
NKCC1 Deficiency in Forming Hippocampal Circuits Triggers Neurodevelopmental Disorder: Role of BDNF-TrkB Signalling


Abstract
:1. Introduction
2. Transcriptional Regulation of Nkcc1
3. SLC12A2 Mutations Reported in Humans and Their Involvement in Neurodevelopmental Disorders
4. Chloride Cotransporter NKCC1 in Animal Models of Neurodevelopmental Disorders
5. Impact of NKCC1 Expression on GABA Shift and Neural Circuits Development
6. The Therapeutical Potential of Re-Establishing E/I Homeostasis of GABAergic Signalling
7. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gamba, G. Molecular Physiology and Pathophysiology of Electroneutral Cation-Chloride Cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Dulin, N.O.; Gusakova, S.V. Cation-chloride cotransporters: Regulation, physiological significance, and role in pathogenesis of arterial hypertension. Biochemistry 2014, 79, 1546–1561. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Forbush, B., III. The Na–K–Cl cotransporter of secretory epithelia. Annu. Rev. Physiol. 2000, 62, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Asp. Med. 2013, 34, 288–298. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium–potassium–chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation–chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [Green Version]
- Delpire, E.; Gagnon, K.B. Water homeostasis and cell volume maintenance and regulation. Curr. Top. Membr. 2018, 81, 3–52. [Google Scholar] [CrossRef]
- Sung, K.-W.; Kirby, M.; McDonald, M.P.; Lovinger, D.M.; Delpire, E. Abnormal GABAA-receptor mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J. Neurosci. 2000, 20, 7531–7538. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Lovinger, D.; Delpire, E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J. Neurophysiol. 2005, 93, 1557–1568. [Google Scholar] [CrossRef] [Green Version]
- Koumangoye, R.; Bastarache, L.; Delpire, E. NKCC1: Newly Found as a Human Disease-Causing Ion Transporter. Function 2021, 2, zqaa028. [Google Scholar] [CrossRef]
- Virtanen, M.A.; Uvarov, P.; Hübner, C.A.; Kaila, K. NKCC1, an Elusive Molecular Target in Brain Development: Making Sense of the Existing Data. Cells 2020, 9, 2607. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G.; Miyanoshita, A.; Lombardi, M.; Lytton, J.; Lee, W.S.; Hediger, M.A.; Hebert, S.C. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 1994, 269, 17713–17722. [Google Scholar] [CrossRef]
- Benarroch, E.E. Cation-chloride cotransporters in the nervous system: General features and clinical correlations. Neurology 2013, 80, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Savardi, A.; Borgogno, M.; De Vivo, M.; Cancedda, L. Pharmacological Tools to Target Nkcc1 in Brain Disorders. Trends Pharmacol. Sci. 2021, 42, 1009–1034. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Fukuda, A. Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 2015, 9, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhou, J.; Zhang, Y.; Liu, T.; Friedel, P.; Zhuo, W.; Somasekharan, S.; Roy, K.; Zhang, L.; Liu, Y.; et al. The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 2021, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Sedmak, G.; Jovanov-Milosevic, N.; Puskarjov, M.; Ulamec, M.; Kruslin, B.; Kaila, K.; Judas, M. Developmental Expression Patterns of KCC2 and Functionally Associated Molecules in the Human Brain. Cereb. Cortex 2016, 26, 4574–4589. [Google Scholar] [CrossRef] [Green Version]
