
Microglial Hemoxygenase-1 Deletion Reduces Inflammation in the Retina of Old Mice with Tauopathy
 ,
,  ,
,  ,
,  ,
, 
 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Groups
2.2. Generation of Hemoxygenase-1 Knockout Animals for the HMO-KO and HMO-KO+TAU Groups
2.3. Generation of Animals with Tauopathy: TAU and HMO-KO+TAU Groups
2.4. Immunohistochemistry
2.5. Morphometric Analysis of Retinal Wholemounts
2.5.1. Tau Protein Expression
2.5.2. Microglial Characterization
Number of Microglia Iba-1+
Number of Microglia Iba-1+/CD68+
Area of Soma and Arbor Area of Iba-1+ Cell
2.5.3. Macroglial Characterization
Retinal Area Occupied by GFAP+ Immunolabeling (AROA)
Retinal Area Occupied by C3+ Immunolabeling (AROC3)
2.6. Statistical Analysis
3. Results
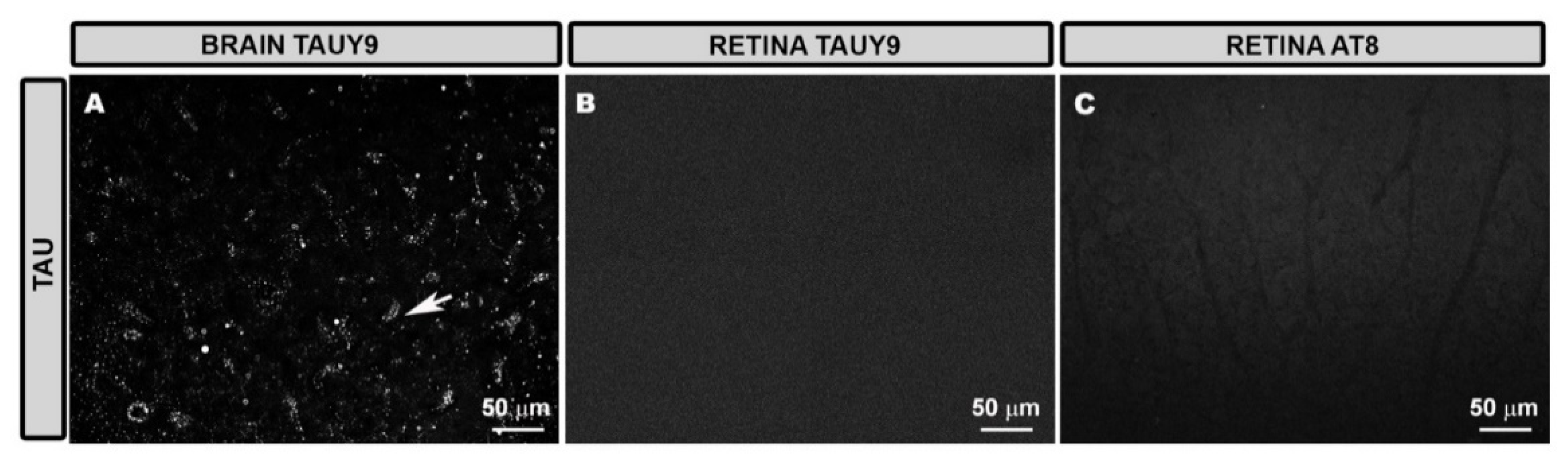
3.1. Tau Expression in Retinal Tissue
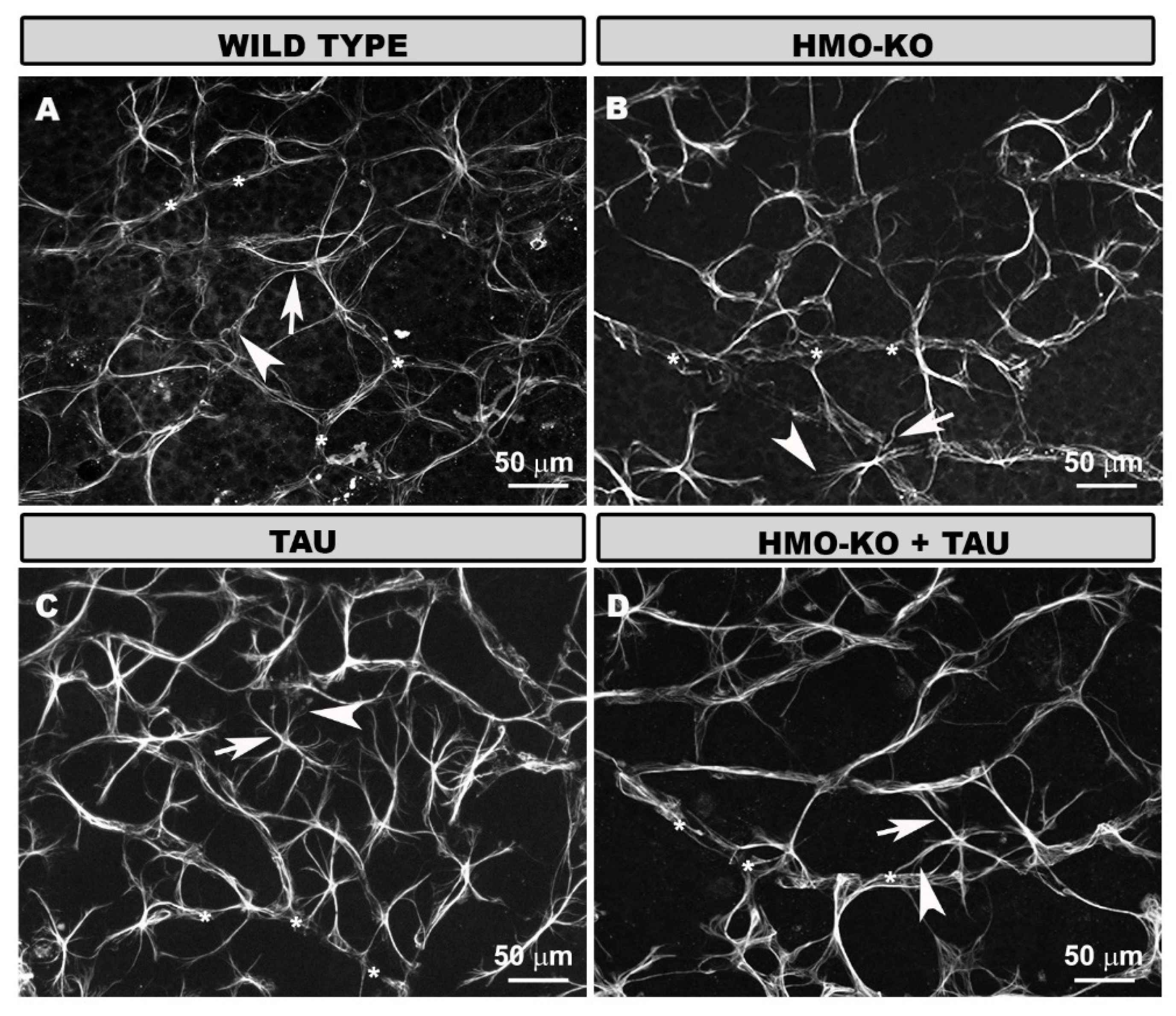
3.2. Morphology and Distribution of Retinal Microglia in the Different Study Groups
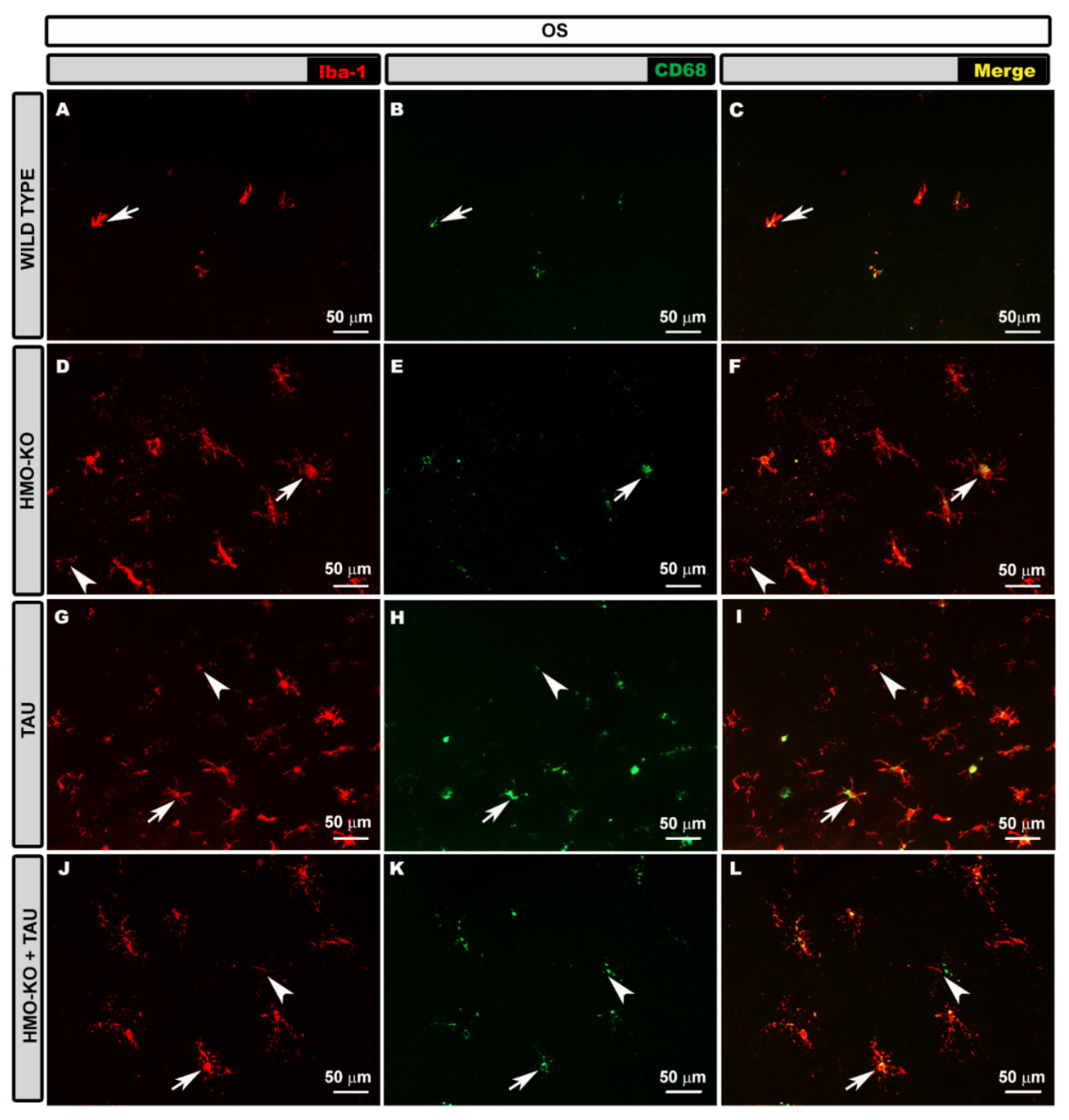
3.2.1. Outer Segment Layer (OS)
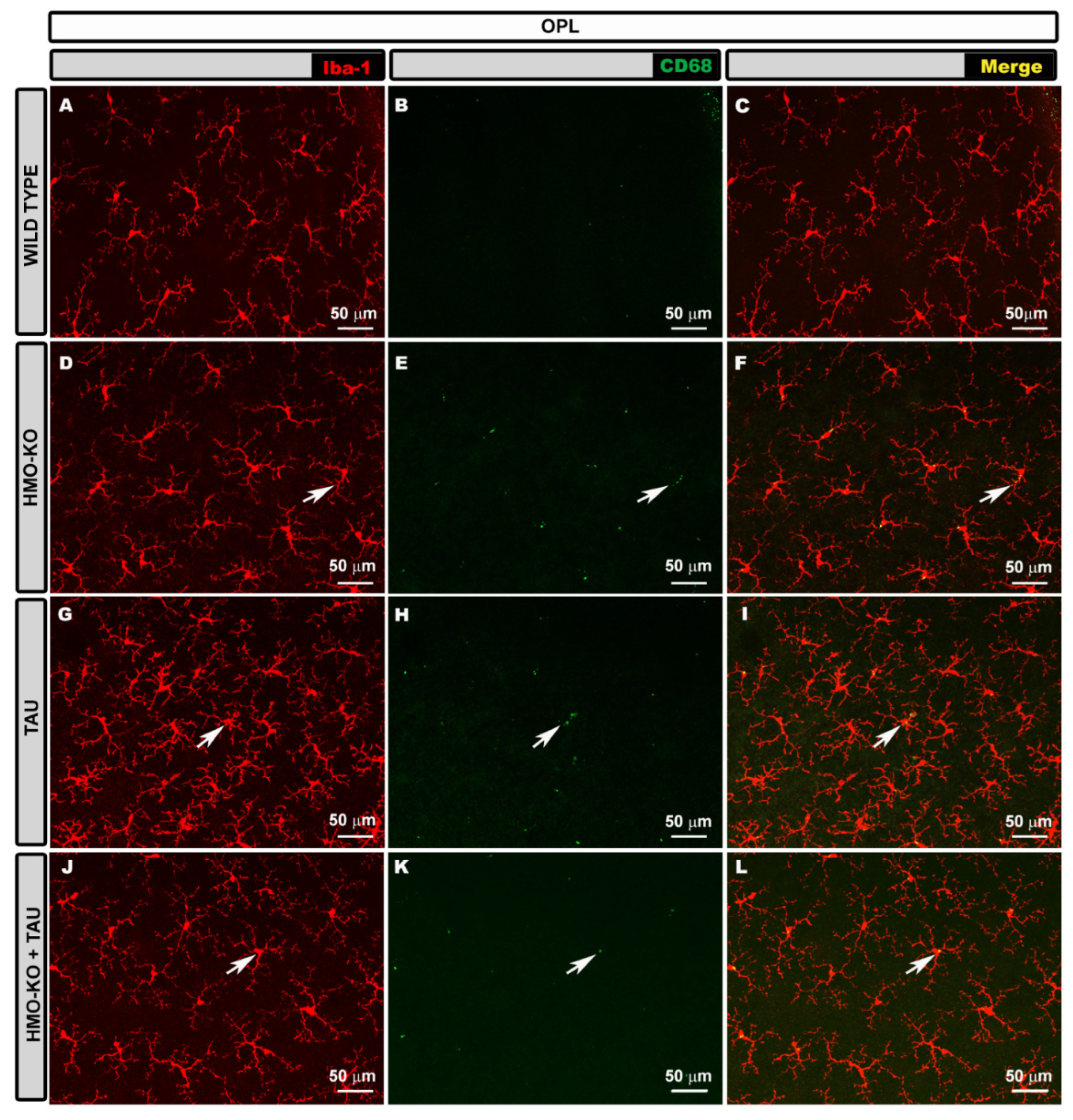
3.2.2. Outer Plexiform Layer (OPL)
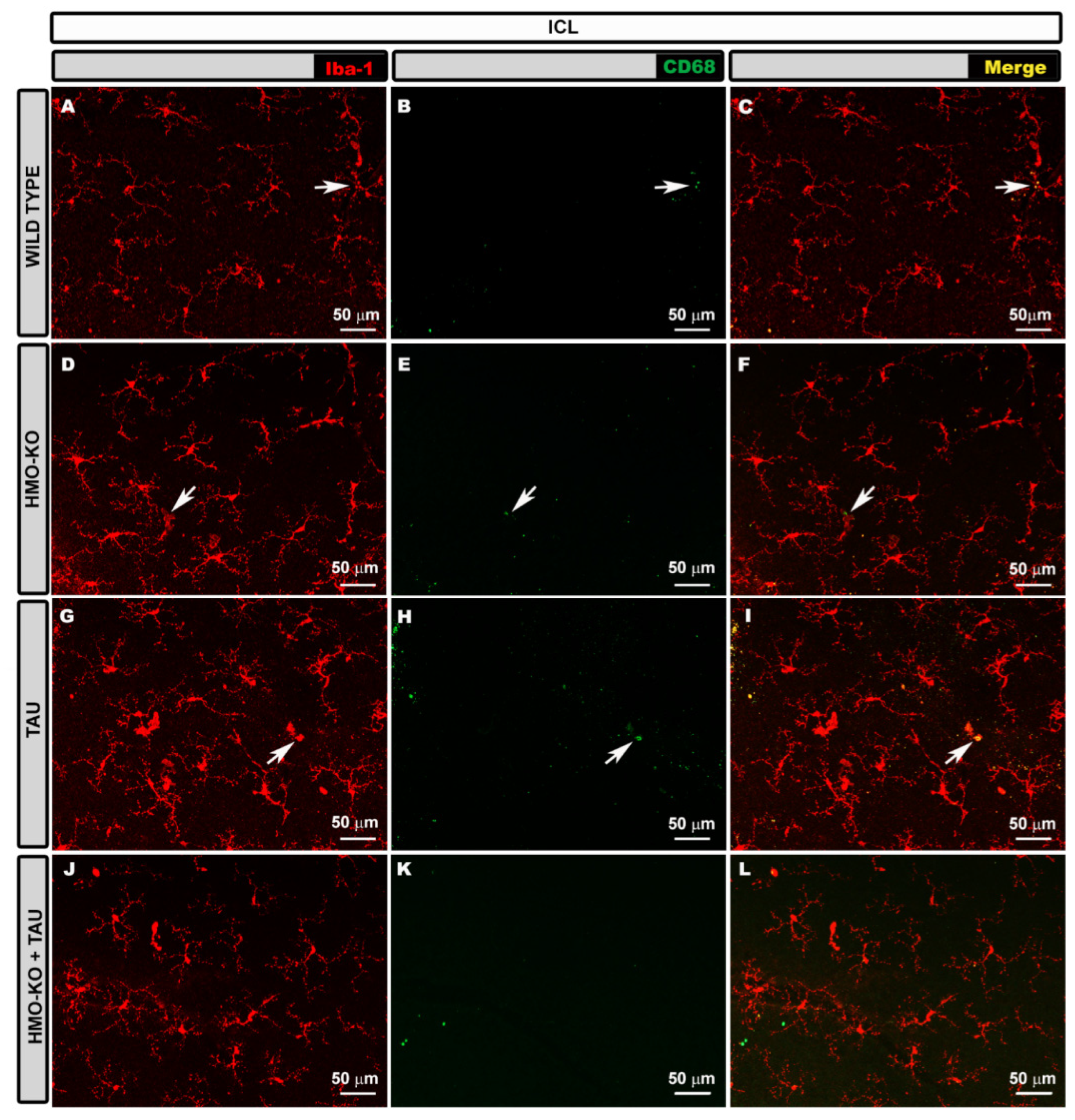
3.2.3. Inner Complex Layers (ICL)
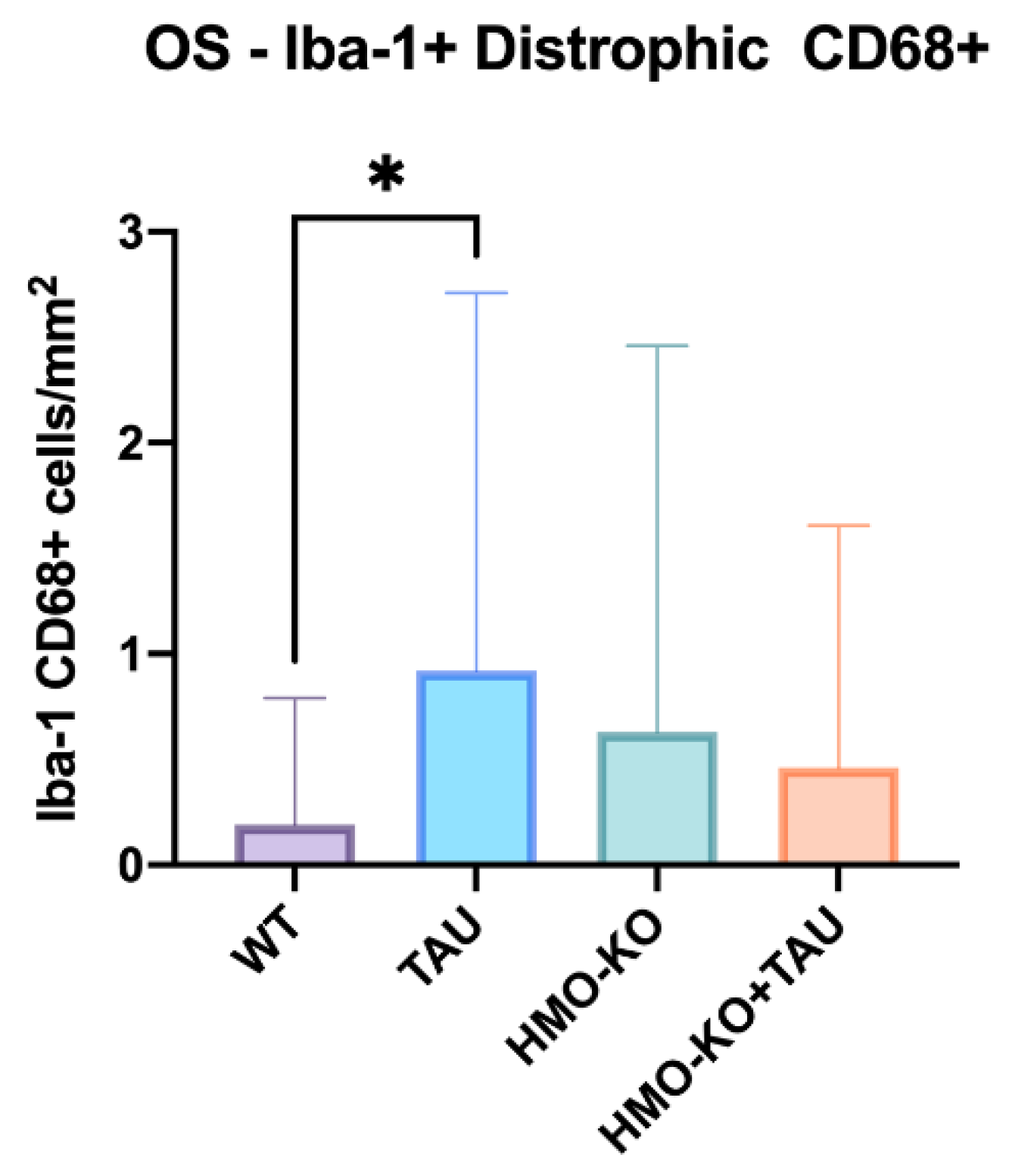
3.2.4. Microglial CD68 Expression
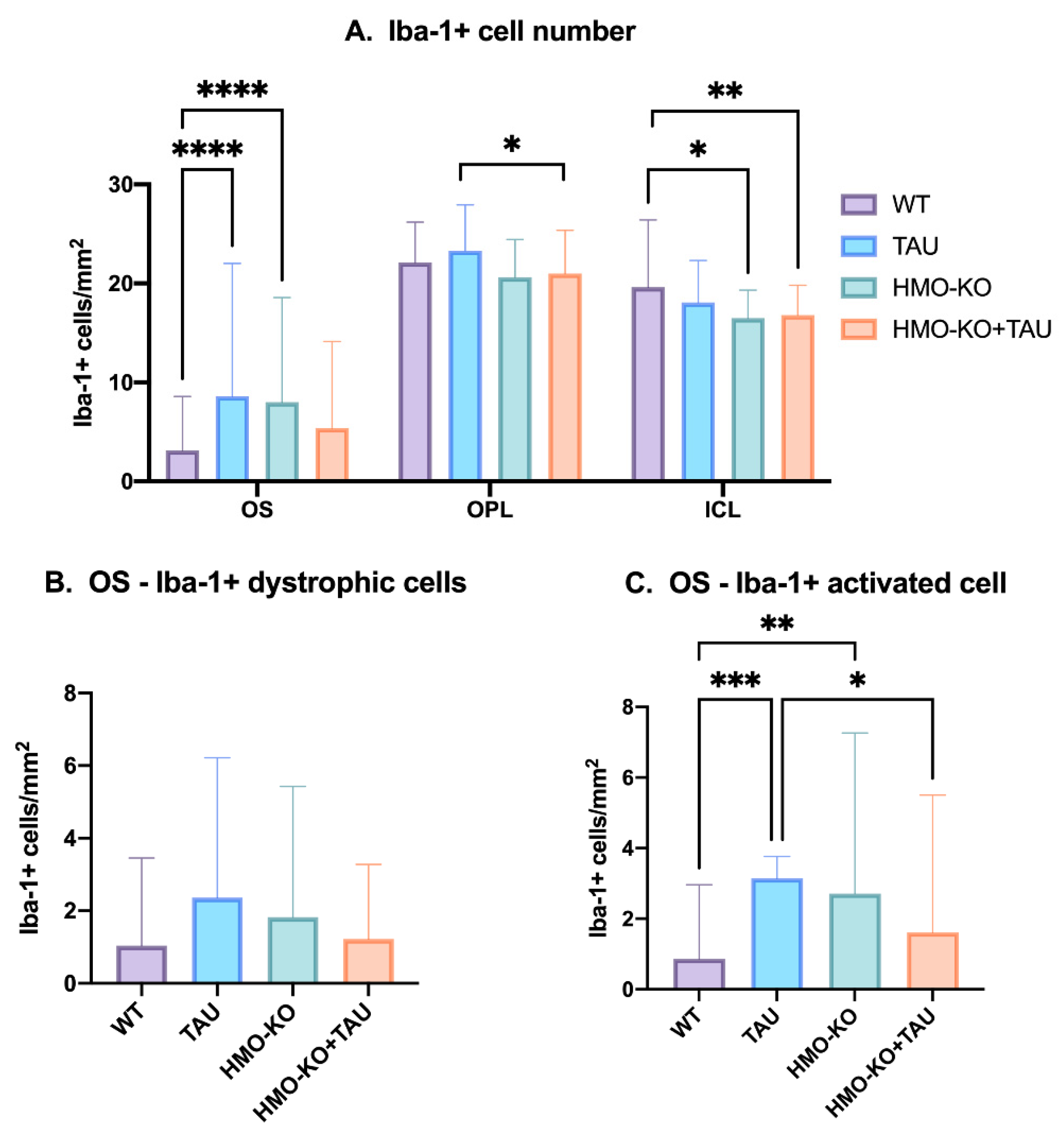
3.2.5. Quantitative Study of Retinal Microglial Cells
Microglial Cell Numbers
Number of Dystrophic Iba-1+ Microglia and Iba-1+ Microglia Activated in OS Layer
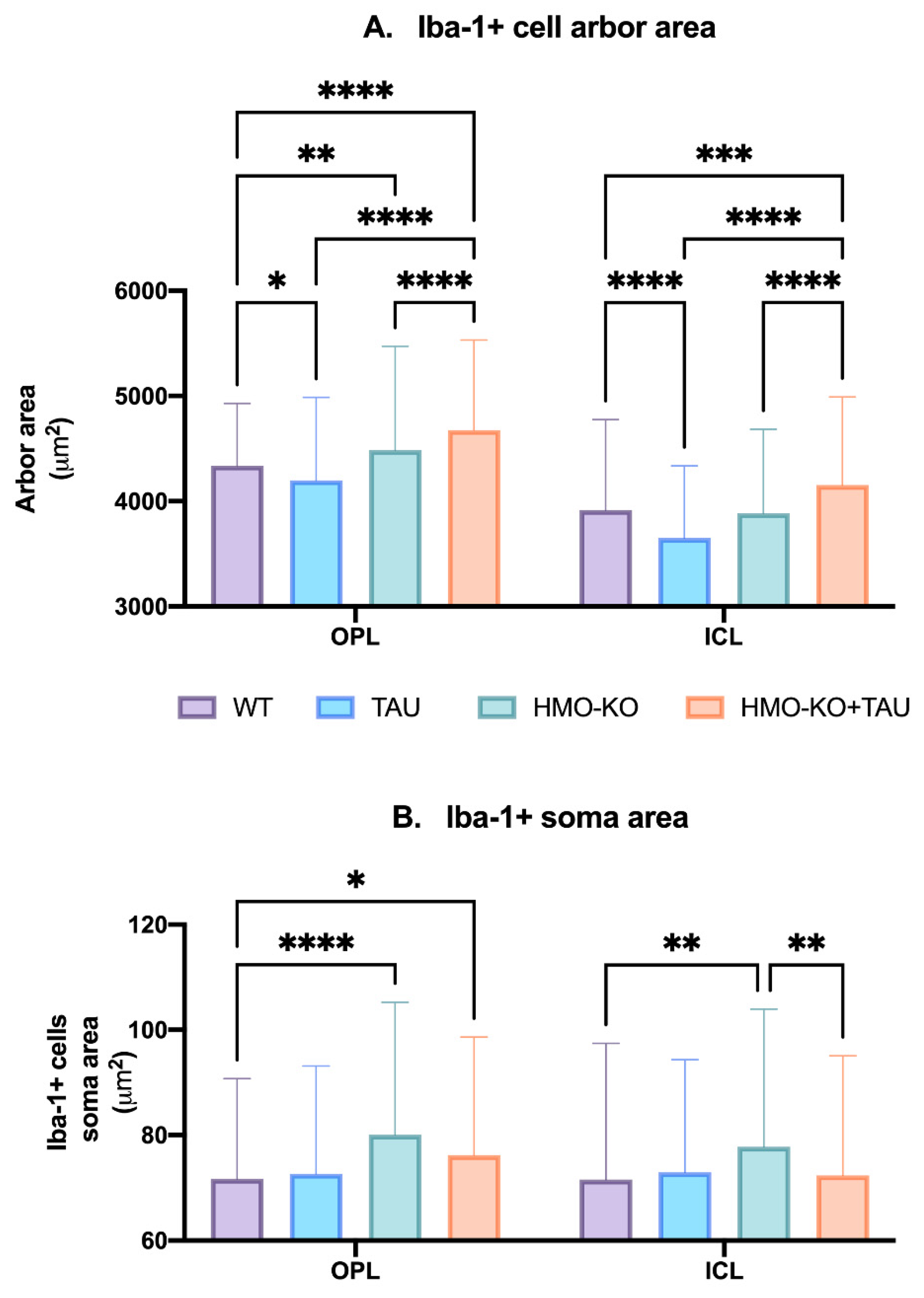
Microglial Arbor Area
Microglial Soma Area
Number of Iba-1+/CD68+ Cells
3.3. Morphology and Distribution of Retinal Macroglia in the Different Study Groups
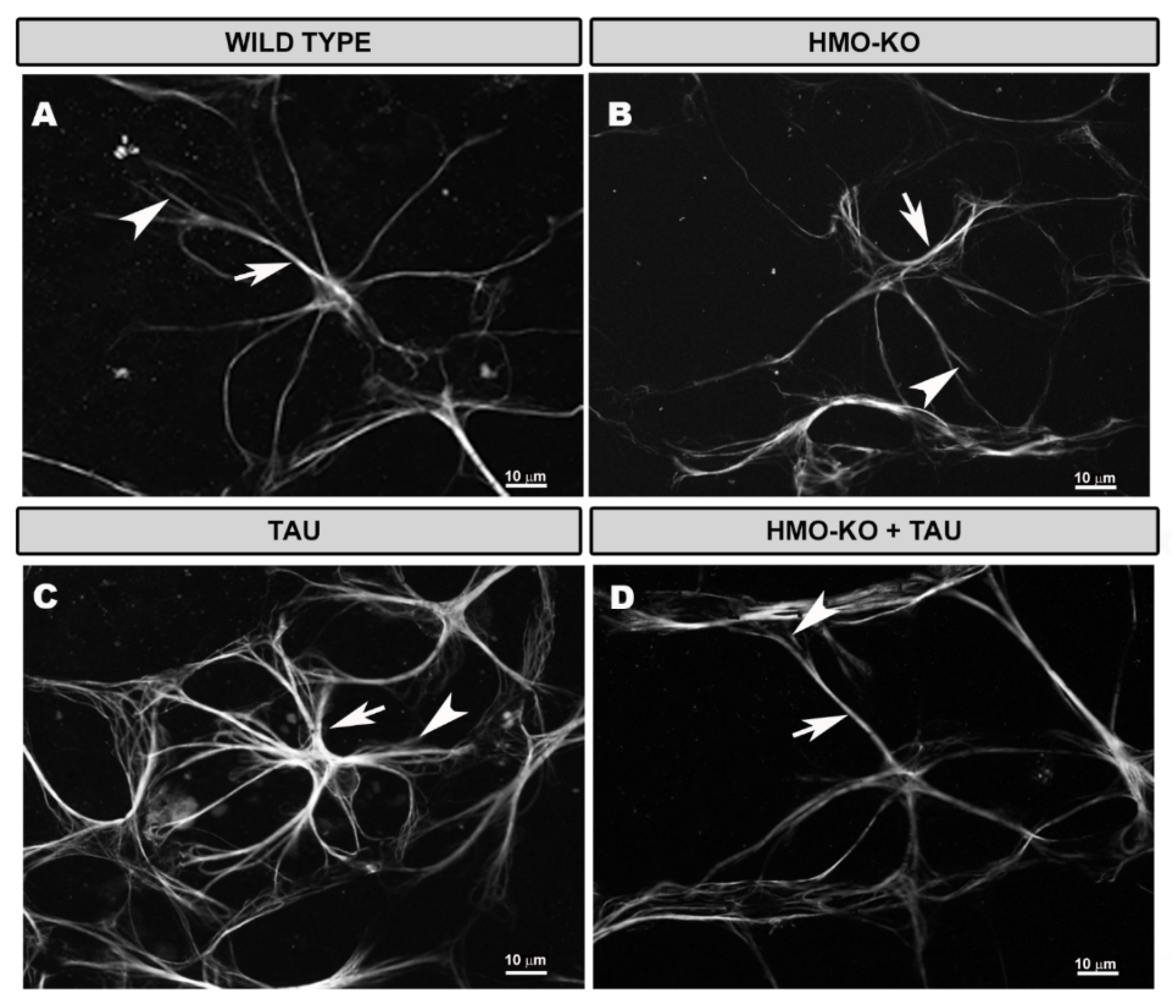
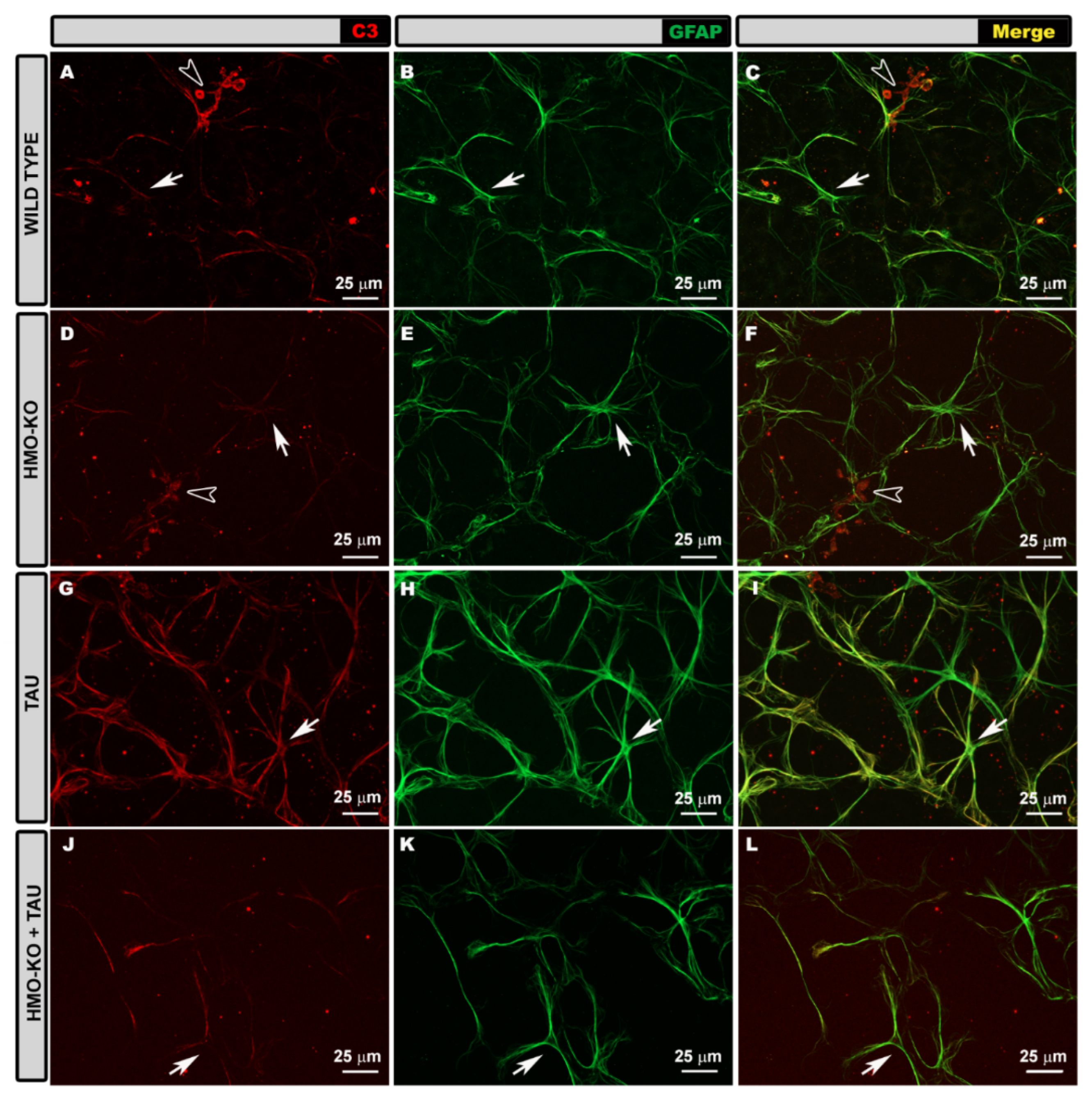
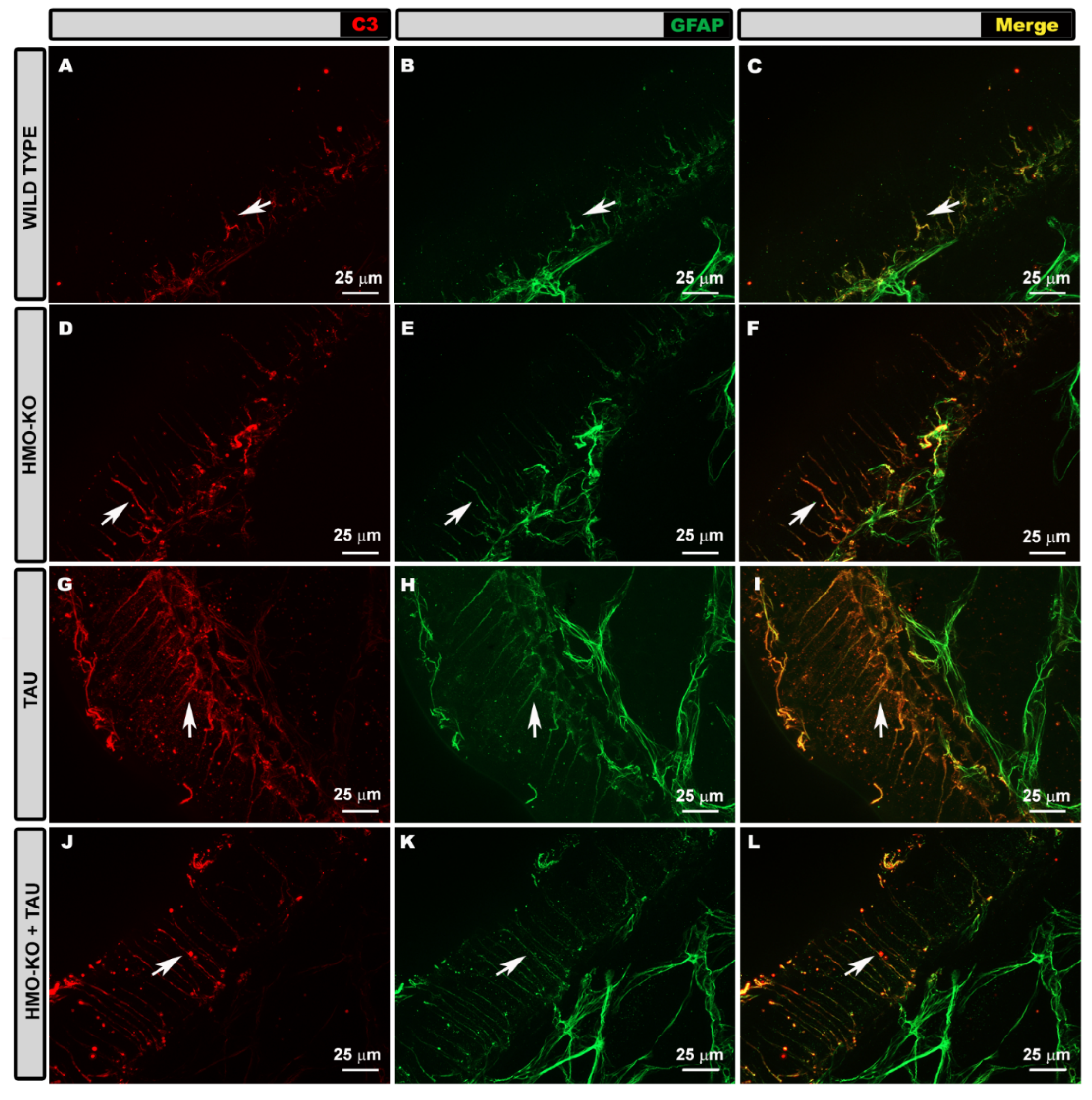
3.3.1. Morphological Study Using GFAP Antibody
3.3.2. Morphological Study Using C3 Antibody
3.3.3. Quantitative Study of Retinal Macroglial Cells
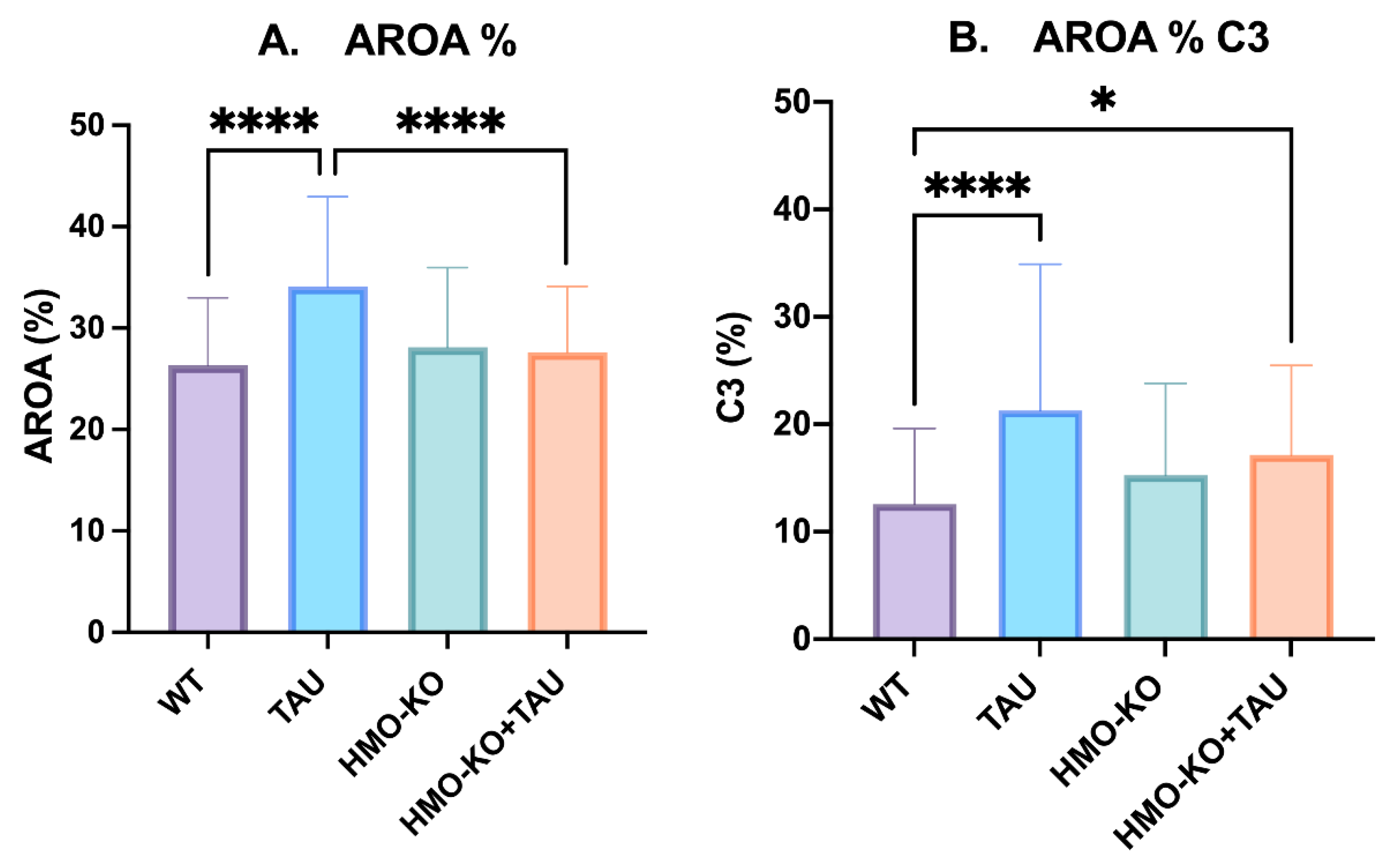
Area of Retina Occupied by GFAP (AROA)
Area of Retina Occupied by C3 (AROC3)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS Neurodegenerative Diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Res 2016, 5, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.O.; Baccarelli, A. Metals and Neurotoxicology. J. Nutr. 2007, 137, 2809–2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T. Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Barrientos, R.M.; Kitt, M.M.; Watkins, L.R.; Maier, S.F. Neuroinflammation in the Normal Aging Hippocampus. Neuroscience 2015, 309, 84–99. [Google Scholar] [CrossRef] [Green Version]
