
Targeting the Cation-Chloride Co-Transporter NKCC1 to Re-Establish GABAergic Inhibition and an Appropriate Excitatory/Inhibitory Balance in Selective Neuronal Circuits: A Novel Approach for the Treatment of Alzheimer’s Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Alterations of GABAergic Signaling in Alzheimer’s Disease and in Animal Models
3. NGF and Alzheimer’s Disease
3.1. NGF and the Cholinergic Hypothesis for AD
3.2. NGF and the Modulation of APP Processing via TrkA and p75 Receptors
3.3. AD11 Anti-NGF Mice: A Link between NGF Deprivation and Alzheimer’s-like Aβ and Tau Neurodegeneration
3.4. Microglia as New Cellular Targets of NGF Actions in the Adult CNS
3.5. proNGF/NGF Dysmetabolism Is a Trigger for Neurodegeneration
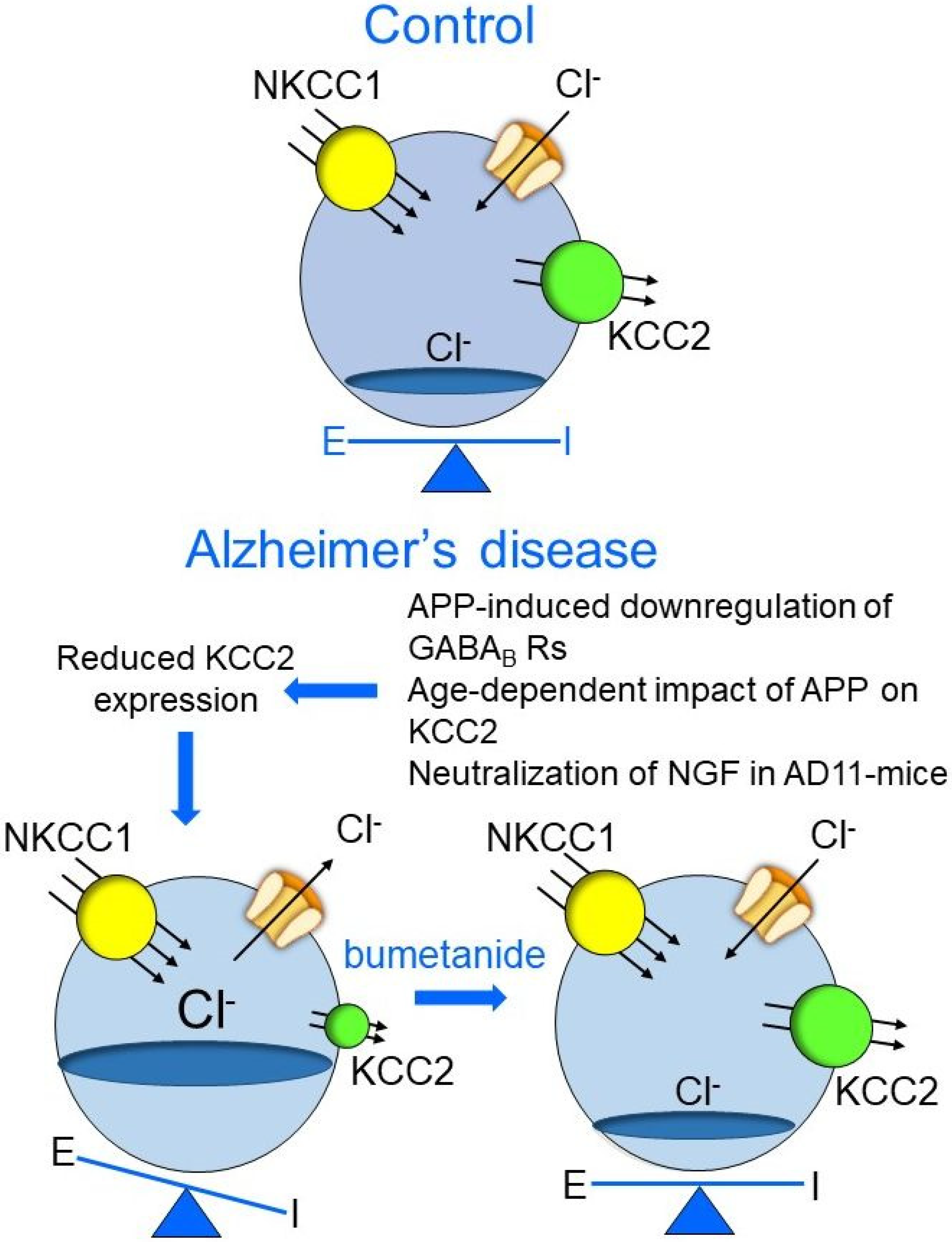
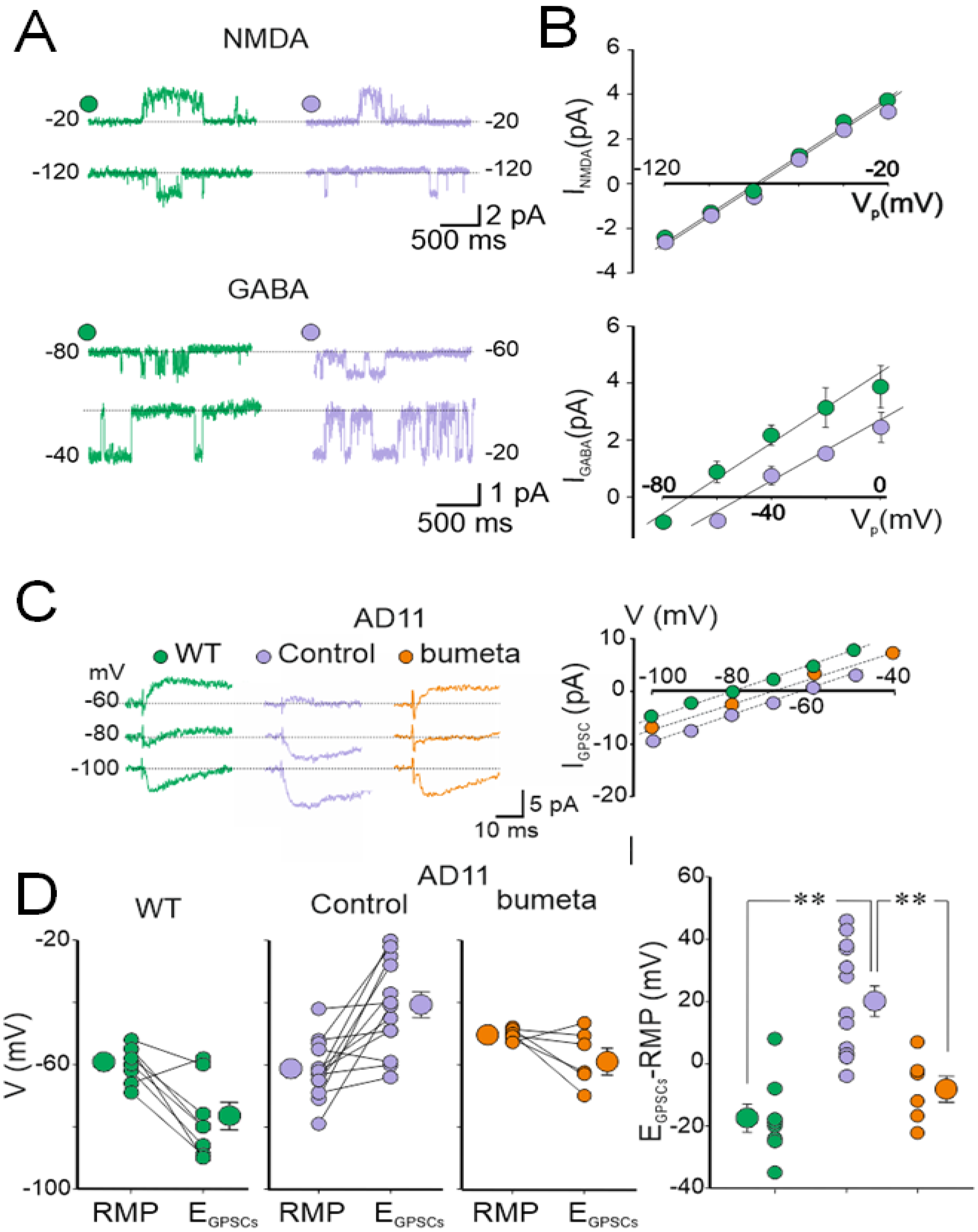
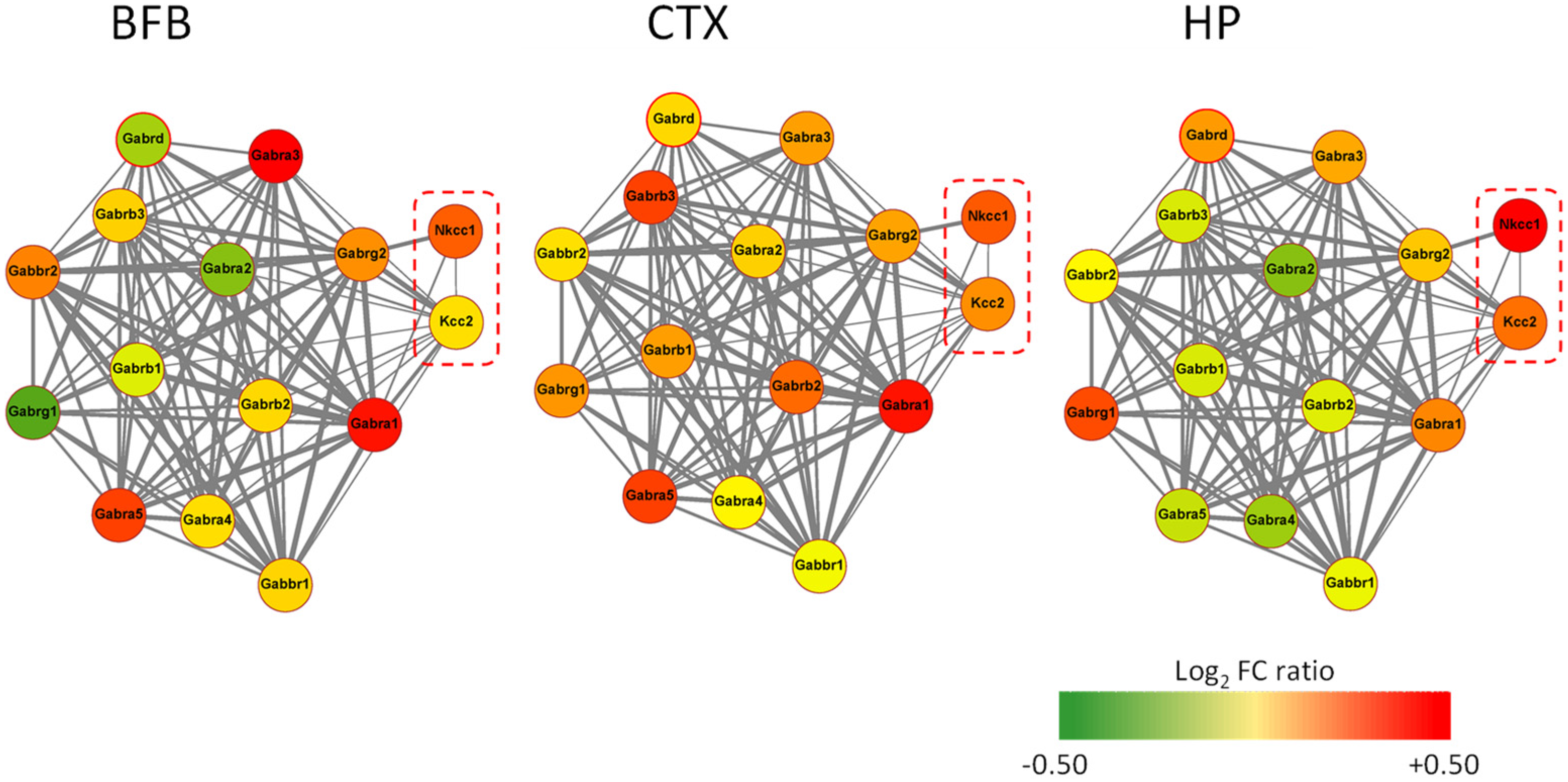
4. AD11 Anti-NGF Mice: A Link between NGF Deprivation, Dysfunction of GABAergic, and Cholinergic Signaling and AD
5. Rescuing a Proper GABAergic Signaling for the Treatment of AD by Repurposed Drugs
6. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semyanov, A.; Walker, M.C.; Kullmann, D.M.; Silver, R.A. Tonically Active GABAA Receptors: Modulating Gain and Maintaining the Tone. Trends Neurosci. 2004, 27, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Nusser, Z. Variations on an Inhibitory Theme: Phasic and Tonic Activation of GABAA Receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Cellot, G.; Cherubini, E. Functional Role of Ambient GABA in Refining Neuronal Circuits Early in Postnatal Development. Front. Neural Circuits 2013, 7, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherubini, E.; Gaiarsa, J.L.; Ben-Ari, Y. GABA: An Excitatory Transmitter in Early Postnatal Life. Trends Neurosci. 1991, 14, 515–519. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA Excitatory/Inhibitory Shift in Brain Maturation and Neurological Disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− Co-Transporter KCC2 Renders GABA Hyperpolarizing during Neuronal Maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-β–Induced Neuronal Dysfunction in Alzheimer’s Disease: From Synapses toward Neural Networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. Neural Synchrony in Brain Disorders: Relevance for Cognitive Dysfunctions and Pathophysiology. Neuron 2006, 52, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid-β Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossor, M.N.; Emson, P.C.; Mountjoy, C.Q.; Roth, M.; Iversen, L.L. Neurotransmitters of the Cerebral Cortex in Senile Dementia of Alzheimer Type. In The Aging Brain; Hoyer, S., Ed.; Experimental Brain Research Supplementum; Springer: Berlin/Heidelberg, Germany, 1982; Volume 5, pp. 153–157. ISBN 978-3-642-68509-5. [Google Scholar]
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villette, V.; Dutar, P. GABAergic Microcircuits in Alzheimer’s Disease Models. Curr. Alzheimer Res. 2016, 14, 30–39. [Google Scholar] [CrossRef]
- Selkoe, D.J. Early Network Dysfunction in Alzheimer’s Disease. Science 2019, 365, 540–541. [Google Scholar] [CrossRef]
- Bakker, A.; Krauss, G.L.; Albert, M.S.; Speck, C.L.; Jones, L.R.; Stark, C.E.; Yassa, M.A.; Bassett, S.S.; Shelton, A.L.; Gallagher, M. Reduction of Hippocampal Hyperactivity Improves Cognition in Amnestic Mild Cognitive Impairment. Neuron 2012, 74, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and Impact of Subclinical Epileptiform Activity in Alzheimer’s Disease: Subclinical Epileptiform Activity in AD. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam Suppresses Neuronal Network Dysfunction and Reverses Synaptic and Cognitive Deficits in an Alzheimer’s Disease Model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic Activity in Alzheimer’s Disease: Causes and Clinical Relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Grouselle, D.; Winsky-Sommerer, R.; David, J.P.; Delacourte, A.; Dournaud, P.; Epelbaum, J. Loss of Somatostatin-like Immunoreactivity in the Frontal Cortex of Alzheimer Patients Carrying the Apolipoprotein Epsilon 4 Allele. Neurosci. Lett. 1998, 255, 21–24. [Google Scholar] [CrossRef]
- Bai, X.; Edden, R.A.E.; Gao, F.; Wang, G.; Wu, L.; Zhao, B.; Wang, M.; Chan, Q.; Chen, W.; Barker, P.B. Decreased γ-Aminobutyric Acid Levels in the Parietal Region of Patients with Alzheimer’s Disease: In Vivo GABA Levels Measurement in AD. J. Magn. Reson. Imaging 2015, 41, 1326–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, C.; Yu, S.; Wong, W.; McGeer, E.G.; McGeer, P.L. GAD65, GAD67, and GABAT Immunostaining in Human Brain and Apparent GAD65 Loss in Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 33, 1073–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Marin, V. Diminished Perisomatic GABAergic Terminals on Cortical Neurons Adjacent to Amyloid Plaques. Front. NeuroaNat. 2009, 3, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Miguel, A.; Hercher, C.; Beasley, C.L.; Barr, A.M.; Bayer, T.A.; Falkai, P.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A.; Honer, W.G. Loss of Munc18-1 Long Splice Variant in GABAergic Terminals Is Associated with Cognitive Decline and Increased Risk of Dementia in a Community Sample. Mol. Neurodegener. 2015, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiz-Sanchez, D.; Ubeda-Bañon, I.; Flores-Cuadrado, A.; Gonzalez-Rodriguez, M.; Villar-Conde, S.; Astillero-Lopez, V.; Martinez-Marcos, A. Somatostatin, Olfaction, and Neurodegeneration. Front. Neurosci. 2020, 14, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [Green Version]