- Peerboom, C.; Wierenga, C.J. The postnatal GABA shift: A developmental perspective. Neurosci. Biobehavio. Rev. 2021, 124, 179–192. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Belluscio, L.; Friedman, B.; Alderson, R.F.; Wiegand, S.J.; Furth, M.E.; Lindsay, R.M.; Yancopoulos, G.D. NT-3, BDNF, and NGF in the developing rat nervous system: Parallel as well as reciprocal patterns of expression. Neuron 1990, 5, 501–509. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minichiello, L.; Korte, M.; Wolfer, D.; Kühn, R.; Unsicker, K.; Cestari, V.; Rossi-Arnaud, C.; Lipp, H.P.; Bonhoeffer, T.; Klein, R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 1999, 24, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Minichiello, L.; Calella, A.M.; Medina, D.L.; Bonhoeffer, T.; Klein, R.; Korte, M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 2002, 36, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Darnieder, L.M.; Deeb, T.Z.; Moss, S.J. Regulation of GABAARs by Phosphorylation. Adv. Pharm. 2014, 72, 97–146. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, J.N.; Thomas, P.; Kittler, J.T.; Smart, T.G.; Moss, S.J. Brain-Derived Neurotrophic Factor Modulates Fast Synaptic Inhibition by Regulating GABAA Receptor Phosphorylation, Activity, and Cell-Surface Stability. J. Neurosci. 2004, 24, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Riffault, B.; Medina, I.; Dumon, C.; Thalman, C.; Ferrand, N.; Friedel, P.; Gaiarsa, J.-L.; Porcher, C. Pro-Brain-Derived Neurotrophic Factor Inhibits GABAergic Neurotransmission by Activating Endocytosis and Repression of GABAA Receptors. J. Neurosci. 2014, 34, 13516–13534. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef] [Green Version]
- Rivera, C.; Voipio, J.; Thomas-Crusells, J.; Li, H.; Emri, Z.; Sipila, S.; Payne, J.A.; Minichiello, L.; Saarma, M.; Kaila, K. Mechanism of Activity-Dependent Downregulation of the Neuron-Specific K-Cl Cotransporter KCC2. J. Neurosci. 2004, 24, 4683–4691. [Google Scholar] [CrossRef] [Green Version]
- Aguado, F.; Carmona, M.A.; Pozas, E.; Aguiló, A.; Martínez-Guijarro, F.J.; Alcantara, S.; Borrell, V.; Yuste, R.; Ibañez, C.F.; Soriano, E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development 2003, 130, 1267–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, A.; Uvarov, P.; Soni, S.; Thomas-Crusells, J.; Airaksinen, M.S.; Rivera, C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 2011, 31, 644–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uvarov, P.; Ludwig, A.; Markkanen, M.; Rivera, C.; Airaksinen, M.S. Upregulation of the Neuron-Specific K+/Cl− Cotransporter Expression by Transcription Factor Early Growth Response 4. J. Neurosci. 