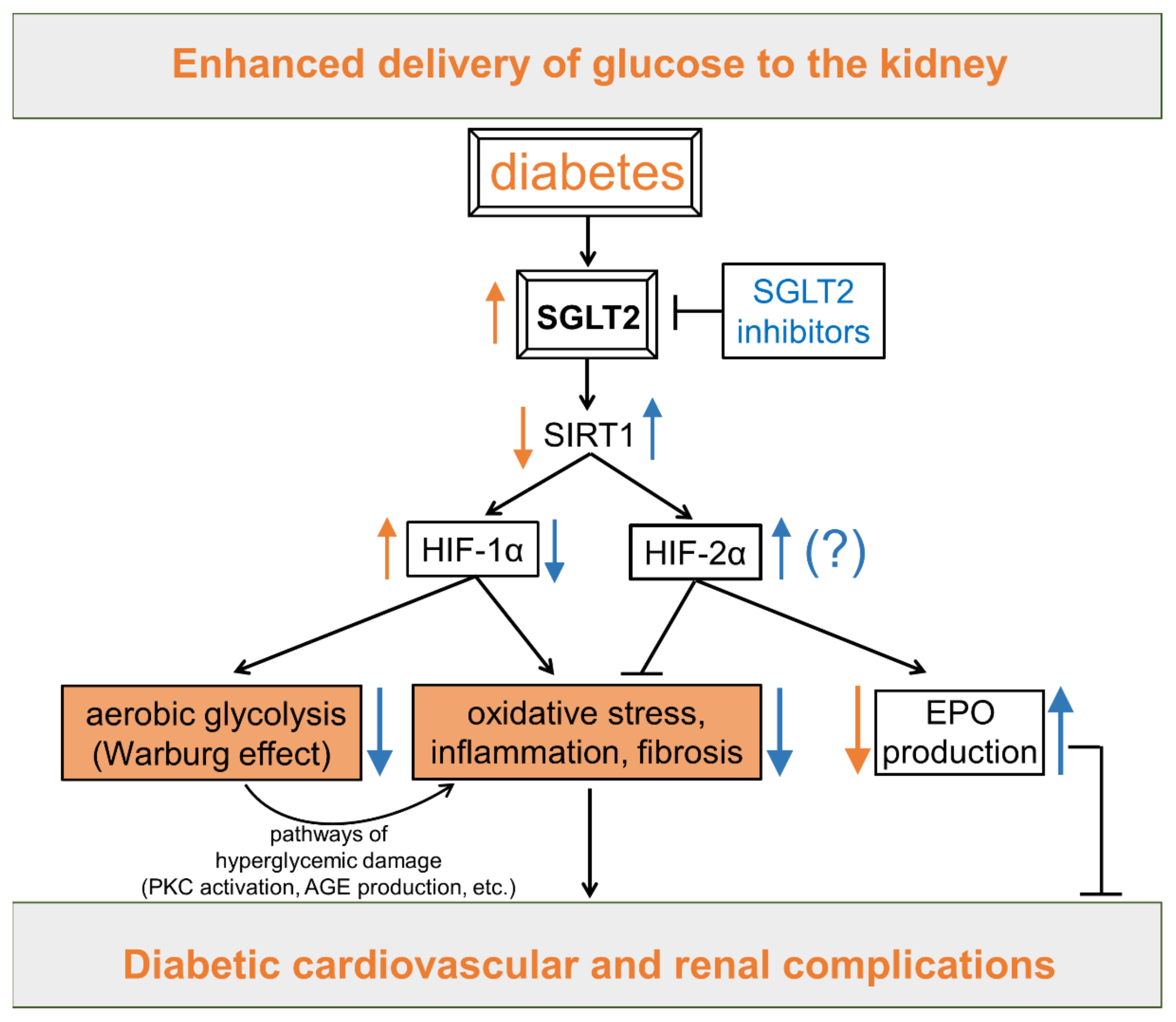
5.2.2. Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Connell, J.M.; Manson, S.M. Understanding the Economic Costs of Diabetes and Prediabetes and What We May Learn About Reducing the Health and Economic Burden of These Conditions. Diabetes Care 2019, 42, 1609–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall, T.M.; Yang, W.; Gillespie, K.; Mocarski, M.; Byrne, E.; Cintina, I.; Beronja, K.; Semilla, A.P.; Iacobucci, W.; Hogan, P.F. The Economic Burden of Elevated Blood Glucose Levels in 2017: Diagnosed and Undiagnosed Diabetes, Gestational Diabetes Mellitus, and Prediabetes. Diabetes Care 2019, 42, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kähm, K.; Laxy, M.; Schneider, U.; Rogowski, W.H.; Lhachimi, S.K.; Holle, R. Health Care Costs Associated with Incident Complications in Patients with Type 2 Diabetes in Germany. Diabetes Care 2018, 41, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbatreek, M.H.; Pachado, M.P.; Cuadrado, A.; Jandeleit-Dahm, K.; Schmidt, H.H.H.W. Reactive Oxygen Comes of Age: Mechanism-Based Therapy of Diabetic End-Organ Damage. Trends Endocrinol. Metab. 2019, 30, 312–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10, 727. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Edelstein, D.; Brownlee, M. The Missing Link: A Single Unifying Mechanism for Diabetic Complications. Kidney Int. 2000, 58, S26–S30. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus-Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Jha, J.C.; Banal, C.; Chow, B.S.M.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef]
- Casas, A.I.; Dao, V.T.V.; Daiber, A.; Maghzal, G.J.; di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catrina, S.B.; Zheng, X. Hypoxia and Hypoxia-Inducible Factors in Diabetes and Its Complications. Diabetologia 2021, 64, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Gunton, J.E. Hypoxia-Inducible Factors and Diabetes. J. Clin. Investig. 2020, 130, 5063–5073. [Google Scholar] [CrossRef]
- Packer, M. Role of Deranged Energy Deprivation Signaling in the Pathogenesis of Cardiac and Renal Disease in States of Perceived Nutrient Overabundance. Circulation 2020, 141, 2095–2105. [Google Scholar] [CrossRef]
- García-Pastor, C.; Benito-Martínez, S.; Moreno-Manzano, V.; Fernández-Martínez, A.B.; Lucio-Cazaña, F.J. Mechanism and Consequences of The Impaired Hif-1α Response to Hypoxia in Human Proximal Tubular HK-2 Cells Exposed to High Glucose. Sci. Rep. 2019, 9, 15868. [Google Scholar] [CrossRef] [Green Version]
- Isoe, T.; Makino, Y.; Mizumoto, K.; Sakagami, H.; Fujita, Y.; Honjo, J.; Takiyama, Y.; Itoh, H.; Haneda, M. High Glucose Activates HIF-1-Mediated Signal Transduction in Glomerular Mesangial Cells through a Carbohydrate Response Element Binding Protein. Kidney Int. 2010, 78, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Uttarwar, L.; Gao, B.; Charbonneau, M.; Shi, Y.; Chan, J.S.D.; Dubois, C.M.; Krepinsky, J.C. High Glucose Up-Regulates ADAM17 through HIF-1α in Mesangial Cells. J. Biol. Chem. 2015, 290, 21603–21614. [Google Scholar] [CrossRef] [Green Version]
- Nayak, B.K.; Shanmugasundaram, K.; Friedrichs, W.E.; Cavaglierii, R.C.; Patel, M.; Barnes, J.; Block, K. HIF-1 Mediates Renal Fibrosis in OVE26 Type 1 Diabetic Mice. Diabetes 2016, 65, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Yu, H.; Yan, N.; Lai, K.; Xiang, M. Hypoxia-Inducible Factor-1α Target Genes Contribute to Retinal Neuroprotection. Front. Cell. Neurosci. 2017, 11, 20. [Google Scholar] [CrossRef]
- Ekberg, N.R.; Eliasson, S.; Li, Y.W.; Zheng, X.; Chatzidionysiou, K.; Falhammar, H.; Gu, H.F.; Catrina, S.B. Protective Effect of the HIF-1A Pro582Ser Polymorphism on Severe Diabetic Retinopathy. J. Diabetes Res. 2019, 2019, 2936962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Lv, F.L.; Wang, G.H. Effects of HIF-1α on Diabetic Retinopathy Angiogenesis and VEGF Expression. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5071–5076. [Google Scholar] [CrossRef] [PubMed]
- Dodd, M.; Fialho, M.D.L.S.; Aparicio, C.N.M.; Kerr, M.; Timm, K.N.; Griffin, J.L.; Luiken, J.J.; Glatz, J.F.; Tyler, D.; Heather, L.C. Fatty Acids Prevent Hypoxia-Inducible Factor-1α Signaling Through Decreased Succinate in Diabetes. JACC Basic Transl. Sci. 2018, 3, 485–498. [Google Scholar] [CrossRef]
- Packer, M. Mutual Antagonism of Hypoxia-Inducible Factor Isoforms in Cardiac, Vascular, and Renal Disorders. JACC Basic Transl. Sci. 2020, 5, 961–968. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Flügel, D.; Becht, S.; Belaiba, R.S.; Bonello, S.; Hess, J.; Kietzmann, T.; Görlach, A. The Hypoxia-Inducible Factor-2α Is Stabilized by Oxidative Stress Involving NOX4. Antioxid. Redox Signal. 2010, 13, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Maintenance of Redox Homeostasis by Hypoxia-Inducible Factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Raval, R.; Moore, J.; Ratcliffe, P.J.; Harris, A.L. Predominant Role of Hypoxia-Inducible Transcription Factor (Hif)-1 versus Hif-2 in Regulation of the Transcriptional Response to Hypoxia 1. Cancer Res. 2003, 63, 6130–6134. [Google Scholar]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding Specificities of the HIF-1α and HIF-2α Transcription Factors in Chromatin. EMBO Rep. 2019, 20, e46401. [Google Scholar] [CrossRef]