- Palop, J.J.; Jones, B.; Kekonius, L.; Chin, J.; Yu, G.-Q.; Raber, J.; Masliah, E.; Mucke, L. Neuronal Depletion of Calcium-Dependent Proteins in the Dentate Gyrus Is Tightly Linked to Alzheimer’s Disease-Related Cognitive Deficits. Proc. Natl. Acad. Sci. USA 2003, 100, 9572–9577. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Brasnjevic, I.; Rutten, B.P.F.; Van Der Kolk, N.; Perl, D.P.; Bouras, C.; Steinbusch, H.W.M.; Schmitz, C.; Hof, P.R.; Dickstein, D.L. Hippocampal Interneuron Loss in an APP/PS1 Double Mutant Mouse and in Alzheimer’s Disease. Brain Struct Funct. 2010, 214, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Li, H.-L.; Tian, N.; Liu, F.; Wang, L.; Yin, Y.; Yue, L.; Ma, L.; Wan, Y.; Wang, J.-Z. Interneuron Accumulation of Phosphorylated Tau Impairs Adult Hippocampal Neurogenesis by Suppressing GABAergic Transmission. Cell Stem Cell 2020, 26, 331–345. [Google Scholar] [CrossRef]
- Soininen, H.; Riekkinen, P.J.; Partanen, J.; Helkala, E.-L.; Laulumaa, V.; Jolkkonen, J.; Reinikainen, K. Cerebrospinal Fluid Somatostatin Correlates with Spectral EEG Variables and with Parietotemporal Cognitive Dysfunction in Alzheimer Patients. Neurosci. Lett. 1988, 85, 131–136. [Google Scholar] [CrossRef]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic System as a Therapeutic Target for Alzheimer’s Disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwakowsky, A.; Calvo-Flores Guzmán, B.; Pandya, M.; Turner, C.; Waldvogel, H.J.; Faull, R.L. GABAA Receptor Subunit Expression Changes in the Human Alzheimer’s Disease Hippocampus, Subiculum, Entorhinal Cortex and Superior Temporal Gyrus. J. Neurochem. 2018, 145, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, M.; Mizukami, K.; Ikonomovic, M.D.; Ishikawa, M.; Abrahamson, E.E.; DeKosky, S.T.; Asada, T. An Immunohistochemical Study of GABAA Receptor Gamma Subunits in Alzheimer’s Disease Hippocampus: Relationship to Neurofibrillary Tangle Progression. Neuropathology 2009, 29, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, M.; Mizukami, K.; Ikonomovic, M.D.; Ishikawa, M.; Hidaka, S.; Abrahamson, E.E.; DeKosky, S.T.; Asada, T. Changes in Hippocampal GABABR1 Subunit Expression in Alzheimer’s Patients: Association with Braak Staging. Acta Neuropathol. 2005, 109, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of Functional GABA A Receptors in the Alzheimer Diseased Brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krantic, S.; Isorce, N.; Mechawar, N.; Davoli, M.A.; Vignault, E.; Albuquerque, M.; Chabot, J.-G.; Moyse, E.; Chauvin, J.-P.; Aubert, I.; et al. Hippocampal GABAergic Neurons Are Susceptible to Amyloid-β Toxicity in Vitro and Are Decreased in Number in the Alzheimer’s Disease TgCRND8 Mouse Model. J. Alzheimer’s Dis. 2012, 29, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.; Baglietto-Vargas, D.; del Rio, J.C.; Moreno-Gonzalez, I.; Santa-Maria, C.; Jimenez, S.; Caballero, C.; Lopez-Tellez, J.F.; Khan, Z.U.; Ruano, D.; et al. Early Neuropathology of Somatostatin/NPY GABAergic Cells in the Hippocampus of a PS1×APP Transgenic Model of Alzheimer’s Disease. Neurobiol. Aging 2006, 27, 1658–1672. [Google Scholar] [CrossRef]
- Oyelami, T.; Bondt, A.D.; den Wyngaert, I.V.; Van Hoorde, K.; Hoskens, L.; Shaban, H.; Kemp, J.A.; Drinkenburg, W.H. Age-dependent Concomitant Changes in Synaptic Function and GABAergic Pathway in the APP/PS1 Mouse Model. Acta Neurobiol. Exp. 2016, 76, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Gonzalez, I.; Baglietto-Vargas, D.; Sanchez-Varo, R.; Jimenez, S.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; del Rio, J.C.; Torres, M.; Romero-Acebal, M.; Ruano, D.; et al. Extracellular Amyloid-β and Cytotoxic Glial Activation Induce Significant Entorhinal Neuron Loss in Young PS1M146L/APP751SL Mice. J. Alzheimer’s Dis. 2009, 18, 755–776. [Google Scholar] [CrossRef]
