
The Effect of Acute Oral Galactose Administration on the Redox System of the Rat Small Intestine

 ,
,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Tissue Collection and Sample Preparation
2.3. Protein Content Determination
2.4. Lipid Peroxidation
2.5. Nitrocellulose Redox Permanganometry
2.6. The Concentration of Low-Molecular-Weight Thiols and Protein Sulfhydryl Content
2.7. Superoxide Dismutase Activity
2.8. Catalase Activity
2.9. Nicotinamide Adenine Dinucleotide Phosphates
2.10. Data Analysis
3. Results
3.1. The Effect of Acute Oral Galactose Administration on Lipid Peroxidation and Reductive Capacity in Duodenum and Ileum
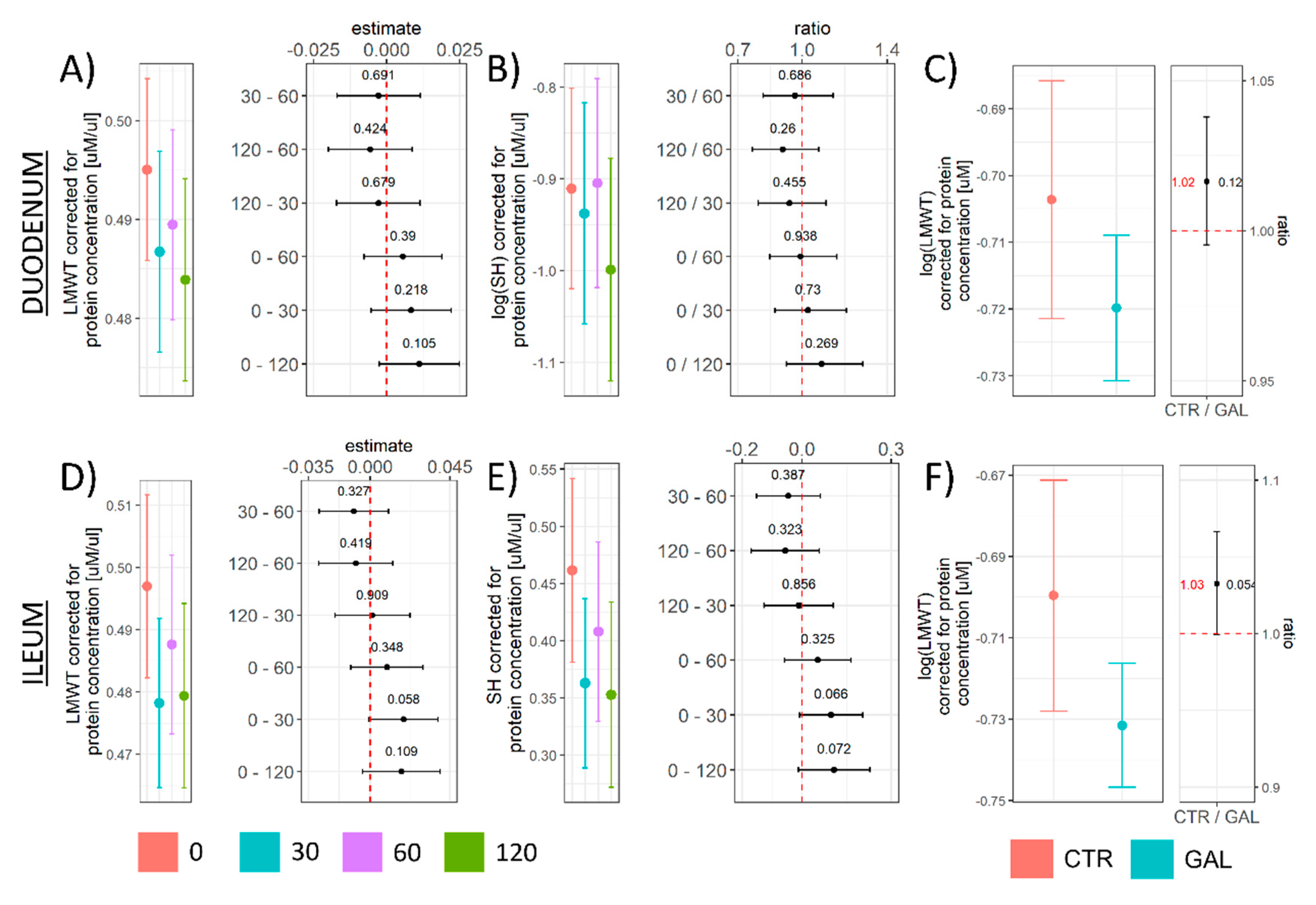
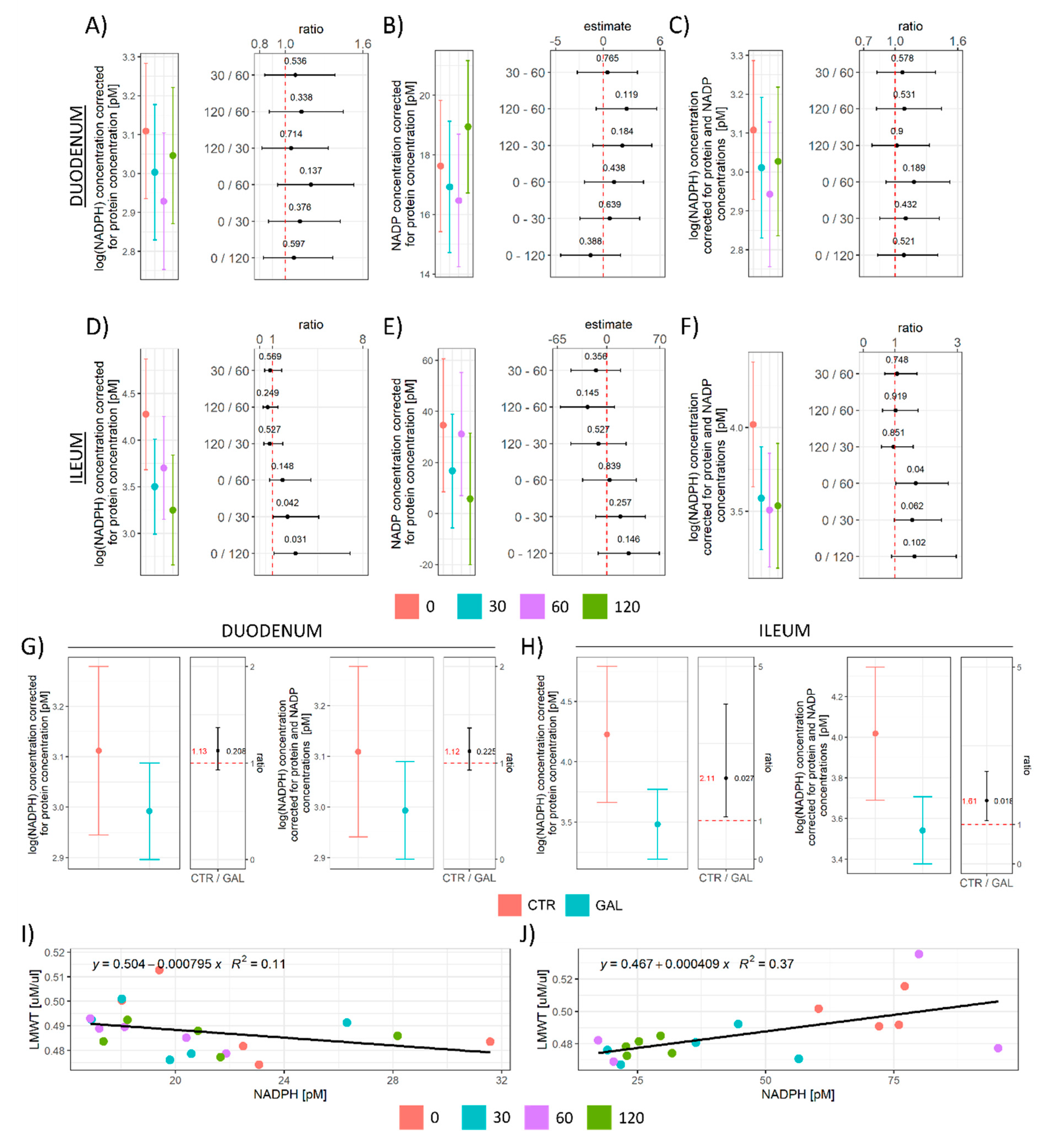
3.2. The Effect of Acute Oral Galactose Administration on Low-Molecular-Weight Thiols, Protein Sulfhydryl Content, and Nicotinamide Adenine Dinucleotide Phosphates
3.3. Hydrogen Peroxide Concentration, Hydrogen Peroxide Dissociation Rate, and Superoxide Dismutase Activity
3.4. Overall Redox-Related Changes in the Duodenum and Ileum following Acute Oral Administration of Galactose
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end products |
| AR | aldehyde reductase |
| CAT | catalase |
| CTR | control |
| Dim1 | 1st dimension |
| Dim2 | 2nd dimension |
| DTNB | 5,5′-dithio-bis(2-nitrobenzoic acid) |
| GAL | galactose |
| GIP | glucose-dependent insulinotropic polypeptide |
| GLP-1 | glucagon-like peptide-1 |
| GO | galactose oxidase |
| GPx | glutathione peroxidase |
| GR | glutathione reductase |
| GSH | glutathione |
| LMWT | low-molecular-weight thiols |
| MDA | malondialdehyde |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NRP | nitrocellulose redox permanganometry |
| OS | oxidative stress |
| PBS | phosphate-buffered saline |
| PCA | principal component analysis |
| sAD | sporadic Alzheimer’s disease |
| SH | sulfhydryl compounds |
| SOD | superoxide dismutase |
| STZ-icv | intracerebroventricularly administered streptozotocin |
| TBARS | thiobarbituric acid reactive substances |
| THB | 1,2,3-trihydroxybenzene |
| TNB | 5-thio-2-nitrobenzoic acid |
References
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose Metabolism and Health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef]
- Lai, K.; Elsas, L.; Wierenga, K. Galactose Toxicity in Animals. IUBMB Life 2009, 61, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Homolak, J.; Babic Perhoc, A.; Knezovic, A.; Kodvanj, I.; Virag, D.; Barilar, J.O.; Riederer, P.; Salkovic-Petrisic, M. Is Galactose a Hormetic Sugar? An Exploratory Study of the Rat Hippocampal Redox Regulatory Network. Mol. Nutr. Food Res. 2021, 65, e2100400. [Google Scholar] [CrossRef]