- Yang, S.L.; Wu, C.; Xiong, Z.F.; Fang, X. Progress on Hypoxia-Inducible Factor-3: Its Structure, Gene Regulation and Biological Function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Berra, E.; Benizri, E.; Ginouvès, A.; Volmat, V.; Roux, D.; Pouysségur, J. HIF Prolyl-Hydroxylase 2 Is the Key Oxygen Sensor Setting Low Steady-State Levels of HIF-1α in Normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 Is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- McNeill, L.A.; Hewitson, K.S.; Claridge, T.D.; Seibel, J.F.; Horsfall, L.E.; Schofield, C.J. Hypoxia-Inducible Factor Asparaginyl Hydroxylase (FIH-1) Catalyses Hydroxylation at the Beta-Carbon of Asparagine-803. Biochem. J. 2002, 367, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A Novel Protein That Interacts with HIF-1α and VHL to Mediate Repression of HIF-1 Transcriptional Activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Hellwig-Bürgel, T.; Rutkowski, K.; Metzen, E.; Fandrey, J.; Jelkmann, W. Interleukin-1β and Tumor Necrosis Factor-α Stimulate DNA Binding of Hypoxia-Inducible Factor-1. Blood 1999, 94, 1561–1567. [Google Scholar] [CrossRef]
- Richard, D.E.; Berra, E.; Pouysségur, J. Nonhypoxic Pathway Mediates the Induction of Hypoxia-Inducible Factor 1α in Vascular Smooth Muscle Cells. J. Biol. Chem. 2000, 275, 26765–26771. [Google Scholar] [CrossRef]
- Kietzmann, T.; Samoylenko, A.; Roth, U.; Jungermann, K. Hypoxia-Inducible Factor-1 and Hypoxia Response Elements Mediate the Induction of Plasminogen Activator Inhibitor-1 Gene Expression by Insulin in Primary Rat Hepatocytes. Blood 2003, 101, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic Induction of the Hypoxia-Inducible Factor 1α by Insulin and Interleukin-1β Involves the Phosphatidylinositol 3-Kinase Pathway. FEBS Lett. 2002, 512, 157–162. [Google Scholar] [CrossRef]
- Doronzo, G.; Russo, I.; Mattiello, L.; Riganti, C.; Anfossi, G.; Trovati, M. Insulin Activates Hypoxia-Inducible Factor-1α in Human and Rat Vascular Smooth Muscle Cells via Phosphatidylinositol-3 Kinase and Mitogen-Activated Protein Kinase Pathways: Impairment in Insulin Resistance Owing to Defects in Insulin Signalling. Diabetologia 2006, 49, 1049–1063. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Vitale, M.; Pugliese, G.; Menini, S. Normalizing Hif-1α Signaling Improves Cellular Glucose Metabolism and Blocks the Pathological Pathways of Hyperglycemic Damage. Biomedicines 2021, 9, 1139. [Google Scholar] [CrossRef]
- Catrina, S.B.; Okamoto, K.; Pereira, T.; Brismar, K.; Poellinger, L. Hyperglycemia Regulates Hypoxia-Inducible Factor-1α Protein Stability and Function. Diabetes 2004, 53, 3226–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouysségur, J.; Mechta-Grigoriou, F. Redox Regulation of the Hypoxia-Inducible Factor. Biol. Chem. 2006, 387, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic ComplicationsA Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Nagai, R.; Murray, D.B.; Metz, T.O.; Baynes, J.W. Chelation: A Fundamental Mechanism of Action of AGE Inhibitors, AGE Breakers, and Other Inhibitors of Diabetes Complications. Diabetes 2012, 61, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.S.; MacFadyen, R.J.; Lip, G.Y. Diabetes Mellitus, the Renin-Angiotensin-Aldosterone System, and the Heart. Arch. Intern. Med. 2004, 164, 1737–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Induced Transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1α during Hypoxia: A Mechanism of O2 Sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen Sensing Requires Mitochondrial ROS but Not Oxidative Phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.S.; Chandel, N.S. The Qo Site of the Mitochondrial Complex III Is Required for the Transduction of Hypoxic Signaling via Reactive Oxygen Species Production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial Complex III Is Required for Hypoxia-Induced ROS Production and Cellular Oxygen Sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin Activates the Hypoxia-Inducible Factor-1 Signaling Pathway in Vascular Smooth Muscle Cells. Circ. Res. 2001, 89, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Haddad, J.J.; Land, S.C. A Non-Hypoxic, ROS-Sensitive Pathway Mediates TNF-α-Dependent Regulation of HIF-1α. FEBS Lett. 2001, 505, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Ding, M.; Zheng, J.Z.; Zhang, Z.; Leonard, S.S.; Liu, K.J.; Shi, X.; Jiang, B.H. Vanadate-Induced Expression of Hypoxia-Inducible Factor 1α and Vascular Endothelial Growth Factor through Phosphatidylinositol 3-Kinase/Akt Pathway and Reactive Oxygen Species. J. Biol. Chem. 2002, 277, 31963–31971. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Kim, T.-Y.; Jong, H.-S.; Kim, T.Y.; Chun, Y.-S.; Park, J.-W.; Lee, C.-T.; Jung, H.C.; Kim, N.K.; Bang, Y.-J. Gastric Epithelial Reactive Oxygen Species Prevent Normoxic Degradation of Hypoxia-Inducible Factor-1 in Gastric Cancer Cells. Clin. Cancer Res. 2003, 9, 433–440. [Google Scholar]