- Arruda, L.F.; Arruda, S.F.; Campos, N.A.; de Valencia, F.F.; de Almeida Siqueira, E.M. Dietary Iron Concentration May Influence Aging Process by Altering Oxidative Stress in Tissues of Adult Rats. PLoS ONE 2013, 8, e61058. [Google Scholar] [CrossRef] [Green Version]
- Salvador, G.A.; Uranga, R.M.; Giusto, N.M. Iron and Mechanisms of Neurotoxicity. Int. J. Alzheimers Dis. 2010, 2011, 720658. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.G. Invited Review: Neuropathology of Tauopathies: Principles and Practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Fong, J.; Ang, L.C.; Yücel, Y.H. Retinal Tau Pathology in Human Glaucomas. Can. J. Ophthalmol. 2008, 43, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, M.; Zweckstetter, M. Upregulated Levels and Pathological Aggregation of Abnormally Phosphorylated Tau-Protein in Children with Neurodevelopmental Disorders. Neurosci. Biobehav. Rev. 2019, 98, 1–9. [Google Scholar] [CrossRef] [PubMed]
- de Hoz, R.; Rojas, B.; Ramírez, A.I.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Retinal Macroglial Responses in Health and Disease. Biomed. Res. Int. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; López-Cuenca, I.; Rojas, P.; Triviño, A.; Ramírez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I. Dystrophic (Senescent) Rather than Activated Microglial Cells Are Associated with Tau Pathology and Likely Precede Neurodegeneration in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.-S.; Streit, W.J. Microglial Pathology in Down Syndrome. Acta Neuropathol. 2011, 122, 455–466. [Google Scholar] [CrossRef]
- Simmons, D.A.; Casale, M.; Alcon, B.; Pham, N.; Narayan, N.; Lynch, G. Ferritin Accumulation in Dystrophic Microglia Is an Early Event in the Development of Huntington’s Disease. Glia 2007, 55, 1074–1084. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S. Microglia in Dementia with Lewy Bodies. Brain Behav. Immun. 2016, 55, 191–201. [Google Scholar] [CrossRef]
- Hoare, M.; Narita, M. Transmitting Senescence to the Cell Neighbourhood. Nat. Cell Biol. 2013, 15, 887–889. [Google Scholar] [CrossRef]