- Conte, F.; van Buuringen, N.; Voermans, N.C.; Lefeber, D.J. Galactose in Human Metabolism, Glycosylation and Congenital Metabolic Diseases: Time for a Closer Look. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129898. [Google Scholar] [CrossRef]
- Azman, K.F.; Zakaria, R. d-Galactose-Induced Accelerated Aging Model: An Overview. Biogerontology 2019, 20, 763–782. [Google Scholar] [CrossRef]
- Shwe, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Role of d-Galactose-Induced Brain Aging and Its Potential Used for Therapeutic Interventions. Exp. Gerontol. 2018, 101, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-C.; Liu, J.-H.; Wu, R.-Y. Establishment of the Mimetic Aging Effect in Mice Caused by d-Galactose. Biogerontology 2003, 4, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Majdi, A.; McCann, S.K.; Mahmoudi, J.; Vafaee, M.S.; Macleod, M.R. d-Galactose-Induced Brain Ageing Model: A Systematic Review and Meta-Analysis on Cognitive Outcomes and Oxidative Stress Indices. PLoS ONE 2017, 12, e0184122. [Google Scholar] [CrossRef]
- Yanar, K.; Aydın, S.; Cakatay, U.; Mengi, M.; Buyukpınarbaşılı, N.; Atukeren, P.; Sitar, M.E.; Sönmez, A.; Uslu, E. Protein and DNA Oxidation in Different Anatomic Regions of Rat Brain in a Mimetic Ageing Model. Basic Clin. Pharm. Toxicol. 2011, 109, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Dvornik, E.; Simard-Duquesne, N.; Krami, M.; Sestanj, K.; Gabbay, K.H.; Kinoshita, J.H.; Varma, S.D.; Merola, L.O. Polyol Accumulation in Galactosemic and Diabetic Rats: Control by an Aldose Reductase Inhibitor. Science 1973, 182, 1146–1148. [Google Scholar] [CrossRef]
- Cui, X.; Wang, L.; Zuo, P.; Han, Z.; Fang, Z.; Li, W.; Liu, J. d-Galactose-Caused Life Shortening in Drosophila Melanogaster and Musca Domestica Is Associated with Oxidative Stress. Biogerontology 2004, 5, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Homolak, J.; Kodvanj, I.; Toljan, K.; Babic Perhoc, A.; Virag, D.; Osmanovic Barilar, J.; Mlinaric, Z.; Smailovic, U.; Trkulja, V.; Hackenberger, B.; et al. Separating Science from Science Fiction: A Non-Existent Enzyme Is a Primary Driver of Pathophysiological Processes in Galactose-Induced Rodent Models of Aging. RG Prepr. 2020. Available online: https://doi.org/10.13140/RG.2.2.31233.89449/1 (accessed on 23 December 2021). [CrossRef]
- Sosicka, P.; Ng, B.G.; Freeze, H.H. Therapeutic Monosaccharides: Looking Back, Moving Forward. Biochemistry 2020, 59, 3064–3077. [Google Scholar] [CrossRef] [PubMed]