- Shatrov, V.A.; Sumbayev, V.V.; Zhou, J.; Brüne, B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1α (HIF-1α) accumulation via redox-dependent mechanisms. Blood 2003, 101, 4847–4849. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Fandrey, J.; Schümann, J.; Schümann, S.; Tiegs, G.; Brüne, B.; Brüne, B.; Zhou, J.; Fandrey, J.; Schü Mann, G.; et al. NO and TNF-Released from Activated Macrophages Stabilize HIF-1 in Resting Tubular LLC-PK 1 Cells. Am. J. Physiol. Cell Physiol. 2003, 284, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.Y.; Rodriguez-Porcel, M.; Bentley, M.D.; Chade, A.R.; Sica, V.; Napoli, C.; Caplice, N.; Ritman, E.L.; Lerman, A.; Lerman, L.O. Antioxidant Intervention Attenuates Myocardial Neovascularization in Hypercholesterolemia. Circulation 2004, 109, 2109–2115. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.A.; Nespereira, B.; Pérez-Ilzarbe, M.; Eguinoa, E.; Páramo, J.A. Vitamins C and E Prevent Endothelial VEGF and VEGFR-2 Overexpression Induced by Porcine Hypercholesterolemic LDL. Cardiovasc. Res. 2005, 65, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-López, E.; López, A.F.; Esteban, V.; Yagüe, S.; Egido, J.; Ruiz-Ortega, M.; Álvarez-Arroyo, M.V. Angiotensin II Regulates Vascular Endothelial Growth Factor via Hypoxia-Inducible Factor-1α Induction and Redox Mechanisms in the Kidney. Antioxid. Redox Signal. 2005, 7, 1275–1284. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Lee, M.H.; Jin, S.M.; Jin, Y.; Yoo, W.H.; Lee, Y.C. Hydrogen Peroxide Induces Vascular Permeability via Regulation of Vascular Endothelial Growth Factor. Am. J. Respir. Cell Mol. Biol. 2006, 35, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Maranchie, J.K.; Zhan, Y. Nox4 Is Critical for Hypoxia-Inducible Factor 2-α Transcriptional Activity in von Hippel-Lindau–Deficient Renal Cell Carcinoma. Cancer Res. 2005, 65, 9190–9193. [Google Scholar] [CrossRef] [Green Version]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of Hypoxia-Inducible Factor-1α Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef] [Green Version]
- Kaewpila, S.; Venkataraman, S.; Buettner, G.R.; Oberley, L.W. Manganese Superoxide Dismutase Modulates Hypoxia-Inducible Factor-1α Induction via Superoxide. Cancer Res. 2008, 68, 2781–2788. [Google Scholar] [CrossRef] [Green Version]
- Sasabe, E.; Yang, Z.; Ohno, S.; Yamamoto, T. Reactive Oxygen Species Produced by the Knockdown of Manganese-Superoxide Dismutase up-Regulate Hypoxia-Inducible Factor-1α Expression in Oral Squamous Cell Carcinoma Cells. Free Radic. Biol. Med. 2010, 48, 1321–1329. [Google Scholar] [CrossRef]
- Brüne, B.; Zhou, J. Hypoxia-Inducible Factor-1α Under the Control of Nitric Oxide. Methods Enzymol. 2007, 435, 463–478. [Google Scholar] [CrossRef]
- Schofield, C.J.; Zhang, Z. Structural and Mechanistic Studies on 2-Oxoglutarate-Dependent Oxygenases and Related Enzymes. Curr. Opin. Struct. Biol. 1999, 9, 722–731. [Google Scholar] [CrossRef]
- Schofield, C.; Ratcliffe, P. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Ho, V.T.; Bunn, H.F. Effects of Transition Metals on the Expression of the Erythropoietin Gene: Further Evidence That the Oxygen Sensor Is a Heme Protein. Biochem. Biophys. Res. Commun. 1996, 223, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Desferrioxamine Induces Erythropoietin Gene Expression and Hypoxia-Inducible Factor 1 DNA-Binding Activity: Implications for Models of Hypoxia Signal Transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Melillo, G.; Taylor, L.S.; Brooks, A.; Musso, T.; Cox, G.W.; Varesio, L. Functional Requirement of the Hypoxia-Responsive Element in the Activation of the Inducible Nitric Oxide Synthase Promoter by the Iron Chelator Desferrioxamine. J. Biol. Chem. 1997, 272, 12236–12243. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Liang, S.; Cheng, Z.; Zhang, X.; Luo, L.; Li, L.; Zhang, W.; Li, S.; Xu, Q.; Zhong, M.; et al. ROS/PI3K/Akt and Wnt/β-Catenin Signalings Activate HIF-1α-Induced Metabolic Reprogramming to Impart 5-Fluorouracil Resistance in Colorectal Cancer. J. Exp. Clin. Cancer Res. 2022, 41, 15. [Google Scholar] [CrossRef]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN Facilitates HIF-1-Mediated Gene Expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [CrossRef]
- Blancher, C.; Moore, J.W.; Robertson, N.; Harris, A.L. Effects of Ras and von Hippel-Lindau (VHL) Gene Mutations on Hypoxia-Inducible Factor (HIF)-1, HIF-2, and Vascular Endothelial Growth Factor Expression and Their Regulation by the Phosphatidylinositol 3-Kinase/Akt Signaling Pathway 1. Cancer Res. 2001, 61, 7349–7355. [Google Scholar]
- Sodhi, A.; Montaner, S.; Miyazaki, H.; Gutkind, J.S. MAPK and Akt Act Cooperatively but Independently on Hypoxia Inducible Factor-1α in RasV12 Upregulation of VEGF. Biochem. Biophys. Res. Commun. 2001, 287, 292–300. [Google Scholar] [CrossRef]
- Akeno, N.; Robins, J.; Zhang, M.; Czyzyk-Krzeska, M.F.; Clemens, T.L. Induction of Vascular Endothelial Growth Factor by IGF-I in Osteoblast-Like Cells Is Mediated by the PI3K Signaling Pathway through the Hypoxia-Inducible Factor-2α. Endocrinology 2002, 143, 420–425. [Google Scholar] [CrossRef]
- Kietzmann, T.; Jungermann, K.; Görlach, A. Regulation of the Hypoxia-Dependent Plasminogen Activator Inhibitor I Expression by MAP Kinases. Thromb. Haemost. 2003, 89, 666–673. [Google Scholar] [CrossRef]
- Qian, D.; Lin, H.Y.; Wang, H.M.; Zhang, X.; Liu, D.L.; Li, Q.L.; Zhu, C. Normoxic Induction of the Hypoxic-Inducible Factor-1α by Interleukin-1β Involves the Extracellular Signal-Regulated Kinase 1/2 Pathway in Normal Human Cytotrophoblast Cells. Biol. Reprod. 2004, 70, 1822–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.H.; Wang, Y.X.; Bingle, L.; Gong, L.H.; Heng, W.J.; Li, Y.; Fang, W.G. In Vitro Study of HIF-1 Activation and VEGF Release by BFGF in the T47D Breast Cancer Cell Line under Normoxic Conditions: Involvement of PI-3K/Akt and MEK1/ERK Pathways. J. Pathol. 2005, 205, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Shen, L.; Zhang, Z.; Leonard, S.S.; He, H.; Zhang, X.G.; Shi, X.; Jiang, B.H. Arsenite Induces HIF-1α and VEGF through PI3K, Akt and Reactive Oxygen Species in DU145 Human Prostate Carcinoma Cells. Mol. Cell. Biochem. 2004, 255, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.W.; Kang, T.S.; Lee, B.K.; Chang, W.; Hwang, K.C.; Rhee, J.H.; Min, P.K.; Hong, B.K.; Rim, S.J.; Kwon, H.M. Pathobiological Role of Advanced Glycation Endproducts via Mitogen-Activated Protein Kinase Dependent Pathway in the Diabetic Vasculopathy. Exp. Mol. Med. 2008, 40, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Nawroth, P.P. Multiple Levels of Regulation Determine the Role of the Receptor for AGE (RAGE) as Common Soil in Inflammation, Immune Responses and Diabetes Mellitus and Its Complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef] [Green Version]