- Li, J.-W.; Zong, Y.; Cao, X.-P.; Tan, L.; Tan, L. Microglial Priming in Alzheimer’s Disease. Ann. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Holmes, C. Microglial Priming in Neurodegenerative Disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and Stress. Neuropsychopharmacology 2016, 42, 318–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, A.I.; Fernández-Albarral, J.A.; de Hoz, R.; López-Cuenca, I.; Salobrar-García, E.; Rojas, P.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; et al. Microglial Changes in the Early Aging Stage in a Healthy Retina and an Experimental Glaucoma Model. Prog. Brain Res. 2020, 256, 125–149. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.R.; Streit, W.J. Microglia in the Aging Brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [PubMed] [Green Version]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the Aged Brain: Primed to Be Activated and Resistant to Regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-Mediated Neuroinflammation in Neurodegenerative Diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Rojas, P.; Ramírez, A.I.; Cadena, M.; Fernández-Albarral, J.A.; Salobrar-García, E.; López-Cuenca, I.; Santos-García, I.; de Lago, E.; Urcelay-Segura, J.L.; Ramírez, J.M.; et al. Retinal Ganglion Cell Loss and Microglial Activation in a SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 1663. [Google Scholar] [CrossRef]
- Ramírez, A.I.; Rojas, B.; de Hoz, R.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Microglia, Inflammation, and Glaucoma. In Glaucoma; SM Group Open Access eBooks: Dover, DE, USA, 2015; pp. 1–16. [Google Scholar]
- Fernández-Albarral, J.A.; Salazar, J.J.; de Hoz, R.; Marco, E.M.; Martín-Sánchez, B.; Flores-Salguero, E.; Salobrar-García, E.; López-Cuenca, I.; Barrios-Sabador, V.; Avilés-Trigueros, M.; et al. Retinal Molecular Changes Are Associated with Neuroinflammation and Loss of RGCs in an Experimental Model of Glaucoma. Int. J. Mol. Sci. 2021, 22, 2066. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A Central Element in Neurological Diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef]