- Li, G.; Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Ring, K.; Halabisky, B.; Deng, C.; Mahley, R.W.; Huang, Y. GABAergic Interneuron Dysfunction Impairs Hippocampal Neurogenesis in Adult Apolipoprotein E4 Knockin Mice. Cell Stem Cell 2009, 5, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Leung, L.; Andrews-Zwilling, Y.; Yoon, S.Y.; Jain, S.; Ring, K.; Dai, J.; Wang, M.M.; Tong, L.; Walker, D.; Huang, Y. Apolipoprotein E4 Causes Age- and Sex-Dependent Impairments of Hilar GABAergic Interneurons and Learning and Memory Deficits in Mice. PLoS ONE 2012, 7, e53569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.M.; Djukic, B.; Arnold, C.; Gillespie, A.K.; Yoon, S.Y.; Wang, M.M.; Zhang, O.; Knoferle, J.; Rubenstein, J.L.R.; Alvarez-Buylla, A.; et al. Inhibitory Interneuron Progenitor Transplantation Restores Normal Learning and Memory in ApoE4 Knock-In Mice without or with Aβ Accumulation. J. Neurosci. 2014, 34, 9506–9515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Gan, J.; Jonas, P. Fast-Spiking, Parvalbumin + GABAergic Interneurons: From Cellular Design to Microcircuit Function. Science 2014, 345, 1255263. [Google Scholar] [CrossRef] [PubMed]
- Zallo, F.; Gardenal, E.; Verkhratsky, A.; Rodríguez, J.J. Loss of Calretinin and Parvalbumin Positive Interneurones in the Hippocampal CA1 of Aged Alzheimer’s Disease Mice. Neurosci. Lett. 2018, 681, 19–25. [Google Scholar] [CrossRef]
- Czapski, G.A.; Strosznajder, J.B. Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1677. [Google Scholar] [CrossRef]
- Kang, J.-Q. Epileptic Mechanisms Shared by Alzheimer’s Disease: Viewed via the Unique Lens of Genetic Epilepsy. Int. J. Mol. Sci. 2021, 22, 7133. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma Frequency Entrainment Attenuates Amyloid Load and Modifies Microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Flores Guzmán, B.; Kim, S.; Chawdhary, B.; Peppercorn, K.; Tate, W.P.; Waldvogel, H.J.; Faull, R.L.; Montgomery, J.; Kwakowsky, A. Amyloid-Beta1-42 -Induced Increase in GABAergic Tonic Conductance in Mouse Hippocampal CA1 Pyramidal Cells. Molecules 2020, 25, 693. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from Reactive Astrocytes Impairs Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Chu, D.C.M.; Penney, J.B.; Young, A.B. Cortical GABAB and GABAA Receptors in Alzheimer’s Disease: A Quantitative Autoradiographic Study. Neurology 1987, 37, 1454. [Google Scholar] [CrossRef]
- Salazar, A.M.; Leisgang, A.M.; Ortiz, A.A.; Murtishaw, A.S.; Kinney, J.W. Alterations of GABA B Receptors in the APP/PS1 Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2021, 97, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Martín-Belmonte, A.; Aguado, C.; Alfaro-Ruíz, R.; Moreno-Martínez, A.E.; de la Ossa, L.; Martínez-Hernández, J.; Buisson, A.; Shigemoto, R.; Fukazawa, Y.; Luján, R. Density of GABAB Receptors Is Reduced in Granule Cells of the Hippocampus in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinamarca, M.C.; Raveh, A.; Schneider, A.; Fritzius, T.; Früh, S.; Rem, P.D.; Stawarski, M.; Lalanne, T.; Turecek, R.; Choo, M.; et al. Complex Formation of APP with GABAB Receptors Links Axonal Trafficking to Amyloidogenic Processing. Nat. Commun. 2019, 10, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.; Newey, S.E.; Ilie, A.; Wefelmeyer, W.; Raimondo, J.V.; Ginham, R.; Mcllhinney, R.A.J.; Akerman, C.J. Neuronal Chloride Regulation via KCC2 Is Modulated through a GABAB Receptor Protein Complex. J. Neurosci. 2017, 37, 5447–5462. [Google Scholar] [CrossRef] [Green Version]
- Doshina, A.; Gourgue, F.; Onizuka, M.; Opsomer, R.; Wang, P.; Ando, K.; Tasiaux, B.; Dewachter, I.; Kienlen-Campard, P.; Brion, J.-P.; et al. Cortical Cells Reveal APP as a New Player in the Regulation of GABAergic Neurotransmission. Sci. Rep. 2017, 7, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Wang, J.; Jiang, J.; Zheng, X.; Justice, N.J.; Wang, K.; Ran, X.; Li, Y.; Huo, Q.; Zhang, J.; et al. APP Modulates KCC2 Expression and Function in Hippocampal GABAergic Inhibition. eLife 2017, 6, e20142. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, E.; Di Cristo, G.; Avoli, M. Dysregulation of GABAergic Signaling in Neurodevelomental Disorders: Targeting Cation-Chloride Co-Transporters to Re-Establish a Proper E/I Balance. Front. Cell. Neurosci. 2022, 15, 813441. [Google Scholar] [CrossRef]
- Fiumelli, H.; Woodin, M.A. Role of Activity-Dependent Regulation of Neuronal Chloride Homeostasis in Development. Curr. Opin. Neurobiol. 2007, 17, 81–86. [Google Scholar] [CrossRef]
- Capsoni, S.; Ugolini, G.; Comparini, A.; Ruberti, F.; Berardi, N.; Cattaneo, A. Alzheimer-like Neurodegeneration in Aged Antinerve Growth Factor Transgenic Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6826–6831. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, P.; Price, D.; Struble, R.; Clark, A.; Coyle, J.; DeLong, M.R. Alzheimer’s Disease and Senile Dementia: Loss of Neurons in the Basal Forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Pontecorvo, M.J.; Flicker, C. The Cholinergic Hypothesis: A Historical Overview, Current Perspective, and Future Directions. Ann. N. Y. Acad. Sci. 1985, 444, 332–358. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Pepeu, G. Sex and Gender Differences in the Brain Cholinergic System and in the Response to Therapy of Alzheimer Disease with Cholinesterase Inhibitors. Curr. Alzheimer Res. 2018, 15, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- DeKosky, D.S.T.; Harbaugh, R.E.; Schmitt, F.A.; Bakay, R.A.; Chui, H.C.; Knopman, D.S.; Reeder, T.M.; Shetter, A.G.; Senter, H.J.; Markesbery, W.R. Cortical Biopsy in Alzheimer’s Disease: Diagnostic Accuracy and Neurochemical, Neuropathological, and Cognitive Correlations. Ann. Neurol. 1992, 32, 625–632. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The Cholinergic Hypothesis of Geriatric Memory Dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- Coyle, J.T.; Price, D.L.; DeLong, M.R. Alzheimer’s Disease: A Disorder of Cortical Cholinergic Innervation. Science 1983, 219, 1184–1190. [Google Scholar] [CrossRef]