- Rai, V.; Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Schmidt, A.M.; Shekhtman, A. Signal Transduction in Receptor for Advanced Glycation End Products (RAGE). J. Biol. Chem. 2012, 287, 5133–5144. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Yu, X.; Yang, H.; Wu, X. Advanced Glycation End Products Induce Human Corneal Epithelial Cells Apoptosis through Generation of Reactive Oxygen Species and Activation of JNK and P38 MAPK Pathways. PLoS ONE 2013, 8, e66781. [Google Scholar] [CrossRef] [Green Version]
- Menini, S.; Iacobini, C.; Ricci, C.; Fantauzzi, C.B.; Pugliese, G. Protection from Diabetes-Induced Atherosclerosis and Renal Disease by d-Carnosine-Octylester: Effects of Early vs. Late Inhibition of Advanced Glycation End-Products in Apoe-Null Mice. Diabetologia 2015, 58, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tang, L.; Chen, S.; Yang, Y.; Chen, M.; Luo, J. Effect of Advanced Glycation End Products on the Expression of Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor Proteins in RF/6A Cells. Exp. Ther. Med. 2013, 5, 1519–1522. [Google Scholar] [CrossRef] [Green Version]
- Poitz, D.M.; Augstein, A.; Weinert, S.; Braun-Dullaeus, R.C.; Strasser, R.H.; Schmeisser, A. OxLDL and Macrophage Survival: Essential and Oxygen-Independent Involvement of the Hif-Pathway. Basic Res. Cardiol. 2011, 106, 761–772. [Google Scholar] [CrossRef]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; van Obberghen, E. Regulation of Vascular Endothelial Growth Factor Expression by Advanced Glycation End Products. J. Biol. Chem. 2001, 276, 43836–43841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Ma, W.Q.; Han, X.Q.; Wang, Y.; Wang, X.; Liu, N.F. Advanced Glycation End Products Accelerate Calcification in VSMCs through HIF-1α/PDK4 Activation and Suppress Glucose Metabolism. Sci. Rep. 2018, 8, 13730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Leleu, D.; Masson, D. Cholesterol and HIF-1α: Dangerous Liaisons in Atherosclerosis. Front. Immunol. 2022, 13, 868958. [Google Scholar] [CrossRef] [PubMed]
- Bondeva, T.; Heinzig, J.; Ruhe, C.; Wolf, G. Advanced Glycated End-Products Affect HIF-Transcriptional Activity in Renal Cells. Mol. Endocrinol. 2013, 27, 1918–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH Oxidase by AGE Links Oxidant Stress to Altered Gene Expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Liu, X.; Cheng, Y.; Yang, J.; Huo, Y.; Zhang, C. Involvement of MicroRNAs in Hydrogen Peroxide-Mediated Gene Regulation and Cellular Injury Response in Vascular Smooth Muscle Cells. J. Biol. Chem. 2009, 284, 7903–7913. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Z.; Li, C.; Chen, Q.; Jing, Y.; Carpenter, R.; Jiang, Y.; Kung, H.F.; Lai, L.; Jiang, B.H. MiR-21 Induced Angiogenesis through AKT and ERK Activation and HIF-1α Expression. PLoS ONE 2011, 6, e19139. [Google Scholar] [CrossRef] [Green Version]
- La Sala, L.; Mrakic-Sposta, S.; Tagliabue, E.; Prattichizzo, F.; Micheloni, S.; Sangalli, E.; Specchia, C.; Uccellatore, A.C.; Lupini, S.; Spinetti, G.; et al. Circulating microRNA-21 is an early predictor of ROS-mediated damage in subjects with high risk of developing diabetes and in drug-naïve T2D. Cardiovasc. Diabetol. 2019, 18, 18. [Google Scholar] [CrossRef] [Green Version]
- Kölling, M.; Kaucsar, T.; Schauerte, C.; Hübner, A.; Dettling, A.; Park, J.K.; Busch, M.; Wulff, X.; Meier, M.; Scherf, K.; et al. Therapeutic MiR-21 Silencing Ameliorates Diabetic Kidney Disease in Mice. Mol. Ther. 2017, 25, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wu, W.; Liao, J.; Tang, F.; Gao, G.; Peng, J.; Fu, X.; Zhan, Y.; Chen, Z.; Xu, W.; et al. MicroRNA-21: A Critical Pathogenic Factor of Diabetic Nephropathy. Front. Endocrinol. 2022, 13, 895010. [Google Scholar] [CrossRef]
- Varma, S.; Lal, B.K.; Zheng, R.; Breslin, J.W.; Saito, S.; Pappas, P.J.; Hobson, R.W.; Durán, W.N. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am. J. Physiol. Circ. Physiol. 2005, 289, H1744–H1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in Innate Immune Responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic Pathways in T Cell Activation and Lineage Differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, J.; Nocera, D.G. Role of Proton-Coupled Electron Transfer in O-O Bond Activation. Acc. Chem. Res. 2007, 40, 543–553. [Google Scholar] [CrossRef]
- Bartz, R.R.; Piantadosi, C.A. Clinical Review: Oxygen as a Signaling Molecule. Crit. Care 2010, 14, 234. [Google Scholar] [CrossRef] [Green Version]