- Lewis, G.P.; Fisher, S.K. Up-Regulation of Glial Fibrillary Acidic Protein in Response to Retinal Injury: Its Potential Role in Glial Remodeling and a Comparison to Vimentin Expression. Int. Rev. Cytol. 2003, 230, 263–290. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.M.; Rodríguez, J.J.; Gutierrez-Lanza, R.; Yates, J.; Verkhratsky, A.; Lutty, G.A. Retinal Macroglia Changes in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Exp. Eye Res. 2014, 127, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Ho, G.; Zhang, Y.; Zhuo, L. In Vivo Imaging of Retinal Gliosis: A Platform for Diagnosis of PD and Screening of Anti-PD Compounds. In Proceedings of the 2010 Annual International Conference of the IEEE Engineering in Medicine and Biology, Buenos Aires, Argentina, 31 August–4 September 2010; pp. 3049–3052. [Google Scholar] [CrossRef]
- Liddelow, S.; Guttenplan, K.; Clarke, L.E.; Bennett, F.; Bohlen, C.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M. v Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The Retina as a Window to the Brain-From Eye Research to CNS Disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Salobrar-Garcia, E.; Hoyas, I.; Leal, M.; Hoz, R.d.; Rojas, B.; Ramirez, A.I.; Salazar, J.J.; Yubero, R.; Gil, P.; Triviño, A.; et al. Analysis of Retinal Peripapillary Segmentation in Early Alzheimer’s Disease Patients. Biomed. Res. Int. 2015, 2015, 636548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, P.; Ramírez, A.I.; Fernández-Albarral, J.A.; López-Cuenca, I.; Salobrar-García, E.; Cadena, M.; Elvira-Hurtado, L.; Salazar, J.J.; de Hoz, R.; Ramírez, J.M. Amyotrophic Lateral Sclerosis: A Neurodegenerative Motor Neuron Disease With Ocular Involvement. Front. Neurosci. 2020, 14, 566858. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martin, E.S.; Rojas, B.; Ramirez, A.I.; de Hoz, R.; Salazar, J.J.; Yubero, R.; Gil, P.; Triviño, A.; Ramirez, J.M. Macular Thickness as a Potential Biomarker of Mild Alzheimer’s Disease. Ophthalmology 2014, 121, 1149–1151.e3. [Google Scholar] [CrossRef] [Green Version]
- López-Cuenca, I.; Salobrar-García, E.; Elvira-Hurtado, L.; Fernández-Albarral, J.A.; Sánchez-Puebla, L.; Salazar, J.J.; Ramírez, J.M.; Ramírez, A.I.; de Hoz, R. The Value of OCT and OCTA as Potential Biomarkers for Preclinical Alzheimer’s Disease: A Review Study. Life 2021, 11, 712. [Google Scholar] [CrossRef]
- Gupta, V.B.; Chitranshi, N.; den Haan, J.; Mirzaei, M.; You, Y.; Lim, J.K.H.; Basavarajappa, D.; Godinez, A.; Angelantonio, S.d.; Sachdev, P.; et al. Retinal Changes in Alzheimer’s Disease—Integrated Prospects of Imaging, Functional and Molecular Advances. Prog. Retin. Eye Res. 2021, 82, 100899. [Google Scholar] [CrossRef]