- Sasaoka, N.; Imamura, H.; Kakizuka, A. A Trace Amount of Galactose, a Major Component of Milk Sugar, Allows Maturation of Glycoproteins during Sugar Starvation. iScience 2018, 10, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Salkovic-Petrisic, M.; Osmanovic-Barilar, J.; Knezovic, A.; Hoyer, S.; Mosetter, K.; Reutter, W. Long-Term Oral Galactose Treatment Prevents Cognitive Deficits in Male Wistar Rats Treated Intracerebroventricularly with Streptozotocin. Neuropharmacology 2014, 77, 68–80. [Google Scholar] [CrossRef]
- Knezovic, A.; Osmanovic Barilar, J.; Babic, A.; Bagaric, R.; Farkas, V.; Riederer, P.; Salkovic-Petrisic, M. Glucagon-like Peptide-1 Mediates Effects of Oral Galactose in Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. Neuropharmacology 2018, 135, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Babic Perhoc, A.; Osmanovic Barilar, J.; Knezovic, A.; Farkas, V.; Bagaric, R.; Svarc, A.; Grünblatt, E.; Riederer, P.; Salkovic-Petrisic, M. Cognitive, Behavioral and Metabolic Effects of Oral Galactose Treatment in the Transgenic Tg2576 Mice. Neuropharmacology 2019, 148, 50–67. [Google Scholar] [CrossRef]
- Chogtu, B.; Arivazhahan, A.; Kunder, S.K.; Tilak, A.; Sori, R.; Tripathy, A. Evaluation of Acute and Chronic Effects of d-Galactose on Memory and Learning in Wistar Rats. Clin. Psychopharmacol Neurosci. 2018, 16, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Budni, J.; Pacheco, R.; da Silva, S.; Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; de Medeiros, J.; Voss, B.C.; Steckert, A.V.; Valvassori, S.d.S.; et al. Oral Administration of d-Galactose Induces Cognitive Impairments and Oxidative Damage in Rats. Behav. Brain Res. 2016, 302, 35–43. [Google Scholar] [CrossRef]
- Budni, J.; Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; da Silva, S.; da Luz, A.P.; Schiavo, G.L.; Batista-Silva, H.; Scaini, G.; Streck, E.L.; et al. The Oral Administration of d-Galactose Induces Abnormalities within the Mitochondrial Respiratory Chain in the Brain of Rats. Metab. Brain Dis. 2017, 32, 811–817. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Homolak, J.; Babic Perhoc, A.; Knezovic, A.; Osmanovic Barilar, J.; Salkovic-Petrisic, M. Failure of the Brain Glucagon-Like Peptide-1-Mediated Control of Intestinal Redox Homeostasis in a Rat Model of Sporadic Alzheimer’s Disease. Antioxidants 2021, 10, 1118. [Google Scholar] [CrossRef]
- Homolak, J.; Babic Perhoc, A.; Knezovic, A.; Osmanovic Barilar, J.; Koc, F.; Stanton, C.; Ross, R.P.; Salkovic-Petrisic, M. Disbalance of the Duodenal Epithelial Cell Turnover and Apoptosis Accompanies Insensitivity of Intestinal Redox Homeostasis to Inhibition of the Brain Glucose-Dependent Insulinotropic Polypeptide Receptors in a Rat Model of Sporadic Alzheimer’s Disease. Neuroendocrinology 2021. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Intestinal Redox Biology and Oxidative Stress. Semin. Cell Dev. Biol. 2012, 23, 729–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Rostami Dovom, M.; Moslehi, N.; Mirmiran, P.; Azizi, F.; Ramezani Tehrani, F. Habitual Dietary Lactose and Galactose Intakes in Association with Age at Menopause in Non-Galactosemic Women. PLoS ONE 2019, 14, e0214067. [Google Scholar] [CrossRef]