- Stegen, S.; van Gastel, N.; Eelen, G.; Ghesquière, B.; D’Anna, F.; Thienpont, B.; Goveia, J.; Torrekens, S.; van Looveren, R.; Luyten, F.P.; et al. HIF-1α Promotes Glutamine-Mediated Redox Homeostasis and Glycogen-Dependent Bioenergetics to Support Post-Implantation Cell Survival. Cell Metab. 2016, 23, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Narayanan, S.; Xu, C.; Angelstig, S.E.; Grünler, J.; Zhao, A.; di Toro, A.; Bernardi, L.; Mazzone, M.; Carmeliet, P.; et al. Repression of Hypoxia-Inducible Factor-1 Contributes to Increased Mitochondrial Reactive Oxygen Species Production in Diabetes. Elife 2022, 11, e70714. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK Dysregulation Promotes Diabetes-Related Reduction of Superoxide and Mitochondrial Function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Sharma, K. Challenging the Dogma of Mitochondrial Reactive Oxygen Species Overproduction in Diabetic Kidney Disease. Kidney Int. 2016, 90, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Zhang, G.; Hall, D.; Oates, P.J.; Maity, S.; Madesh, M.; Han, X.; Sharma, K. Restoring Mitochondrial Superoxide Levels with Elamipretide (MTP-131) Protects Db/Db Mice against Progression of Diabetic Kidney Disease. J. Biol. Chem. 2020, 295, 7249–7260. [Google Scholar] [CrossRef] [Green Version]
- La Sala, L.; Pujadas, G.; de Nigris, V.; Canivell, S.; Novials, A.; Genovese, S.; Ceriello, A. Oscillating Glucose and Constant High Glucose Induce Endoglin Expression in Endothelial Cells: The Role of Oxidative Stress. Acta Diabetol. 2015, 52, 505–512. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Okada, R.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; et al. Rho-Kinase Inhibition Prevents the Progression of Diabetic Nephropathy by Downregulating Hypoxia-Inducible Factor 1α. Kidney Int. 2013, 84, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Pirri, D.; Fragiadaki, M.; Evans, P.C. Diabetic Atherosclerosis: Is There a Role for the Hypoxia-Inducible Factors? Biosci. Rep. 2020, 40, BSR20200026. [Google Scholar] [CrossRef]
- Liu, D.; Lei, L.; Desir, M.; Huang, Y.; Cleman, J.; Jiang, W.; Fernandez-Hernando, C.; di Lorenzo, A.; Sessa, W.C.; Giordano, F.J. Smooth Muscle Hypoxia-Inducible Factor 1α Links Intravascular Pressure and Atherosclerosis-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 442–445. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Bowden, N.; Fragiadaki, M.; Souilhol, C.; Hsiao, S.; Mahmoud, M.; Allen, S.; Pirri, D.; Ayllon, B.T.; Akhtar, S.; et al. Mechanical Activation of Hypoxia-Inducible Factor 1a Drives Endothelial Dysfunction at Atheroprone Sites. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Huang, R.T.; Hamanaka, R.B.; Krause, M.; Oh, M.J.; Kuo, C.H.; Nigdelioglu, R.; Meliton, A.Y.; Witt, L.; Dai, G.; et al. HIF-1α Is Required for Disturbed Flow-Induced Metabolic Reprogramming in Human and Porcine Vascular Endothelium. Elife 2017, 6, e25217. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Wang, J.F.; Chan, P.; Lee, H.M. Angiotensin II stimulates hypoxia-inducible factor 1alpha accumulation in glomerular mesangial cells. Ann. N. Y. Acad. Sci. 2005, 1042, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1α Expression in Macrophages Promotes Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1782–1790. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of Oxidative Stress in Diabetic Complications: A New Perspective on an Old Paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Darshi, M.; Sharma, K. The Warburg Effect in Diabetic Kidney Disease. Semin. Nephrol. 2018, 38, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate Kinase M2 Activation May Protect against the Progression of Diabetic Glomerular Pathology and Mitochondrial Dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. JBIC J. Biol. Inorg. Chem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Pipino, C.; Shah, H.; Prudente, S.; di Pietro, N.; Zeng, L.; Park, K.; Trischitta, V.; Pennathur, S.; Pandolfi, A.; Doria, A. Association of the 1q25 Diabetes-Specific Coronary Heart Disease Locus with Alterations of the γ-Glutamyl Cycle and Increased Methylglyoxal Levels in Endothelial Cells. Diabetes 2020, 69, 2206–2216. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Ting, J.P.Y.; O’Neill, L.A.J. A Role for the NLRP3 Inflammasome in Metabolic Diseases and Did Warburg Miss Inflammation? Nat. Immunol. 2012, 13, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Katavetin, P.; Miyata, T.; Inagi, R.; Tanaka, T.; Sassa, R.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. High Glucose Blunts Vascular Endothelial Growth Factor Response to Hypoxia via the Oxidative Stress-Regulated Hypoxia-Inducible Factor/Hypoxia-Responsible Element Pathway. J. Am. Soc. Nephrol. 2006, 17, 1405–1413. [Google Scholar] [CrossRef]
- Yu, X.; Fang, Y.; Liu, H.; Zhu, J.; Zou, J.; Xu, X.; Jiang, S.; Ding, X. The Balance of Beneficial and Deleterious Effects of Hypoxia-Inducible Factor Activation by Prolyl Hydroxylase Inhibitor in Rat Remnant Kidney Depends on the Timing of Administration. Nephrol. Dial. Transplant. 2012, 27, 3110–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Peng, Y.-J.; Reddy, V.D.; Makarenko, V.V.; Nanduri, J.; Khan, S.A.; Garcia, J.A.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Mutual antagonism between hypoxia-inducible factors 1α and 2α regulates oxygen sensing and cardio-respiratory homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, E1788–E1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.-J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.A.; et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Ban, J.-J.; Ruthenborg, R.J.; Cho, K.W.; Kim, J. Regulation of Obesity and Insulin Resistance by Hypoxia-Inducible Factors. Hypoxia 2014, 2, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential Activation and Antagonistic Function of HIF-α Isoforms in Macrophages Are Essential for NO Homeostasis. Genes Dev. 2010, 24, 491. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, N.; Segawa, R.; Tobita, R.; Asakawa, S.; Mizuno, N.; Hiratsuka, M.; Hirasawa, N. Hypoxia Inhibits TNF-α-Induced TSLP Expression in Keratinocytes. PLoS ONE 2019, 14, e0224705. [Google Scholar] [CrossRef] [Green Version]