- Gupta, V.K.; Chitranshi, N.; Gupta, B.; Golzan, M.; Dheer, Y.; Wall, R.v.; Georgevsky, D.; King, A.E.; Vickers, J.C.; Chung, R.; et al. Amyloid Accumulation and Inner Retinal Degenerative Changes in Alzheimer’s Disease Transgenic Mouse. Neurosci. Lett. 2016, 623, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; vande Velde, C.; Panayi, F.; Louis, C.; di Polo, A. Tau Accumulation in the Retina Promotes Early Neuronal Dysfunction and Precedes Brain Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, P.J.; Bórquez, D.A.; Núñez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef]
- Matsui, T.; Masaki, U.; Masao, I.S. Heme Oxygenase Reveals Its Strategy for Catalyzing Three Successive Oxygenation Reactions. Acc. Chem. Res. 2010, 43, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 Overexpression and Underexpression: Clinical Implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef] [PubMed]
- Zeynalov, E.; Shah, Z.A.; chi Li, R.; Doré, S. Heme Oxygenase 1 Is Associated with Ischemic Preconditioning-Induced Protection against Brain Ischemia. Neurobiol. Dis. 2009, 35, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, M.; Deshmukh, R.; Kaundal, M.; Reddy, B.V.K. Pharmacological Induction of Hemeoxygenase-1 Activity Attenuates Intracerebroventricular Streptozotocin Induced Neurocognitive Deficit and Oxidative Stress in Rats. Eur. J. Pharmacol. 2016, 772, 43–50. [Google Scholar] [CrossRef]
- Li, L.; Du, G.P.; Wang, D.J.; Zhou, J.; Jiang, G.; Jiang, H. Overexpression of Heme Oxygenase-1 in Mesenchymal Stem Cells Augments Their Protection on Retinal Cells in Vitro and Attenuates Retinal Ischemia/Reperfusion Injury in Vivo against Oxidative Stress. Stem. Cells Int. 2017, 2017, 4985323. [Google Scholar] [CrossRef] [Green Version]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The Sinister Face of Heme Oxygenase-1 in Brain Aging and Disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [Green Version]
- Kutty, R.K.; Kutty, G.; Wiggert, B.; Chader, G.J.; Darrow, R.M.; Organisciak, D.T. Induction of Heme Oxygenase 1 in the Retina by Intense Visible Light: Suppression by the Antioxidant Dimethylthiourea. Proc. Natl. Acad. Sci. USA 1995, 92, 1177–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Mendívil, C.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N.; Lopez, M.G. Aging and Progression of Beta-Amyloid Pathology in Alzheimer’s Disease Correlates with Microglial Heme-Oxygenase-1 Overexpression. Antioxidants 2020, 9, 644. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mendívil, C.; Luengo, E.; Trigo-Alonso, P.; García-Magro, N.; Negredo, P.; López, M.G. Protective Role of Microglial HO-1 Blockade in Aging: Implication of Iron Metabolism. Redox Biol. 2021, 38, 101789. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.K.D.; Godard, P.; de Saint-Hubert, M.; Morrhaye, G.; Swine, C.; Geenen, V.; Martens, H.J.; Debacq-Chainiaux, F.; Toussaint, O. Transcriptomic Biomarkers of the Response of Hospitalized Geriatric Patients with Infectious Diseases. Immun. Ageing 2010, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yang, H.; Yahui, P.; Tang, L.; Jin, J.; He, R.; Li, Y.; Zhang, S.; Li, L.; Zhou, Y.; et al. Overexpression of Heme Oxygenase 1 Causes Cognitive Decline and Affects Pathways for Tauopathy in Mice. J. Alzheimer’s Dis. 2015, 43, 519–534. [Google Scholar] [CrossRef]
- Gozzelino, R.; Arosio, P. Iron Homeostasis in Health and Disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Sengupta, P. Men and Mice: Relating Their Ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-Related Alterations in the Dynamic Behavior of Microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Lucin, K.M.; Wyss-Coray, T. Immune Activation in Brain Aging and Neurodegeneration: Too Much or Too Little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef]
- Sierra, A.; Gottfried-Blackmore, A.C.; Mcewen, B.S.; Bulloch, K. Microglia Derived from Aging Mice Exhibit an Altered Inflammatory Profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef]
- Frost, B.; Götz, J.; Feany, M.B. Connecting the Dots between Tau Dysfunction and Neurodegeneration. Trends Cell Biol. 2015, 25, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau Activates Microglia via the PQBP1-CGAS-STING Pathway to Promote Brain Inflammation. Nat. Commun. 2021, 12, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalfe, M.J.; Figueiredo-Pereira, M.E. Relationship Between Tau Pathology and Neuroinflammation in Alzheimer’s Disease. Mt. Sinai. J. Med. 2010, 77, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Whittle, T.B.; Crain, C.N.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimers Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.