2006, 26, 13463–13473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulis Sato, S.; Artoni, P.; Landi, S.; Cozzolino, O.; Parra, R.; Pracucci, E.; Trovato, F.; Szczurkowska, J.; Luin, S.; Arosio, D.; et al. Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E8770–E8779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Colonnese, M.T. GABAergic interneurons excite neonatal hippocampus in vivo. Sci. Adv. 2020, 6, eaba1430. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Goh, E.L.K.; Sailor, K.A.; Kitabatake, Y.; Ming, G.; Song, H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 2006, 439, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Eftekhari, S.; Mehrabi, S.; Soleimani, M.; Hassanzadeh, G.; Shahrokhi, A.; Mostafavi, H.; Hayat, P.; Barati, M.; Mehdizadeh, H.; Rahmanzadeh, R.; et al. BDNF modifies hippocampal KCC2 and NKCC1 expression in a temporal lobe epilepsy model. Acta. Neurobiol. Exp. 2014, 74, 276–287. [Google Scholar]
- Badurek, S.; Griguoli, M.; Asif-Malik, A.; Zonta, B.; Guo, F.; Middei, S.; Lagostena, L.; Jurado-Parras, M.T.; Gillingwater, T.H.; Gruart, A.; et al. Immature dentate granule cells require Ntrk2/Trkb for the formation of functional hippocampal circuitry. iScience 2020, 23, 101078. [Google Scholar] [CrossRef]
- Tyzio, R.; Cossart, R.; Khalilov, I.; Minlebaev, M.; Hubner, C.A.; Represa, A.; Ben-Ari, Y.; Khazipov, R. Maternal oxytocin triggers a transient inhibitory shift in GABA signaling in the fetal brain during delivery. Science 2006, 314, 1788–1792. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyaya, B.; Cristo, G.D. GABAergic Circuit Dysfunctions in Neurodevelopmental Disorders. Front. Psych. 2012, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Braat, S.; Kooy, R.F. The GABAA Receptor as a Therapeutic Target for Neurodevelopmental Disorders. Neuron 2015, 86, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, L.; Torrella Barrufet, J.; Heine, V.M. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropatho. Comm. 2019, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Nomura, T.; Xu, J.; Contractor, A. The Developmental Shift in GABA Polarity Is Delayed in Fragile X Mice. J. Neurosci. 2014, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Rajagopal, L.; Meltzer, H.Y.; Martina, M. Depolarizing GABAA current in the prefrontal cortex is linked with cognitive impairment in a mouse model relevant for schizophrenia. Sci. Adv. 2021, 7, eaba5032. [Google Scholar] [CrossRef]
- Hyde, T.M.; Lipska, B.K.; Ali, T.; Mathew, S.V.; Law, A.J.; Metitiri, O.E.; Straub, R.E.; Ye, T.; Colantuoni, C.; Herman, M.M.; et al. Expression of GABA Signaling Molecules KCC2, NKCC1, and GAD1 in Cortical Development and Schizophrenia. J. Neurosci. 2011, 31, 11088–11095. [Google Scholar] [CrossRef] [Green Version]