- Charron, C.E.; Chou, P.C.; Coutts, D.J.C.; Kumar, V.; To, M.; Akashi, K.; Pinhu, L.; Griffiths, M.; Adcock, I.M.; Barnes, P.J.; et al. Hypoxia-Inducible Factor 1α Induces Corticosteroid-Insensitive Inflammation via Reduction of Histone Deacetylase-2 Transcription. J. Biol. Chem. 2009, 284, 36047–36054. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.S.; Lin, Y.L.; Wang, Y.A.; Mo, S.T.; Chi, P.Y.; Lai, A.C.Y.; Pan, H.Y.; Chang, Y.J.; Lai, M.Z. HIF-2α Is Indispensable for Regulatory T Cell Function. Nat. Commun. 2020, 11, 5005. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Tang, L.; Zhu, Q.; Yi, F.; Zhang, F.; Li, P.L.; Li, N. Contribution of Hypoxia Inducible Factor-1α to the Profibrotic Action of Angiotensin II in Cultured Renal Medullary Interstitial Cells. Kidney Int. 2011, 79, 300–310. [Google Scholar] [CrossRef]
- Ishizuka, S.; Sakai, T.; Hiraiwa, H.; Hamada, T.; Knudson, W.; Omachi, T.; Ono, Y.; Nakashima, M.; Matsukawa, T.; Oda, T.; et al. Hypoxia-Inducible Factor-2α Induces Expression of Type X Collagen and Matrix Metalloproteinases 13 in Osteoarthritic Meniscal Cells. Inflamm. Res. 2016, 65, 439–448. [Google Scholar] [CrossRef]
- Xu, X.; Song, N.; Zhang, X.; Jiao, X.; Hu, J.; Liang, M.; Teng, J.; Ding, X. Renal Protection Mediated by Hypoxia Inducible Factor-1α Depends on Proangiogenesis Function of MiR-21 by Targeting Thrombospondin 1. Transplantation 2017, 101, 1811–1819. [Google Scholar] [CrossRef] [Green Version]
- Conde, E.; Giménez-Moyano, S.; Martín-Gómez, L.; Rodríguez, M.; Ramos, M.E.; Aguado-Fraile, E.; Blanco-Sanchez, I.; Saiz, A.; Garciá-Bermejo, M.L. HIF-1α Induction during Reperfusion Avoids Maladaptive Repair after Renal Ischemia/Reperfusion Involving MiR127-3p. Sci. Rep. 2017, 7, 41099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Zhang, H.; Zhong, Y.; Ding, X. Prolyl Hydroxylase 2 (PHD2) Inhibition Protects Human Renal Epithelial Cells and Mice Kidney from Hypoxia Injury. Oncotarget 2016, 7, 54317–54328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Li, J.; Li, Y.; Zhang, Y.; Du, Q.; Hao, P.; Li, J.; Cao, X.; Li, L. Berberine Protects Against Simulated Ischemia/Reperfusion Injury-Induced H9C2 Cardiomyocytes Apoptosis In Vitro and Myocardial Ischemia/Reperfusion-Induced Apoptosis In Vivo by Regulating the Mitophagy-Mediated HIF-1α/BNIP3 Pathway. Front. Pharmacol. 2020, 11, 367. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, L.; Wang, J.; Chen, N.; Yan, J.; Pan, X. HIF-2α and Oct4 Have Synergistic Effects on Survival and Myocardial Repair of Very Small Embryonic-like Mesenchymal Stem Cells in Infarcted Hearts. Cell Death Dis. 2017, 8, e2548. [Google Scholar] [CrossRef] [Green Version]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 Mediates Protection and Recovery from Ischemic Kidney Injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Luo, G.; Fang, Q.; Sun, Z. Stable Expression of Hypoxia-Inducible Factor-1α in Human Renal Proximal Tubular Epithelial Cells Promotes Epithelial to Mesenchymal Transition. Transplant. Proc. 2014, 46, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Lv, L.L.; Tang, T.T.; Wang, B.; Feng, Y.; Zhou, L.T.; Cao, J.Y.; Tang, R.N.; Wu, M.; Liu, H.; et al. HIF-1α Inducing Exosomal MicroRNA-23a Expression Mediates the Cross-Talk between Tubular Epithelial Cells and Macrophages in Tubulointerstitial Inflammation. Kidney Int. 2019, 95, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia Promotes Fibrogenesis in Vivo via HIF-1 Stimulation of Epithelial-to-Mesenchymal Transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Hayashida, T.; Liang, X.; Schnaper, H.W. Hypoxia-Inducible Factor-1α Promotes Glomerulosclerosis and Regulates COL1A2 Expression through Interactions with Smad3. Kidney Int. 2016, 90, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, W.; Zhang, F.; Li, P.L.; Boini, K.M.; Yi, F.; Li, N. Hypoxia Inducible Factor-1α Mediates the Profibrotic Effect of Albumin in Renal Tubular Cells. Sci. Rep. 2017, 7, 15878. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zeng, H.; Chen, S.T.; Zhou, L.; Xie, X.J.; He, X.; Tao, Y.K.; Tuo, Q.H.; Deng, C.; Liao, D.F.; et al. Ablation of Endothelial Prolyl Hydroxylase Domain Protein-2 Promotes Renal Vascular Remodelling and Fibrosis in Mice. J. Cell. Mol. Med. 2017, 21, 1967–1978. [Google Scholar] [CrossRef]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, S.; Hartmann, P.; Karshovska, E.; Rinderknecht, F.A.; Subramanian, P.; Gremse, F.; Grommes, J.; Jacobs, M.; Kiessling, F.; Weber, C.; et al. Endothelial Hypoxia-Inducible Factor-1α Promotes Atherosclerosis and Monocyte Recruitment by Upregulating MicroRNA-19a. Hypertension 2015, 66, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Lu, W.H.; Ai, R.; Yang, J.H.; Chen, F.; Tang, Z.Z. The Relationship between Serum Hypoxia-Inducible Factor 1α and Coronary Artery Calcification in Asymptomatic Type 2 Diabetic Patients. Cardiovasc. Diabetol. 2014, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Y.; Wang, P.; Zhang, S.Y.; Dong, Y.; Zeng, G.; Yan, Y.; Sun, L.; Wu, Q.; Liu, H.; et al. Adipocyte Hypoxia-Inducible Factor 2α Suppresses Atherosclerosis by Promoting Adipose Ceramide Catabolism. Cell Metab. 2019, 30, 937–951.e5. [Google Scholar] [CrossRef]