-Y.; Troncoso, J.C.; Knox, D.; Stark, W.; Eberhart, C.G.; Eberhart, C. Beta-Amyloid, Phospho-Tau and Alpha-Synuclein Deposits Similar to Those in the Brain Are Not Identified in the Eyes of Alzheimer’s and Parkinson’s Disease Patients. Brain Pathol. 2014, 24, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.A.; McGuone, D.; Frosch, M.P.; Hyman, B.T.; Laver, N.; Stemmer-Rachamimov, A. Absence of Alzheimer Disease Neuropathologic Changes in Eyes of Subjects With Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2017, 76, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.J.; Lucassen, P.J.; van Dam, A.M. Microglial Phenotypes and Toll-like Receptor 2 in the Substantia Nigra and Hippocampus of Incidental Lewy Body Disease Cases and Parkinson’s Disease Patients. Acta Neuropathol. Commun. 2014, 2, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Shahidehpour, R.K.; Higdon, R.E.; Crawford, N.G.; Neltner, J.H.; Lghodaro, E.T.; Patel, E.; Price, D.; Nelson, P.T.; Bachstetter, A.D. Dystrophic Microglia Are Associated with Neurodegenerative Disease and Not Healthy Aging in the Human Brain. Neurobiol. Aging 2021, 99, 19–27. [Google Scholar] [CrossRef]
- Rodrigues-Neves, A.C.; Carecho, R.; Correia, S.C.; Carvalho, C.; Campos, E.J.; Baptista, F.I.; Moreira, P.I.; Ambrósio, A.F. Retina and Brain Display Early and Differential Molecular and Cellular Changes in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3043–3060. [Google Scholar] [CrossRef]
- Grimaldi, A.; Brighi, C.; Peruzzi, G.; Ragozzino, D.; Bonanni, V.; Limatola, C.; Ruocco, G.; Angelantonio, S. di Inflammation, Neurodegeneration and Protein Aggregation in the Retina as Ocular Biomarkers for Alzheimer’s Disease in the 3xTg-AD Mouse Model. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, A.; Pediconi, N.; Oieni, F.; Pizzarelli, R.; Rosito, M.; Giubettini, M.; Santini, T.; Limatola, C.; Ruocco, G.; Ragozzino, D.; et al. Neuroinflammatory Processes, A1 Astrocyte Activation and Protein Aggregation in the Retina of Alzheimer’s Disease Patients, Possible Biomarkers for Early Diagnosis. Front. Neurosci. 2019, 13, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanguilder, H.D.; Bixler, G.v.; Brucklacher, R.M.; Farley, J.A.; Yan, H.; Warrington, J.P.; Sonntag, W.E.; Freeman, W.M. Concurrent Hippocampal Induction of MHC II Pathway Components and Glial Activation with Advanced Aging Is Not Correlated with Cognitive Impairment. J. Neuroinflammation 