- Macnamara, E.F.; Koehler, A.E.; D’Souza, P.; Estwick, T.; Lee, P.; Vezina, G.; Undiagnosed Diseases Network; Fauni, H.; Braddock, S.R.; Torti, E.; et al. Kilquist Syndrome: A Novel Syndromic Hearing Loss Disorder Caused by Homozygous Deletion of SLC12A2. Hum. Mutation. 2019, 40, 532–538. [Google Scholar] [CrossRef]
- Stödberg, T.; Magnusson, M.; Lesko, N.; Wredenberg, A.; Martin Munoz, D.; Stranneheim, H.; Wedell, A. SLC12A2 mutations cause NKCC1 deficiency with encephalopathy and impaired secretory epithelia. Neurol. Genet. 2020, 6, e478. [Google Scholar] [CrossRef]
- McNeill, A.; Aurora, P.; Rajput, K.; Nash, R.; Stals, K.; Robinson, H.; Wakeling, E. Dominant and recessive SLC12A2-syndrome. Am. J. Med. Genet. 2021, 188, 996–999. [Google Scholar] [CrossRef]
- Bilal Shamsi, M.; Saleh, M.; Almuntashri, M.; Alharby, E.; Samman, M.; Peake, R.W.A.; Al-Fadhli, F.M.; Alasmari, A.; Faqeih, E.A.; Almontashiri, N.A.M. Clinical characterization and further confirmation of the autosomal recessive SLC12A2 disease. J. Hum. Genet. 2021, 66, 689–695. [Google Scholar] [CrossRef]
- McNeill, A.; Iovino, E.; Mansard, L.; Vache, C.; Baux, D.; Bedoukian, E.; Cox, H.; Dean, J.; Goudie, D.; Kumar, A.; et al. SLC12A2 variants cause a neurodevelopmental disorder or cochleovestibular defect. Brain 2020, 143, 2380–2387. [Google Scholar] [CrossRef]
- Vibat, C.R.T.; Holland, M.J.; Kang, J.J.; Putney, L.K.; O’Donnell, M.E. Quantitation of Na+-K+-2Cl− Cotransport Splice Variants in Human Tissues Using Kinetic Polymerase Chain Reaction. Analyt. Biochem. 2001, 298, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Gazzard, J.; Chaudhry, S.S.; Sampson, N.; Schulte, B.A.; Steel, K.P. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum. Mol. Genet. 1999, 8, 1579–1584. [Google Scholar] [CrossRef] [Green Version]
- Mutai, H.; Wasano, K.; Momozawa, Y.; Kamatani, Y.; Miya, F.; Masuda, S.; Morimoto, N.; Nara, K.; Takahashi, S.; Tsunoda, T.; et al. Variants encoding a restricted carboxy-terminal domain of SLC12A2 cause hereditary hearing loss in humans. PLoS Genet. 2020, 16, e1008643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchese, M.; Valvo, G.; Moro, F.; Sicca, F.; Santorelli, F.M. Targeted Gene Resequencing (Astrochip) to Explore the Tripartite Synapse in Autism–Epilepsy Phenotype with Macrocephaly. NeuroMol. Med. 2015, 18, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Mercado, A.; Khanna, A.R.; Hodgkinson, A.; Bruat, V.; Awadalla, P.; Gamba, G.; Rouleau, G.A.; Kahle, K.T. Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J. Psych. Res. 2016, 77, 22–26. [Google Scholar] [CrossRef]
- Delpire, E.; Wolfe, L.; Flores, B.; Koumangoye, R.; Schornak, C.C.; Omer, S.; Pusey, B.; Lau, C.; Markello, T.; Adams, D.R. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, A.J.; Madden, V.J.; Henson, O.W.J.; Koller, B.H.; Henson, M.M. Ultrastructure of the inner ear of NKCC1-deficient mice. Hear Res 2001, 156, 17–30. [Google Scholar] [CrossRef]