- Inada, A.; Inada, O.; Yasunami, Y.; Arakawa, K.; Nabeshima, Y.; Fukatsu, A. Amelioration of Murine Diabetic Nephropathy with a SGLT2 Inhibitor Is Associated with Suppressing Abnormal Expression of Hypoxia-Inducible Factors. Am. J. Pathol. 2022, 192, 1028–1052. [Google Scholar] [CrossRef]
- Rahtu-Korpela, L.; Määttä, J.; Dimova, E.Y.; Hörkkö, S.; Gylling, H.; Walkinshaw, G.; Hakkola, J.; Kivirikko, K.I.; Myllyharju, J.; Serpi, R.; et al. Hypoxia-Inducible Factor Prolyl 4-Hydroxylase-2 Inhibition Protects Against Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Natale, P.; Palmer, S.C.; Jaure, A.; Hodson, E.M.; Ruospo, M.; Cooper, T.E.; Hahn, D.; Saglimbene, V.M.; Craig, J.C.; Strippoli, G.F. Hypoxia-inducible Factor Stabilisers for the Anaemia of Chronic Kidney Disease. Cochrane Database Syst. Rev. 2022, 2022, 8. [Google Scholar] [CrossRef]
- Hasegawa, S.; Tanaka, T.; Saito, T.; Fukui, K.; Wakashima, T.; Susaki, E.A.; Ueda, H.R.; Nangaku, M. The Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Enarodustat Counteracts Alterations in Renal Energy Metabolism in the Early Stages of Diabetic Kidney Disease. Kidney Int. 2020, 97, 934–950. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Tanaka, S.; Tanaka, T.; Saito, H.; Ishimoto, Y.; Wakashima, T.; Ueda, M.; Fukui, K.; Shimizu, A.; Inagi, R.; et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. J. Am. Soc. Nephrol. 2020, 31, 560–577. [Google Scholar] [CrossRef]
- Ohtomo, S.; Nangaku, M.; Izuhara, Y.; Takizawa, S.; de Strihou, C.V.Y.; Miyata, T. Cobalt Ameliorates Renal Injury in an Obese, Hypertensive Type 2 Diabetes Rat Model. Nephrol. Dial. Transplant. 2008, 23, 1166–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordquist, L.; Friederich-Persson, M.; Fasching, A.; Liss, P.; Shoji, K.; Nangaku, M.; Hansell, P.; Palm, F. Activation of Hypoxia-Inducible Factors Prevents Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Rahtu-Korpela, L.; Karsikas, S.; Hörkkö, S.; Sequeiros, R.B.; Lammentausta, E.; Mäkelä, K.A.; Herzig, K.H.; Walkinshaw, G.; Kivirikko, K.I.; Myllyharju, J.; et al. HIF Prolyl 4-Hydroxylase-2 Inhibition Improves Glucose and Lipid Metabolism and Protects Against Obesity and Metabolic Dysfunction. Diabetes 2014, 63, 3324–3333. [Google Scholar] [CrossRef] [Green Version]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.; Klaus, S.J.; Bachmann, S. Hypoxia-Inducible Factor-2α-Expressing Interstitial Fibroblasts Are the Only Renal Cells That Express Erythropoietin under Hypoxia-Inducible Factor Stabilization. Kidney Int. 2010, 77, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Ochiai, N.; Takano, H.; Io, F.; Takayama, N.; Koretsune, H.; Kunioka, E.; Uchida, S.; Yamamoto, K. TP0463518, a Novel Prolyl Hydroxylase Inhibitor, Specifically Induces Erythropoietin Production in the Liver. J. Pharmacol. Exp. Ther. 2019, 371, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Das, F.; Ghosh-Choudhury, N.; Maity, S.; Kasinath, B.S.; Choudhury, G.G. Oncoprotein DJ-1 Interacts with MTOR Complexes to Effect Transcription Factor Hif1α-Dependent Expression of Collagen I (A2) during Renal Fibrosis. J. Biol. Chem. 2022, 298, 102246. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Li, L.; Zhou, X.; Xue, C.; Livingston, M.J.; Wei, Q.; Dai, B.; Mao, Z.; Mei, C.; Dong, Z. Susceptibility of Renal Fibrosis in Diabetes: Role of Hypoxia Inducible Factor-1. FASEB J. 2022, 36, e22477. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Beitner-Johnson, D.; Millhorn, D.E. Hypoxia-Inducible Factor 2α Binds to Cobalt in Vitro. Biochem. Biophys. Res. Commun. 2001, 288, 849–854. [Google Scholar] [CrossRef]
- Balaiya, S.; Khetpal, V.; Chalam, K.V. Hypoxia Initiates Sirtuin1-Mediated Vascular Endothelial Growth Factor Activation in Choroidal Endothelial Cells through Hypoxia Inducible Factor–2α. Mol. Vis. 2012, 18, 114–120. [Google Scholar]
- Cioffi, C.L.; Liu, X.Q.; Kosinski, P.A.; Garay, M.; Bowen, B.R. Differential Regulation of HIF-1α Prolyl-4-Hydroxylase Genes by Hypoxia in Human Cardiovascular Cells. Biochem. Biophys. Res. Commun. 2003, 303, 947–953. [Google Scholar] [CrossRef]
- Aprelikova, O.; Chandramouli, G.V.R.; Wood, M.; Vasselli, J.R.; Riss, J.; Maranchie, J.K.; Linehan, W.M.; Barrett, J.C. Regulation of HIF Prolyl Hydroxylases by Hypoxia-Inducible Factors. J. Cell. Biochem. 2004, 92, 491–501. [Google Scholar] [CrossRef]
- Miikkulainen, P.; Högel, H.; Seyednasrollah, F.; Rantanen, K.; Elo, L.L.; Jaakkola, P.M. Hypoxia-Inducible Factor (HIF)-Prolyl Hydroxylase 3 (PHD3) Maintains High HIF2A MRNA Levels in Clear Cell Renal Cell Carcinoma. J. Biol. Chem. 2019, 294, 3760–3771. [Google Scholar] [CrossRef] [Green Version]
- Fukui, K.; Shinozaki, Y.; Kobayashi, H.; Deai, K.; Yoshiuchi, H.; Matsui, T.; Matsuo, A.; Matsushita, M.; Tanaka, T.; Nangaku, M. JTZ-951 (Enarodustat), a Hypoxia-Inducibe Factor Prolyl Hydroxylase Inhibitor, Stabilizes HIF-α Protein and Induces Erythropoiesis without Effects on the Function of Vascular Endothelial Growth Factor. Eur. J. Pharmacol. 2019, 859, 172532. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in Mammals: Insights into Their Biological Function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Powis, G. Passing the Baton: The HIF Switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Kanasaki, K.; Koya, D. Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin. Sci. 2012, 124, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic CKD Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. 2020, 31, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. SGLT2 Inhibitors Produce Cardiorenal Benefits by Promoting Adaptive Cellular Reprogramming to Induce a State of Fasting Mimicry: A Paradigm Shift in Understanding Their Mechanism of Action. Diabetes Care 2020, 43, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Zhao, W.; Xu, S.; Weng, J. Resveratrol in Treating Diabetes and Its Cardiovascular Complications: A Review of Its Mechanisms of Action. Antioxidants 2022, 11, 1085. [Google Scholar] [CrossRef]