- Levi-Montalcini, R. Effects of Mouse Tumor Transplantation on the Nervous System. Ann. N. Y. Acad. Sci. 1952, 55, 330–344. [Google Scholar] [CrossRef]
- Dreyfus, C.F. Effects of Nerve Growth Factor on Cholinergic Brain Neurons. Trends Pharmacol. Sci. 1989, 10, 145–149. [Google Scholar] [CrossRef]
- Levi-Montalcini, R. The Nerve Growth Factor 35 Years Later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef]
- Lindsay, R.M.; Harmar, A.J. Nerve Growth Factor Regulates Expression of Neuropeptide Genes in Adult Sensory Neurons. Nature 1989, 337, 362–364. [Google Scholar] [CrossRef]
- McAllister, A.K. Neurotrophins and Neuronal Differentiation in the Central Nervous System. Cell Mol. Life Sci. 2001, 58, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Mobley, W.C.; Rutkowski, J.L.; Tennekoon, G.I.; Gemski, J.; Buchanan, K.; Johnston, M.V. Nerve Growth Factor Increases Choline Acetyltransferase Activity in Developing Basal Forebrain Neurons. Mol. Brain Res. 1986, 1, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Hefti, F. Nerve Growth Factor Promotes Survival of Septal Cholinergic Neurons after Fimbrial Transections. J. Neurosci. 1986, 6, 2155–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayer-LeLievre, C.; Olson, L.; Ebendal, T.; Seiger, A.; Persson, H. Expression of the Beta-Nerve Growth Factor Gene in Hippocampal Neurons. Science 1988, 240, 1339–1341. [Google Scholar] [CrossRef]
- Korsching, S.; Auburger, G.; Heumann, R.; Scott, J.; Thoenen, H. Levels of Nerve Growth Factor and Its MRNA in the Central Nervous System of the Rat Correlate with Cholinergic Innervation. EMBO J. 1985, 4, 1389–1393. [Google Scholar] [CrossRef]
- Large, T.H.; Bodary, S.C.; Clegg, D.O.; Weskamp, G.; Otten, U.; Reichardt, L.F. Nerve Growth Factor Gene Expression in the Developing Rat Brain. Science 1986, 234, 352–355. [Google Scholar] [CrossRef]
- Whittemore, S.R.; Ebendal, T.; Lärkfors, L.; Olson, L.; Seiger, A.; Strömberg, I.; Persson, H. Development and Regional Expression of Beta Nerve Growth Factor Messenger RNA and Protein in the Rat Central Nervous System. Proc. Natl. Acad. Sci. USA 1986, 83, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Seiler, M.; Schwab, M.E. Specific Retrograde Transport of Nerve Growth Factor (NGF) from Neocortex to Nucleus Basalis in the Rat. Brain Res. 1984, 300, 33–39. [Google Scholar] [CrossRef]
- Fagan, A.M.; Garber, M.; Barbacid, M.; Silos-Santiago, I.; Holtzman, D.M. A Role for TrkA during Maturation of Striatal and Basal Forebrain Cholinergic Neurons In Vivo. J. Neurosci. 1997, 17, 7644–7654. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Holtzman, D.M.; Kromer, L.F.; Kaplan, D.R.; Chua-Couzens, J.; Clary, D.O.; Knusel, B.; Mobley, W.C. Regulation of TrkA and ChAT Expression in Developing Rat Basal Forebrain: Evidence That Both Exogenous and Endogenous NGF Regulate Differentiation of Cholinergic Neurons. J. Neurosci. 1995, 15, 2888–2905. [Google Scholar] [CrossRef] [Green Version]
- Sobreviela, T.; Clary, D.O.; Reichardt, L.F.; Brandabur, M.M.; Kordower, J.H.; Mufson, E.J. TrkA-immunoreactive Profiles in the Central Nervous System: Colocalization with Neurons Containing P75 Nerve Growth Factor Receptor, Choline Acetyltransferase, and Serotonin. J. Comp. Neurol. 1994, 350, 587–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnahn, H.; Hefti, F.; Heumann, R.; Schwab, M.E.; Thoenen, H. NGF-Mediated Increase of Choline Acetyltransferase (ChAT) in the Neonatal Rat Forebrain: Evidence for a Physiological Role of NGF in the Brain? Dev. Brain Res. 1983, 9, 45–52. [Google Scholar] [CrossRef]
- Williams, L.R.; Varon, S.; Peterson, G.M.; Wictorin, K.; Fischer, W.; Bjorklund, A.; Gage, F.H. Continuous Infusion of Nerve Growth Factor Prevents Basal Forebrain Neuronal Death after Fimbria Fornix Transection. Proc. Natl. Acad. Sci. USA 1986, 83, 9231–9235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kromer, L.F. Nerve Growth Factor Treatment After Brain Injury Prevents Neuronal Death. Science 1987, 235, 214–216. [Google Scholar] [CrossRef]
- Fischer, W.; Wictorin, K.; Björklund, A.; Williams, L.R.; Varon, S.; Gage, F.H. Amelioration of Cholinergic Neuron Atrophy and Spatial Memory Impairment in Aged Rats by Nerve Growth Factor. Nature 1987, 329, 65–68. [Google Scholar] [CrossRef]