- Flagella, M.; Clarke, L.L.; Miller, M.L.; Erway, L.C.; Giannella, R.A.; Andringa, A.; Gawenis, L.R.; Kramer, J.; Duffy, J.J.; Doetschman, T.; et al. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 1999, 274, 26946–26955. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.L.; Park, K.; Turner, R.J.; Watson, G.E.; Nguyen, H.V.; Dennett, M.R.; Hand, A.R.; Flagella, M.; Shull, G.E.; Melvin, J.E. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J. Biol. Chem. 2000, 275, 26720–26726. [Google Scholar] [CrossRef]
- Grubb, B.R.; Lee, E.; Pace, A.J.; Koller, B.H.; Boucher, R.C. Intestinal ion transport in NKCC1-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G707–G718. [Google Scholar] [CrossRef] [PubMed]
- Grubb, B.R.; Pace, A.J.; Lee, E.; Koller, B.H.; Boucher, R.C. Alterations in airway ion transport in NKCC1-deficient mice. Am. J. Physiol. Cell Physiol. 2001, 281, C615–C623. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.M.; Knepper, M.A.; Hassell, K.A.; Fischer, M.P.; Shodeinde, A.; Shin, W.; Pham, T.D.; Meyer, J.W.; Lorenz, J.N.; Beierwaltes, W.H.; et al. Hypotension in NKCC1 null mice: Role of the kidneys. Am. J. Physiol. Ren. Physiol. 2006, 290, F409–F416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, A.J.; Lee, E.; Athirakul, K.; Coffman, T.M.; O’Brien, D.A.; Koller, B.H. Failure of spermatogenesis in mouse lines deficient in the Na(+)-K(+)-2Cl(−) cotransporter. J. Clin. Investig. 2000, 105, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Fame, R.M.; Sadegh, C.; Sutin, J.; Naranjo, C.; Syau, D.; Cui, J.; Shipley, F.B.; Vernon, A.; Gao, F.; et al. Choroid plexus NKCC1 mediates cerebrospinal fluid clearance during mouse early postnatal development. Nat. Commun. 2021, 12, 447. [Google Scholar] [CrossRef]
- Laird, J.M.; García-Nicas, E.; Delpire, E.J.; Cervero, F. Presynaptic inhibition and spinal pain processing in mice: A possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci. Lett. 2004, 361, 200–203. [Google Scholar] [CrossRef]
- Haydar, T.F.; Wang, F.; Schwartz, M.L.; Rakic, P. Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J. Neurosci. 2000, 20, 5764–5774. [Google Scholar] [CrossRef] [Green Version]
- Luhmann, H.J.; Fukuda, A.; Kilb, W. Control of cortical neuronal migration by glutamate and GABA. Front. Cell. Neurosci. 2015, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Sernagor, E. GABAergic control of neurite outgrowth and remodeling during development and adult neurogenesis: General rules and differences in diverse systems. Fronti. Cell. Neurosci. 2010, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; Jedlicka, P.; Luhmann, H.J.; Kilb, W. Giant Depolarizing Potentials Trigger Transient Changes in the Intracellular Cl- Concentration in CA3 Pyramidal Neurons of the Immature Mouse Hippocampus. Front. Cell. Neurosci. 2018, 12, 420. [Google Scholar] [CrossRef]