- Homolak, J.; Kodvanj, I.; Perhoc, A.B.; Virag, D.; Knezovic, A.; Barilar, J.O.; Riederer, P.; Salkovic-Petrisic, M. Nitrocellulose Redox Permanganometry: A Simple Method for Reductive Capacity Assessment. MethodsX 2021, 101611. [Google Scholar] [CrossRef]
- Homolak, J. The Effect of a Color Tattoo on the Local Skin Redox Regulatory Network: An N-of-1 Study. Free Radic. Res. 2021, 55, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E. Determination of Glutathione and Glutathione Disulfide in Biological Samples. Methods Enzym. 1985, 113, 548–555. [Google Scholar] [CrossRef]
- Li, X. Improved Pyrogallol Autoxidation Method: A Reliable and Cheap Superoxide-Scavenging Assay Suitable for All Antioxidants. J. Agric. Food Chem. 2012, 60, 6418–6424. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.; Marklund, G. Involvement of the Superoxide Anion Radical in the Autoxidation of Pyrogallol and a Convenient Assay for Superoxide Dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver: Cu,Zn-SOD IN MITOCHONDRIA. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Whitney, P. Use of Cyanide and Diethyldithiocarbamate in the Assay of Superoxide Dismutases. Free Radic. Biol. Med. 1991, 10, 69–77. [Google Scholar] [CrossRef]
- Ma, X.; Deng, D.; Chen, W. Inhibitors and Activators of SOD, GSH-Px, and CAT. In Enzyme Inhibitors and Activators; Şentürk, M., Ed.; IntechOpen: London, UK, 2017; ISBN 978-953-51-3058-1. [Google Scholar]
- Hadwan, M.H. Simple Spectrophotometric Assay for Measuring Catalase Activity in Biological Tissues. BMC Biochem. 2018, 19, 7. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting Animal Research: Explanation and Elaboration for the ARRIVE Guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]
- Blastland, M.; Freeman, A.L.J.; van der Linden, S.; Marteau, T.M.; Spiegelhalter, D. Five Rules for Evidence Communication. Nature 2020, 587, 362–364. [Google Scholar] [CrossRef]
- Cichoń, M. Reporting Statistical Methods and Outcome of Statistical Analyses in Research Articles. Pharmacol. Rep. 2020, 72, 481–485. [Google Scholar] [CrossRef]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Chang, F.Y.; Lee, S.D.; Yeh, G.H.; Wang, P.S. Comparison of Two Orogastric Feeding Markers for Measuring Gastrointestinal Motor Functions in Rats. Pharmacology 1994, 49, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Gromova, L.V.; Fetissov, S.O.; Gruzdkov, A.A. Mechanisms of Glucose Absorption in the Small Intestine in Health and Metabolic Diseases and Their Role in Appetite Regulation. Nutrients 2021, 13, 2474. [Google Scholar] [CrossRef]