- Costantini, C.; Scrable, H.; Puglielli, L. An Aging Pathway Controls the TrkA to P75NTR Receptor Switch and Amyloid Beta-Peptide Generation. EMBO J. 2006, 25, 1997–2006. [Google Scholar] [CrossRef] [Green Version]
- Yaar, M.; Zhai, S.; Fine, R.E.; Eisenhauer, P.B.; Arble, B.L.; Stewart, K.B.; Gilchrest, B.A. Amyloid β Binds Trimers as Well as Monomers of the 75-KDa Neurotrophin Receptor and Activates Receptor Signaling. J. Biol. Chem. 2002, 277, 7720–7725. [Google Scholar] [CrossRef] [Green Version]
- Frade, J.M. Nuclear Translocation of the P75 Neurotrophin Receptor Cytoplasmic Domain in Response to Neurotrophin Binding. J. Neurosci. 2005, 25, 1407–1411. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF Signaling Control Amyloidogenic Route and Abeta Production in Hippocampal Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Nuydens, R.; Dispersyn, G.; de Jong, M.; Van Den Kieboom, G.; Borgers, M.; Geerts, H. Aberrant Tau Phosphorylation and Neurite Retraction during NGF Deprivation in PC12 Cells. Biochem. Biophys. Res. Commun. 1997, 240, 687–691. [Google Scholar] [CrossRef]
- Davis, P.K.; Johnson, G.V.W. The Microtubule Binding of Tau and High Molecular Weight Tau in Apoptotic PC12 Cells Is Impaired Because of Altered Phosphorylation. J. Biol. Chem. 1999, 274, 35686–35692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelton, S.B.; Johnson, G.V.W. Tau and HMW Tau Phosphorylation and Compartmentalization in Apoptotic Neuronal PC12 Cells. J. Neurosci. Res. 2001, 66, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Ciotti, M.T.; Florenzano, F.; Capsoni, S.; Amato, G.; Calissano, P. Endogenous Aβ Causes Cell Death via Early Tau Hyperphosphorylation. Neurobiol. Aging 2011, 32, 969–990. [Google Scholar] [CrossRef]
- Matrone, C.; Di Luzio, A.; Meli, G.; D’Aguanno, S.; Severini, C.; Ciotti, M.T.; Cattaneo, A.; Calissano, P. Activation of the Amyloidogenic Route by NGF Deprivation Induces Apoptotic Death in PC12 Cells. J. Alzheimer’s Dis. 2008, 13, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Rapposelli, B.; Calissano, P. Three Distinct Types of Monoclonal Antibodies After Long-Term Immunization of Rats with Mouse Nerve Growth Factor. J. Neurochem. 1988, 50, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.; Tongiorgi, E.; Avignone, E.; Gonfloni, S.; Ruberti, F.; Domenici, L.; Cattaneo, A. The Effects of Anti-Nerve Growth Factor Monoclonal Antibodies on Developing Basal Forebrain Neurons Are Transient and Reversible: Transient Effects of Early Postnatal NGF Neutralization. Eur. J. Neurosci. 1998, 10, 3127–3140. [Google Scholar] [CrossRef] [PubMed]
- Ruberti, F.; Capsoni, S.; Comparini, A.; Di Daniel, E.; Franzot, J.; Gonfloni, S.; Rossi, G.; Berardi, N.; Cattaneo, A. Phenotypic Knockout of Nerve Growth Factor in Adult Transgenic Mice Reveals Severe Deficits in Basal Forebrain Cholinergic Neurons, Cell Death in the Spleen, and Skeletal Muscle Dystrophy. J. Neurosci. 2000, 20, 2589–2601. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, R.; Garcia, A.A.; Braschi, C.; Capsoni, S.; Maffei, L.; Berardi, N.; Cattaneo, A. Intranasal Administration of Nerve Growth Factor (NGF) Rescues Recognition Memory Deficits in AD11 Anti-NGF Transgenic Mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3811–3816. [Google Scholar] [CrossRef] [Green Version]
- Capsoni, S.; Brandi, R.; Arisi, I.; D’Onofrio, M.; Cattaneo, A. A Dual Mechanism Linking NGF/ProNGF Imbalance and Early Inflammation to Alzheimer’s Disease Neurodegeneration in the AD11 Anti-NGF Mouse Model. CNS Neurol. Disord.-Drug Targets 2011, 10, 635–647. [Google Scholar] [CrossRef]
- Capsoni, S.; Giannotta, S.; Cattaneo, A. β-Amyloid Plaques in a Model for Sporadic Alzheimer’s Disease Based on Transgenic Anti-Nerve Growth Factor Antibodies. Mol. Cell. Neurosci. 2002, 21, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Pesavento, E.; Capsoni, S.; Domenici, L.; Cattaneo, A. Acute Cholinergic Rescue of Synaptic Plasticity in the Neurodegenerating Cortex of Anti-Nerve-Growth-Factor Mice: Decreased Synaptic Plasticity in Anti-NGF Mice. Eur. J. Neurosci. 2002, 15, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Origlia, N.; Capsoni, S.; Domenici, L.; Cattaneo, A. Time Window in Cholinomimetic Ability to Rescue Long-Term Potentiation in Neurodegenerating Anti-Nerve Growth Factor Mice. J. Alzheimer’s Dis. 2006, 9, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Houeland, G.; Romani, A.; Marchetti, C.; Amato, G.; Capsoni, S.; Cattaneo, A.; Marie, H. Transgenic Mice with Chronic NGF Deprivation and Alzheimer’s Disease-Like Pathology Display Hippocampal Region-Specific Impairments in Short- and Long-Term Plasticities. J. Neurosci. 2010, 30, 13089–13094. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D.; Mahady, L.; Perez, S.E.; Massa, S.M.; Longo, F.M.; Ikonomovic, M.D. Nerve Growth Factor Pathobiology during the Progression of Alzheimer’s Disease. Front. Neurosci. 2019, 13, 533. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The Origins of Alzheimer Disease: A Is for Amyloid. JAMA 2000, 283, 1615–1617. [Google Scholar] [CrossRef]