- Schmidt, H.H.H.W.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, J.; Wang, L.; Li, A.; Qiu, Z.; Qi, L.W.; Kou, J.; Liu, K.; Liu, B.; Huang, F. The Role of Metformin and Resveratrol in the Prevention of Hypoxia-inducible Factor 1α Accumulation and Fibrosis in Hypoxic Adipose Tissue. Br. J. Pharmacol. 2016, 173, 2001–2015. [Google Scholar] [CrossRef]
- Ryu, D.R.; Yu, M.R.; Kong, K.H.; Kim, H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Noh, H. Sirt1–Hypoxia-inducible Factor-1α Interaction Is a Key Mediator of Tubulointerstitial Damage in the Aged Kidney. Aging Cell 2019, 18, e12904. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Lv, C.; Wu, C.; Zhou, Y.; Wang, Q. Mir-217 promotes inflammation and fibrosis in high glucose cultured rat glomerular mesangial cells via Sirt1/HIF-1α signaling pathway. Diabetes/Metab. Res. Rev. 2016, 32, 534–543. [Google Scholar] [CrossRef]
- Wang, S.; Qian, Y.; Gong, D.; Zhang, Y.; Fan, Y. Resveratrol Attenuates Acute Hypoxic Injury in Cardiomyocytes: Correlation with Inhibition of INOS–NO Signaling Pathway. Eur. J. Pharm. Sci. 2011, 44, 416–421. [Google Scholar] [CrossRef]
- Pektaş, M.B.; Sadi, G.; Koca, H.B.; Yuksel, Y.; Vurmaz, A.; Koca, T.; Tosun, M. Resveratrol Ameliorates the Components of Hepatic Inflammation and Apoptosis in a Rat Model of Streptozotocin-Induced Diabetes. Drug Dev. Res. 2016, 77, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of Hypoxia-Inducible Factor 2alpha Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Barutta, F.; Bernardi, S.; Gargiulo, G.; Durazzo, M.; Gruden, G. SGLT2 inhibition to address the unmet needs in diabetic nephropathy. Diabetes/Metab. Res. Rev. 2019, 35, e3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. Role of Ketogenic Starvation Sensors in Mediating the Renal Protective Effects of SGLT2 Inhibitors in Type 2 Diabetes. J. Diabetes Its Complicat. 2020, 34, 107647. [Google Scholar] [CrossRef]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Kanasaki, K. Sodium–Glucose Cotransporter-2 Inhibitors for Diabetic Kidney Disease: Targeting Warburg Effects in Proximal Tubular Cells. Diabetes Metab. 2020, 46, 353–361. [Google Scholar] [CrossRef]
- Bessho, R.; Takiyama, Y.; Takiyama, T.; Kitsunai, H.; Takeda, Y.; Sakagami, H.; Ota, T. Hypoxia-Inducible Factor-1α Is the Therapeutic Target of the SGLT2 Inhibitor for Diabetic Nephropathy. Sci. Rep. 2019, 9, 14754. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef]
- Chang, D.Y.; Li, X.Q.; Chen, M.; Zhao, M.H. Dapagliflozin Ameliorates Diabetic Kidney Disease via Upregulating Crry and Alleviating Complement Over-Activation in Db/Db Mice. Front. Pharmacol. 2021, 12, 729334. [Google Scholar] [CrossRef]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium–Glucose Cotransporter 2 Inhibition Suppresses HIF-1α-Mediated Metabolic Switch from Lipid Oxidation to Glycolysis in Kidney Tubule Cells of Diabetic Mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef]
- Packer, M. Cardioprotective Effects of Sirtuin-1 and Its Downstream Effectors. Circ. Heart Fail. 2020, 13, E007197. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Goto, S. Possible mechanism of hematocrit elevation by sodium glucose cotransporter 2 inhibitors and associated beneficial renal and cardiovascular effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, B.N.; Coleman, A.E.; Schmiedt, C.W.; Brown, C.A.; Rissi, D.R.; Stanton, J.B.; Giguère, S.; Berghaus, R.D.; Brown, S.A.; Tarigo, J.L. Profibrotic Gene Transcription in Renal Tissues from Cats with Ischemia-Induced Chronic Kidney Disease. Am. J. Vet. Res. 2020, 81, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.; Palm, F. Hypoxia-Inducible Factor Activation in Diabetic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 345–350. [Google Scholar] [CrossRef]
- Gu, H.F.; Zheng, X.; Seman, N.A.; Gu, T.; Botusan, I.R.; Sunkari, V.G.; Lokman, E.F.; Brismar, K.; Catrina, S.B. Impact of the Hypoxia-Inducible Factor-1 α (HIF1A) Pro582Ser Polymorphism on Diabetes Nephropathy. Diabetes Care 2013, 36, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, I.; Giorgino, F. SGLT-2 Inhibitors as Cardio-Renal Protective Agents. Metabolism 2022, 127, 154937. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobini, C.; Vitale, M.; Haxhi, J.; Pesce, C.; Pugliese, G.; Menini, S. Mutual Regulation between Redox and Hypoxia-Inducible Factors in Cardiovascular and Renal Complications of Diabetes. Antioxidants 2022, 11, 2183. https://doi.org/10.3390/antiox11112183
Iacobini C, Vitale M, Haxhi J, Pesce C, Pugliese G, Menini S. Mutual Regulation between Redox and Hypoxia-Inducible Factors in Cardiovascular and Renal Complications of Diabetes. Antioxidants. 2022; 11(11):2183. https://doi.org/10.3390/antiox11112183
Chicago/Turabian StyleIacobini, Carla, Martina Vitale, Jonida Haxhi, Carlo Pesce, Giuseppe Pugliese, and Stefano Menini. 2022. "Mutual Regulation between Redox and Hypoxia-Inducible Factors in Cardiovascular and Renal Complications of Diabetes" Antioxidants 11, no. 11: 2183. https://doi.org/10.3390/antiox11112183