- Rudyk, O.; Eaton, P. Biochemical Methods for Monitoring Protein Thiol Redox States in Biological Systems. Redox Biol. 2014, 2, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Role of Metabolic H2O2 Generation. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [Green Version]
- Szypowska, A.A.; Burgering, B.M.T. The Peroxide Dilemma: Opposing and Mediating Insulin Action. Antioxid. Redox Signal. 2011, 15, 219–232. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.-C.; Nakanishi, I.; Matsumoto, K.-I.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A Mitochondrial Superoxide Theory for Oxidative Stress Diseases and Aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, J.; Saez-Atienzar, S.; Atienzar-Aroca, S.; Solesio, M.E.; Galindo, M.F. Pharmacology and Pathology of Superoxide Dismutases (SOD). In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 457–474. ISBN 978-3-642-30018-9. [Google Scholar]
- Kubo, E.; Urakami, T.; Fatma, N.; Akagi, Y.; Singh, D.P. Polyol Pathway-Dependent Osmotic and Oxidative Stresses in Aldose Reductase-Mediated Apoptosis in Human Lens Epithelial Cells: Role of AOP2. Biochem. Biophys. Res. Commun. 2004, 314, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M. Radical Explanations for Old Observations. Gastroenterology 1987, 92, 2026–2028. [Google Scholar] [CrossRef]
- Kohút, A.; Mojzis, J. Effect of Allopurinol and Superoxide Dismutase on Indomethacin-Induced Gastric Lesions in the Rat. Physiol. Res. 1993, 42, 273–276. [Google Scholar]
- Naito, Y.; Yoshikawa, T.; Ando, T.; Kishi, A.; Ueda, S.; Oyamada, H.; Kondo, M. Changes in Superoxide Dismutase Activity in the Gastric Mucosa of Peptic Ulcer Patients. J. Clin. Gastroenterol. 1992, 14, S131–S134. [Google Scholar] [CrossRef]
- Wingler, K.; Müller, C.; Schmehl, K.; Florian, S.; Brigelius-Flohé, R. Gastrointestinal Glutathione Peroxidase Prevents Transport of Lipid Hydroperoxides in CaCo-2 Cells. Gastroenterology 2000, 119, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Müller, C.; Menard, J.; Florian, S.; Schmehl, K.; Wingler, K. Functions of GI-GPx: Lessons from Selenium-Dependent Expression and Intracellular Localization. Biofactors 2001, 14, 101–106. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Aranda, R.; Martín, M.G.; Doroshow, J.H.; Binder, S.W.; Chu, F.F. Mice with Combined Disruption of Gpx1 and Gpx2 Genes Have Colitis. Am. J. Physiol. Gastrointest. Liver. Physiol. 2001, 281, G848–G855. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, Y.; Zhang, X.; Lu, Y.; Chen, H. New Insights in Intestinal Oxidative Stress Damage and the Health Intervention Effects of Nutrients: A Review. J. Funct. Foods 2020, 75, 104248. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox Homeostasis: The Golden Mean of Healthy Living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Homolak, J. Redox Homeostasis in Alzheimer’s Disease. In Redox Signaling and Biomarkers in Ageing; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Sochocka, M.; Donskow-Łysoniewska, K.; Diniz, B.S.; Kurpas, D.; Brzozowska, E.; Leszek, J. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease—A Critical Review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omar, N.A.M.; Frank, J.; Kruger, J.; Bello, F.D.; Medana, C.; Collino, M.; Zamaratskaia, G.; Michaelsson, K.; Wolk, A.; Landberg, R. Effects of High Intakes of Fructose and Galactose, with or without Added Fructooligosaccharides, on Metabolic Factors, Inflammation, and Gut Integrity in a Rat Model. Mol. Nutr. Food Res. 2021, 65, 2001133. [Google Scholar] [CrossRef]
- Council, N.R. Nutrient requirements of the laboratory rat. In Nutrient Requirements of Laboratory Animals, 4th ed.; National Academy Press: Washington, DC, USA, 1995. [Google Scholar]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and Regulation of the Mucus Barrier in the Gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, P.; Cani, P.D. Mucus Barrier, Mucins and Gut Microbiota: The Expected Slimy Partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Herath, M.; Hosie, S.; Bornstein, J.C.; Franks, A.E.; Hill-Yardin, E.L. The Role of the Gastrointestinal Mucus System in Intestinal Homeostasis: Implications for Neurological Disorders. Front. Cell. Infect. Microbiol. 2020, 10, 248. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homolak, J.; Babic Perhoc, A.; Knezovic, A.; Osmanovic Barilar, J.; Virag, D.; Joja, M.; Salkovic-Petrisic, M. The Effect of Acute Oral Galactose Administration on the Redox System of the Rat Small Intestine. Antioxidants 2022, 11, 37. https://doi.org/10.3390/antiox11010037
Homolak J, Babic Perhoc A, Knezovic A, Osmanovic Barilar J, Virag D, Joja M, Salkovic-Petrisic M. The Effect of Acute Oral Galactose Administration on the Redox System of the Rat Small Intestine. Antioxidants. 2022; 11(1):37. https://doi.org/10.3390/antiox11010037
Chicago/Turabian StyleHomolak, Jan, Ana Babic Perhoc, Ana Knezovic, Jelena Osmanovic Barilar, Davor Virag, Mihovil Joja, and Melita Salkovic-Petrisic. 2022. "The Effect of Acute Oral Galactose Administration on the Redox System of the Rat Small Intestine" Antioxidants 11, no. 1: 37. https://doi.org/10.3390/antiox11010037