- Capsoni, S.; Cattaneo, A. On the Molecular Basis Linking Nerve Growth Factor (NGF) to Alzheimer’s Disease. Cell. Mol. Neurobiol. 2006, 26, 617–631. [Google Scholar] [CrossRef]
- Cattaneo, A.; Capsoni, S.; Paoletti, F. Towards Non Invasive Nerve Growth Factor Therapies for Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 15, 255–283. [Google Scholar] [CrossRef] [Green Version]
- D’Onofrio, M.; Arisi, I.; Brandi, R.; Di Mambro, A.; Felsani, A.; Capsoni, S.; Cattaneo, A. Early Inflammation and Immune Response MRNAs in the Brain of AD11 Anti-NGF Mice. Neurobiol. Aging 2011, 32, 1007–1022. [Google Scholar] [CrossRef]
- De Simone, R.; Ambrosini, E.; Carnevale, D.; Ajmone-Cat, M.A.; Minghetti, L. NGF Promotes Microglial Migration through the Activation of Its High Affinity Receptor: Modulation by TGF-β. J. Neuroimmunol. 2007, 190, 53–60. [Google Scholar] [CrossRef]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF Steers Microglia toward a Neuroprotective Phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, F.; Covaceuszach, S.; Konarev, P.V.; Gonfloni, S.; Malerba, F.; Schwarz, E.; Svergun, D.I.; Cattaneo, A.; Lamba, D. Intrinsic Structural Disorder of Mouse ProNGF. Proteins Struct. Funct. Bioinform. 2009, 75, 990–1009. [Google Scholar] [CrossRef] [PubMed]
- Tiveron, C.; Fasulo, L.; Capsoni, S.; Malerba, F.; Marinelli, S.; Paoletti, F.; Piccinin, S.; Scardigli, R.; Amato, G.; Brandi, R. ProNGF\NGF Imbalance Triggers Learning and Memory Deficits, Neurodegeneration and Spontaneous Epileptic-like Discharges in Transgenic Mice. Cell Death Differ. 2013, 20, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Tiveron, C.; Vignone, D.; Amato, G.; Cattaneo, A. Dissecting the Involvement of Tropomyosin-Related Kinase A and P75 Neurotrophin Receptor Signaling in NGF Deficit-Induced Neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 12299–12304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuello, A.C.; Pentz, R.; Hall, H. The Brain NGF Metabolic Pathway in Health and in Alzheimer’s Pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahnestock, M.; Shekari, A. ProNGF and Neurodegeneration in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef] [Green Version]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The Human Brain NGF Metabolic Pathway Is Impaired in the Pre-Clinical and Clinical Continuum of Alzheimers Disease. Mol. Psychiatry 2020, 26, 6023–6037. [Google Scholar] [CrossRef]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The Precursor Pro-Nerve Growth Factor Is the Predominant Form of Nerve Growth Factor in Brain and Is Increased in Alzheimer’s Disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Fahnestock, M.; Scott, S.A.; Jetté, N.; Weingartner, J.A.; Crutcher, K.A. Nerve Growth Factor MRNA and Protein Levels Measured in the Same Tissue from Normal and Alzheimer’s Disease Parietal Cortex. Mol. Brain Res. 1996, 42, 175–178. [Google Scholar] [CrossRef]
- Mufson, E.J.; Conner, J.M.; Kordower, J.H. Nerve Growth Factor in Alzheimer’s Disease: Defective Retrograde Transport to Nucleus Basalis. Neuroreport 1995, 6, 1063–1066. [Google Scholar] [CrossRef]
- Shooter, E.M. Early Days of the Nerve Growth Factor Proteins. Annu. Rev. Neurosci. 2001, 24, 601–629. [Google Scholar] [CrossRef]
- Fahnestock, M.; Yu, G.; Coughlin, M.D. ProNGF: A Neurotrophic or an Apoptotic Molecule? Prog. Brain Res. 2004, 146, 101–110. [Google Scholar] [PubMed]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of Cell Survival by Secreted Proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouri, A.; Nomoto, H.; Furukawa, S. Processing of Nerve Growth Factor: The Role of Basic Amino Acid Clusters in the pro-Region. Biochem. Biophys. Res. Commun. 2007, 353, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Cuello, A.C. Activity-Dependent Release of Precursor Nerve Growth Factor, Conversion to Mature Nerve Growth Factor, and Its Degradation by a Protease Cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 6735–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidah, N.G.; Benjannet, S.; Pareek, S.; Savaria, D.; Hamelin, J.; Goulet, B.; Laliberté, J.; Lazure, C.; Chrétien, M.; Murphy, R.A. Cellular Processing of the Nerve Growth Factor Precursor by the Mammalian Pro-Protein Convertases. Biochem. J. 1996, 314, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, U.; Heymach, J.V., Jr.; Shooter, E.M. Two Conserved Domains in the NGF Propeptide Are Necessary and Sufficient for the Biosynthesis of Correctly Processed and Biologically Active NGF. EMBO J. 1991, 10, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Arisi, I.; Cattaneo, A.; Rosato, V. Parameter Estimate of Signal Transduction Pathways. BMC Neurosci. 2006, 7, S6. [Google Scholar] [CrossRef]
- Hempstead, B.L. Dissecting the Diverse Actions of Pro- and Mature Neurotrophins. Curr. Alzheimer Res. 2006, 3, 19–24. [Google Scholar] [CrossRef]