2011, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, G.M.; Peterson, D.A. Phenotypic and Gene Expression Modification with Normal Brain Aging in GFAP-Positive Astrocytes and Neural Stem Cells. Aging Cell 2011, 10, 466–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian Features in Aging GFAP.HMOX1 Transgenic Mice Overexpressing Human HO-1 in the Astroglial Compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Concentration | Host | Reference | Company | |
|---|---|---|---|---|---|
| Primary antibodies | Iba-1 | 1:600 | Rabbit | 01919741 | Wako |
| AT8 | 1:200 | Mouse | MN1020 | Thermo Fisher | |
| CD68 | 1:40 | Rat | MCA1957GA | Bio Rad | |
| TAUY9 | 1:400 | Rabbit | BML-TA3119-0100 | Enzo lifesciences | |
| GFAP-GA5 | 1:150 | Mouse | MAB3402 | Millipore | |
| C3 | 1:15 | Rat | HM1045 | Hycult Biotech | |
| Secondary antibodies | Alexa Fluor® 405 Anti-Mouse | 1:150 | Goat | 31553 | Invitrogen |
| Alexa Fluor® 405 Anti-Rabbit | 1:100 | Goat | A31556 | Invitrogen | |
| Alexa Fluor® 488 Anti-Rat | 1:150 | Donkey | A21208 | Invitrogen | |
| Alexa Fluor® 488 Anti-Mouse | 1:200 | Goat | A11001 | Invitrogen | |
| Alexa Fluor® 594 Anti-Rabbit | 1:800 | Donkey | A21207 | Invitrogen | |
| Alexa Fluor® 594 Anti-Rat | 1:500 | Goat | 405422 | Biolegend |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Albarral, J.A.; Salobrar-García, E.; Matamoros, J.A.; Fernández-Mendívil, C.; del Sastre, E.; Chen, L.; de Hoz, R.; López-Cuenca, I.; Sánchez-Puebla, L.; Ramírez, J.M.; et al. Microglial Hemoxygenase-1 Deletion Reduces Inflammation in the Retina of Old Mice with Tauopathy. Antioxidants 2022, 11, 2151. https://doi.org/10.3390/antiox11112151
Fernández-Albarral JA, Salobrar-García E, Matamoros JA, Fernández-Mendívil C, del Sastre E, Chen L, de Hoz R, López-Cuenca I, Sánchez-Puebla L, Ramírez JM, et al. Microglial Hemoxygenase-1 Deletion Reduces Inflammation in the Retina of Old Mice with Tauopathy. Antioxidants. 2022; 11(11):2151. https://doi.org/10.3390/antiox11112151
Chicago/Turabian StyleFernández-Albarral, José A., Elena Salobrar-García, José A. Matamoros, Cristina Fernández-Mendívil, Eric del Sastre, Lejing Chen, Rosa de Hoz, Inés López-Cuenca, Lidia Sánchez-Puebla, José M. Ramírez, and et al. 2022. "Microglial Hemoxygenase-1 Deletion Reduces Inflammation in the Retina of Old Mice with Tauopathy" Antioxidants 11, no. 11: 2151. https://doi.org/10.3390/antiox11112151