- Spoljaric, I.; Spoljaric, A.; Mavrovic, M.; Seja, P.; Puskarjov, M.; Kaila, K. KCC2-Mediated Cl− Extrusion Modulates Spontaneous Hippocampal Network Events in Perinatal Rats and Mice. Cell Rep. 2019, 26, 1073–1081.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cellot, G.; Maggi, L.; Di Castro, M.; Catalano, M.; Migliore, R.; Migliore, M.; Scattoni, M.L.; Calamandrei, G.; Cherubini, E. Premature changes in neuronal excitability account for hippocampal network impairment and autistic-like behavior in neonatal BTBR T+tf/J mice. Sci. Rep. 2016, 6, 31696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalilov, I.; Minlebaev, M.; Mukhtarov, M.; Khazipov, R. Dynamic changes from depolarizing to hyperpolarizing GABAergic actions during giant depolarizing potentials in the neonatal rat Hippocampus. J. Neurosci. 2015, 35, 12635–12642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzhala, V.I.; Talos, D.M.; Sdrulla, D.A.; Brumback, A.C.; Mathews, G.C.; Benke, T.A.; Delpire, E.; Jensen, J.; Staley, K.J. NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 2005, 11, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.E.; Porter, J.T.; Agmon, A. GABAergic inhibition suppresses paroxysmal network activity in the neonatal rodent hippocampus and neocortex. J. Neurosci. 2000, 20, 8822–8830. [Google Scholar] [CrossRef] [Green Version]
- Sipilä, S.T.; Schuchmann, S.; Voipio, J.; Yamada, J.; Kaila, K. The cation-chloride cotransporter NKCC1 promotes sharp waves in the neonatal rat hippocampus. J. Physiol. 2006, 573, 765–773. [Google Scholar] [CrossRef]
- Galanopoulou, A.S. Dissociated Gender-Specific Effects of Recurrent Seizures on GABA Signaling in CA1 Pyramidal Neurons: Role of GABAA Receptors. J. Neurosci. 2008, 28, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, J.L.; McCarthy, M.M. Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus. Dev. Neurobiol. 2007, 67, 1879–1890. [Google Scholar] [CrossRef] [Green Version]
- Kyrozis, A.; Chudomel, O.; Moshé, S.L.; Galanopoulou, A.S. Sex-dependent maturation of GABAA receptor-mediated synaptic events in rat substantia nigra reticulata. Neurosci. Lett. 2006, 398, 1–5. [Google Scholar] [CrossRef]
- Roux, S.; Lohof, A.; Ben-Ari, Y.; Poulain, B.; Bossu, J.-L. Maturation of GABAergic transmission in cerebellar purkinje cells is sex dependent and altered in the valproate model of autism. Front. Cell. Neurosci. 2018, 12, 1–14. [Google Scholar] [CrossRef]
- Wang, D.D.; Kriegstein, A.R. GABA Regulates Excitatory Synapse Formation in the Neocortex via NMDA Receptor Activation. J. Neurosci. 2008, 28, 5547–5558. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Kriegstein, A.R. Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb. Cortex. 2011, 21, 574–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, P.N.; Amegandjin, C.A.; Szczurkowska, J.; Carriço, J.N.; Fernandes do Nascimento, A.S.; Baho, E.; Chattopadhyaya, B.; Cancedda, L.; Carmant, L.; Di Cristo, G. KCC2 Regulates Dendritic Spine Formation in a Brain-Region Specific and BDNF Dependent Manner. Cereb. Cortex. 2018, 28, 4049–4062. [Google Scholar] [CrossRef] [PubMed]
- Akerman, C.J.; Cline, H.T. Depolarizing GABAergic Conductances Regulate the Balance of Excitation to Inhibition in the Developing Retinotectal Circuit In Vivo. J. Neurosci. 2006, 26, 5117–5130. [Google Scholar] [CrossRef] [PubMed]