- Twiss, J.L.; Chang, J.H.; Schanen, N.C. Pathophysiological Mechanisms for Actions of the Neurotrophins. Brain Pathol. 2006, 16, 320–332. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and Modulators of Pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P.; Madsen, P.; Nielsen, M.S.; Jacobsen, C.; Kliemannel, M.; Schwarz, E.; Willnow, T.E. Sortilin Is Essential for ProNGF-Induced Neuronal Cell Death. Nature 2004, 427, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Schweigreiter, R. The Dual Nature of Neurotrophins. Bioessays 2006, 28, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Masoudi, R.; Ioannou, M.S.; Coughlin, M.D.; Pagadala, P.; Neet, K.E.; Clewes, O.; Allen, S.J.; Dawbarn, D.; Fahnestock, M. Biological Activity of Nerve Growth Factor Precursor Is Dependent upon Relative Levels of Its Receptors. J. Biol. Chem. 2009, 284, 18424–18433. [Google Scholar] [CrossRef] [Green Version]
- Coulson, E.J.; Reid, K.; Baca, M.; Shipham, K.A.; Hulett, S.M.; Kilpatrick, T.J.; Bartlett, P.F. Chopper, a New Death Domain of the P75 Neurotrophin Receptor That Mediates Rapid Neuronal Cell Death. J. Biol. Chem. 2000, 275, 30537–30545. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.O.; Casaccia-Bonnefil, P.; Carter, B.; Chao, M.V. Competitive Signaling between TrkA and P75 Nerve Growth Factor Receptors Determines Cell Survival. J. Neurosci. 1998, 18, 3273–3281. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.M.; Sternberg, D.W.; Hempstead, B.L.; Chao, M.V. The Low-Affinity P75 Nerve Growth Factor (NGF) Receptor Mediates NGF-Induced Tyrosine Phosphorylation. Proc. Natl. Acad. Sci. USA 1991, 88, 7106–7110. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, B.L.; Martin-Zanca, D.; Kaplan, D.R.; Parada, L.F.; Chao, M.V. High-Affinity NGF Binding Requires Coexpression of the Trk Proto-Oncogene and the Low-Affinity NGF Receptor. Nature 1991, 350, 678–683. [Google Scholar] [CrossRef]
- Landreth, G.E.; Shooter, E.M. Nerve Growth Factor Receptors on PC12 Cells: Ligand-Induced Conversion from Low-to High-Affinity States. Proc. Natl. Acad. Sci. USA 1980, 77, 4751–4755. [Google Scholar] [CrossRef] [Green Version]
- Gargano, N.; Levi, A.; Alema’, S. Modulation of Nerve Growth Factor Internalization by Direct Interaction between P75 and TrkA Receptors. J. Neurosci. Res. 1997, 50, 1–12. [Google Scholar] [CrossRef]
- Mahadeo, D.; Kaplan, L.; Chao, M.V.; Hempstead, B.L. High Affinity Nerve Growth Factor Binding Displays a Faster Rate of Association than P140trk Binding. Implications for Multi-Subunit Polypeptide Receptors. J. Biol. Chem. 1994, 269, 6884–6891. [Google Scholar] [CrossRef]
- Conroy, J.N.; Coulson, E.J. High-Affinity TrkA and P75 Neurotrophin Receptor Complexes: A Twisted Affair. J. Biol. Chem. 2022, 298, 101568. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Fine, A.; Hunt, S.P.; Ullrich, A. Nerve Growth Factor MRNA in Peripheral and Central Rat Tissues and in the Human Central Nervous System: Lesion Effects in the Rat Brain and Levels in Alzheimer’s Disease. Mol. Brain Res. 1986, 1, 85–92. [Google Scholar] [CrossRef]
- Jette, N.; Cole, M.S.; Fahnestock, M. NGF MRNA Is Not Decreased in Frontal Cortex from Alzheimer’s Disease Patients. Mol. Brain Res. 1994, 25, 242–250. [Google Scholar] [CrossRef]
- Pedraza, C.E.; Podlesniy, P.; Vidal, N.; Arévalo, J.C.; Lee, R.; Hempstead, B.; Ferrer, I.; Iglesias, M.; Espinet, C. Pro-NGF Isolated from the Human Brain Affected by Alzheimer’s Disease Induces Neuronal Apoptosis Mediated by P75NTR. Am. J. Pathol. 2005, 166, 533–543. [Google Scholar] [CrossRef]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Videla, L.; Benejam, B.; Carmona-Iragui, M.; Blesa, R.; Lleó, A.; Fortea, J.; Cuello, A.C. Nerve Growth Factor (NGF) Pathway Biomarkers in Down Syndrome Prior to and after the Onset of Clinical Alzheimer’s Disease: A Paired CSF and Plasma Study. Alzheimer’s Dement. 2021, 17, 605–617. [Google Scholar] [CrossRef]
- Fasulo, L.; Brandi, R.; Arisi, I.; La Regina, F.; Berretta, N.; Capsoni, S.; D’Onofrio, M.; Cattaneo, A. ProNGF Drives Localized and Cell Selective Parvalbumin Interneuron and Perineuronal Net Depletion in the Dentate Gyrus of Transgenic Mice. Front. Mol. Neurosci. 2017, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights from Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef]
- Sola, E.; Capsoni, S.; Rosato-Siri, M.; Cattaneo, A.; Cherubini, E. Failure of Nicotine-Dependent Enhancement of Synaptic Efficacy at Schaffer-Collateral CA1 Synapses of AD11 Anti-Nerve Growth Factor Transgenic Mice. Eur. J. Neurosci. 2006, 24, 1252–1264. [Google Scholar] [CrossRef]