- Moretto, E.; Murru, L.; Martano, G.; Sassone, J.; Passafaro, M. Glutamatergic synapses in neurodevelopmental disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 84(Pt B), 328–342. [Google Scholar] [CrossRef]
- Nakanishi, K.; Yamada, J.; Takayama, C.; Oohira, A.; Fukuda, A. NKCC1 activity modulates formation of functional inhibitory synapses in cultured neocortical neurons. Synapse 2007, 61, 138–149. [Google Scholar] [CrossRef]
- Chudotvorova, I.; Ivanov, A.; Rama, S.; Pellegrino, C.; Hübner, C.A.; Ben-Ari, Y.; Medina, I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 2005, 566, 671–679. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Musumeci, G.; Sciarretta, C.; Rodriguez-Moreno, A.; Al Banchaabouchi, M.; Negrete-Diaz, V.; Costanzi, M.; Berno, V.; Egorov, A.V.; von Bohlen und Halbach, O.; Cestari, V.; et al. TrkB Modulates Fear Learning and Amygdalar Synaptic Plasticity by Specific Docking Sites. J. Neurosci. 2009, 29, 10131–10143. [Google Scholar] [CrossRef]
- Berg, D.A.; Su, Y.; Jimenez-Cyrus, D.; Patel, A.; Huang, N.; Morizet, D.; Lee, S.; Shah, R.; Rojas Ringeling, F.; Jain, R.; et al. A Common Embryonic Origin of Stem Cells Drives Developmental and Adult Neurogenesis. Cell 2019, 177, 654–668.e15. [Google Scholar] [CrossRef] [Green Version]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front Neurosci. 2019, 13, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Tyzio, R.; Nardou, R.; Ferrari, D.C.; Tsintsadze, T.; Shahrokhi, A.; Eftekhari, S.; Khalilov, I.; Tsintsadze, V.; Brouchoud, C.; Chazal, G.; et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 2014, 343, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Savardi, A.; Borgogno, M.; Narducci, R.; La Sala, G.; Ortega, J.A.; Summa, M.; Armirotti, A.; Bertorelli, R.; Contestabile, A.; De Vivo, M.; et al. Discovery of a Small Molecule Drug Candidate for Selective NKCC1 Inhibition in Brain Disorders. Chem 2020, 6, 2073–2096. [Google Scholar] [CrossRef]
- Lozovaya, N.; Nardou, R.; Tyzio, R.; Chiesa, M.; Pons-Bennaceur, A.; Eftekhari, S.; Bui, T.-T.; Billon-Grand, M.; Rasero, J.; Bonifazi, P.; et al. Early alterations in a mouse model of Rett syndrome: The GABA developmental shift is abolished at birth. Sci. Rep. 2019, 9, 9276. [Google Scholar] [CrossRef] [Green Version]
- Larimore, J.; Zlatic, S.A.; Arnold, M.; Singleton, K.S.; Cross, R.; Rudolph, H.; Bruegge, M.V.; Sweetman, A.; Garza, C.; Whisnant, E.; et al. Dysbindin Deficiency Modifies the Expression of GABA Neuron and Ion Permeation Transcripts in the Developing Hippocampus. Front. Genet. 2017, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Ragot, A.; Luhmann, H.J.; Dipper-Wawra, M.; Heinemann, U.; Holtkamp, M.; Fidzinski, P. Pathology-selective antiepileptic effects in the focal freeze-lesion rat model of malformation of cortical development. Exp. Neurol. 2021, 343, 113776. [Google Scholar] [CrossRef]
- Römermann, K.; Fedrowitz, M.; Hampel, P.; Kaczmarek, E.; Töllner, K.; Erker, T.; Sweet, D.H.; Löscher, W. Multiple blood-brain barrier transport mechanisms limit bumetanide accumulation, and therapeutic potential, in the mammalian brain. Neuropharmacology 2017, 117, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.; Heel, R.C. Bumetanide. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use. Drugs 1984, 28, 426–464. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cherubini, E. The GABA Polarity Shift and Bumetanide Treatment: Making Sense Requires Unbiased and Undogmatic Analysis. Cells 2022, 11, 396. [Google Scholar] [CrossRef]
- Töllner, K.; Brandt, C.; Töpfer, M.; Brunhofer, G.; Erker, T.; Gabriel, M.; Feit, P.W.; Lindfors, J.; Kaila, K.; Löscher, W. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann. Neurol. 2014, 75, 550–562. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Panisi, C.; Marini, M. The Alteration of Chloride Homeostasis/GABAergic Signaling in Brain Disorders: Could Oxidative Stress Play a Role? Antioxidants 2021, 10, 1316. [Google Scholar] [CrossRef]


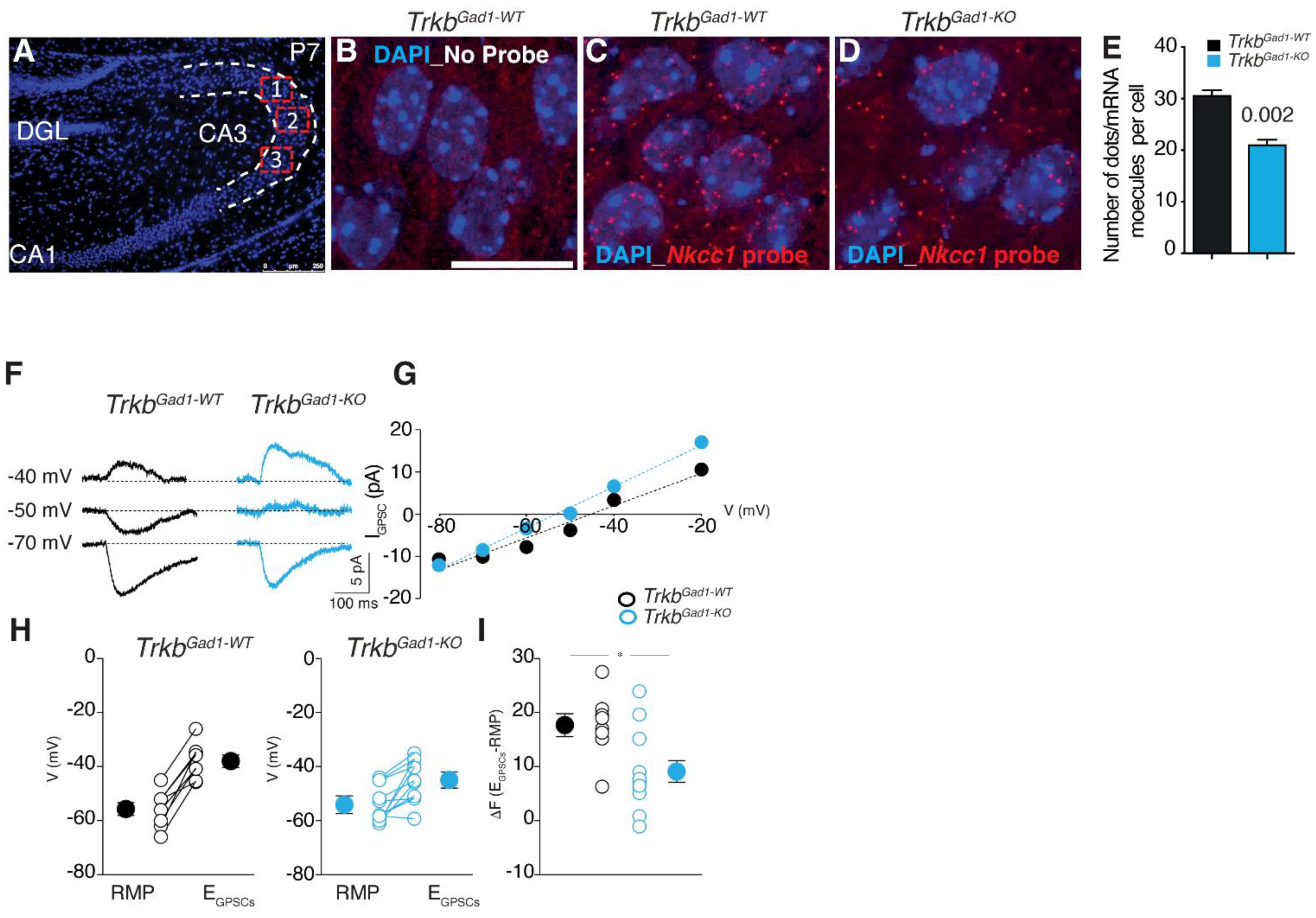{kind=link}
| Type of Mutation | Age | Effect of the Mutation on the Expression of SLC12A2 | Phenotype | Reference |
|---|---|---|---|---|
| 22 kb deletion affecting exons 2–7 of SLC12A2. chr5:127441491–127471419 | 5-year-old | complete absence of NKCC1 expression | multisystem disorder phenotype including intellectual disability | [47] |
| Biallelic loss-of-function variant in SLC12A2 c.2006-1G>A and c.1431delT | 2-month-old and 9-year-old | NKCC1 deficiency | multisystem disorder phenotype including intellectual disability | [48] |
| Biallelic loss-of-function variant in SLC12A2 c.940C>T, p.Q314* and c.1536+4_1536+7del, p.? | 1-year-old | NKCC1 deficiency | multisystem disorder phenotype including intellectual disability | [49] |
| de novo mutations, single nucleotide variants (SNVs): g.127450305C>T, p.A327V (exon 4); g.12746683A>T, p.N376I (exon 5); g.127420201dup, p.H186fs16 (exon 1); g.127469897G>A, p.R410Q (exon 6); g.127503511G>A, p.W892* (exon 18); g.127466845G>C, p.A379L (exon 5). | 1-year-old, 3-year-old, 6-year-old, 9-year-old, 15-year-old, 21-year-old | NKCC1 deficiency | Mild to severe intellectual disability or developmental delay. Some were diagnosed with autism and spastic paraparesis and some have had feeding difficulties | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymanski, J.; Minichiello, L. NKCC1 Deficiency in Forming Hippocampal Circuits Triggers Neurodevelopmental Disorder: Role of BDNF-TrkB Signalling. Brain Sci. 2022, 12, 502. https://doi.org/10.3390/brainsci12040502
Szymanski J, Minichiello L. NKCC1 Deficiency in Forming Hippocampal Circuits Triggers Neurodevelopmental Disorder: Role of BDNF-TrkB Signalling. Brain Sciences. 2022; 12(4):502. https://doi.org/10.3390/brainsci12040502
Chicago/Turabian StyleSzymanski, Jacek, and Liliana Minichiello. 2022. "NKCC1 Deficiency in Forming Hippocampal Circuits Triggers Neurodevelopmental Disorder: Role of BDNF-TrkB Signalling" Brain Sciences 12, no. 4: 502. https://doi.org/10.3390/brainsci12040502