- Rosato-Siri, M.; Cattaneo, A.; Cherubini, E. Nicotine-Induced Enhancement of Synaptic Plasticity at CA3-CA1 Synapses Requires GABAergic Interneurons in Adult Anti-NGF Mice. J. Physiol. 2006, 576, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Lagostena, L.; Rosato-Siri, M.; D’Onofrio, M.; Brandi, R.; Arisi, I.; Capsoni, S.; Franzot, J.; Cattaneo, A.; Cherubini, E. In the Adult Hippocampus, Chronic Nerve Growth Factor Deprivation Shifts GABAergic Signaling from the Hyperpolarizing to the Depolarizing Direction. J. Neurosci. 2010, 30, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Arisi, I.; D’Onofrio, M.; Brandi, R.; Felsani, A.; Capsoni, S.; Drovandi, G.; Felici, G.; Weitschek, E.; Bertolazzi, P.; Cattaneo, A. Gene Expression Biomarkers in the Brain of a Mouse Model for Alzheimer’s Disease: Mining of Microarray Data by Logic Classification and Feature Selection. J. Alzheimer’s Dis. 2011, 24, 721–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisi, I.; D’Onofrio, M.; Brandi, R.; Cattaneo, A.; Bertolazzi, P.; Cumbo, F.; Felici, G.; Guerra, C. Time Dynamics of Protein Complexes in the AD11 Transgenic Mouse Model for Alzheimer’s Disease like Pathology. BMC Neurosci. 2015, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug Repositioning and Repurposing for Alzheimer Disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Chumakov, I.; Nabirotchkin, S.; Cholet, N.; Milet, A.; Boucard, A.; Toulorge, D.; Pereira, Y.; Graudens, E.; Traoré, S.; Foucquier, J.; et al. Combining Two Repurposed Drugs as a Promising Approach for Alzheimer’s Disease Therapy. Sci. Rep. 2015, 5, 7608. [Google Scholar] [CrossRef] [PubMed]
- Boussicault, L.; Laffaire, J.; Schmitt, P.; Rinaudo, P.; Callizot, N.; Nabirotchkin, S.; Hajj, R.; Cohen, D. Combination of Acamprosate and Baclofen (PXT864) as a Potential New Therapy for Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2020, 98, 2435–2450. [Google Scholar] [CrossRef]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef]
- Savardi, A.; Borgogno, M.; Narducci, R.; La Sala, G.; Ortega, J.A.; Summa, M.; Armirotti, A.; Bertorelli, R.; Contestabile, A.; De Vivo, M.; et al. Discovery of a Small Molecule Drug Candidate for Selective NKCC1 Inhibition in Brain Disorders. Chem 2020, 6, 2073–2096. [Google Scholar] [CrossRef]
- Taubes, A.; Nova, P.; Zalocusky, K.A.; Kosti, I.; Bicak, M.; Zilberter, M.Y.; Hao, Y.; Yoon, S.Y.; Oskotsky, T.; Pineda, S.; et al. Experimental and Real-World Evidence Supporting the Computational Repurposing of Bumetanide for APOE4-Related Alzheimer’s Disease. Nat. Aging 2021, 1, 932–947. [Google Scholar] [CrossRef]
- Bie, B.; Wu, J.; Lin, F.; Naguib, M.; Xu, J. Suppression of Hippocampal GABAergic Transmission Impairs Memory in Rodent Models of Alzheimer’s Disease. Eur. J. Pharmacol. 2022, 917, 174771. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, M.A.; Uvarov, P.; Hübner, C.A.; Kaila, K. NKCC1, an Elusive Molecular Target in Brain Development: Making Sense of the Existing Data. Cells 2020, 9, 2607. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.; Bergeron, M.J.; Lavertu, G.; Castonguay, A.; Tripathy, S.; Bonin, R.P.; Perez-Sanchez, J.; Boudreau, D.; Wang, B.; Dumas, L.; et al. Chloride Extrusion Enhancers as Novel Therapeutics for Neurological Diseases. Nat. Med. 2013, 19, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [Green Version]
- Amadoro, G.; Corsetti, V.; Sancesario, G.M.; Lubrano, A.; Melchiorri, G.; Bernardini, S.; Calissano, P.; Sancesario, G. Cerebrospinal Fluid Levels of a 20–22 KDa NH2 Fragment of Human Tau Provide a Novel Neuronal Injury Biomarker in Alzheimer’s Disease and Other Dementias. J. Alzheimer’s Dis. 2014, 42, 211–226. [Google Scholar] [CrossRef]
- Scopa, C.; Marrocco, F.; Latina, V.; Ruggeri, F.; Corvaglia, V.; La Regina, F.; Ammassari-Teule, M.; Middei, S.; Amadoro, G.; Meli, G.; et al. Impaired Adult Neurogenesis Is an Early Event in Alzheimer’s Disease Neurodegeneration, Mediated by Intracellular Aβ Oligomers. Cell Death Differ. 2020, 27, 934–948. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capsoni, S.; Arisi, I.; Malerba, F.; D’Onofrio, M.; Cattaneo, A.; Cherubini, E. Targeting the Cation-Chloride Co-Transporter NKCC1 to Re-Establish GABAergic Inhibition and an Appropriate Excitatory/Inhibitory Balance in Selective Neuronal Circuits: A Novel Approach for the Treatment of Alzheimer’s Disease. Brain Sci. 2022, 12, 783. https://doi.org/10.3390/brainsci12060783
Capsoni S, Arisi I, Malerba F, D’Onofrio M, Cattaneo A, Cherubini E. Targeting the Cation-Chloride Co-Transporter NKCC1 to Re-Establish GABAergic Inhibition and an Appropriate Excitatory/Inhibitory Balance in Selective Neuronal Circuits: A Novel Approach for the Treatment of Alzheimer’s Disease. Brain Sciences. 2022; 12(6):783. https://doi.org/10.3390/brainsci12060783
Chicago/Turabian StyleCapsoni, Simona, Ivan Arisi, Francesca Malerba, Mara D’Onofrio, Antonino Cattaneo, and Enrico Cherubini. 2022. "Targeting the Cation-Chloride Co-Transporter NKCC1 to Re-Establish GABAergic Inhibition and an Appropriate Excitatory/Inhibitory Balance in Selective Neuronal Circuits: A Novel Approach for the Treatment of Alzheimer’s Disease" Brain Sciences 12, no. 6: 783. https://doi.org/10.3390/brainsci12060783