
Understanding the Formation of Complex Phases: The Case of FeSi2
 and
and
Abstract
:1. Introduction
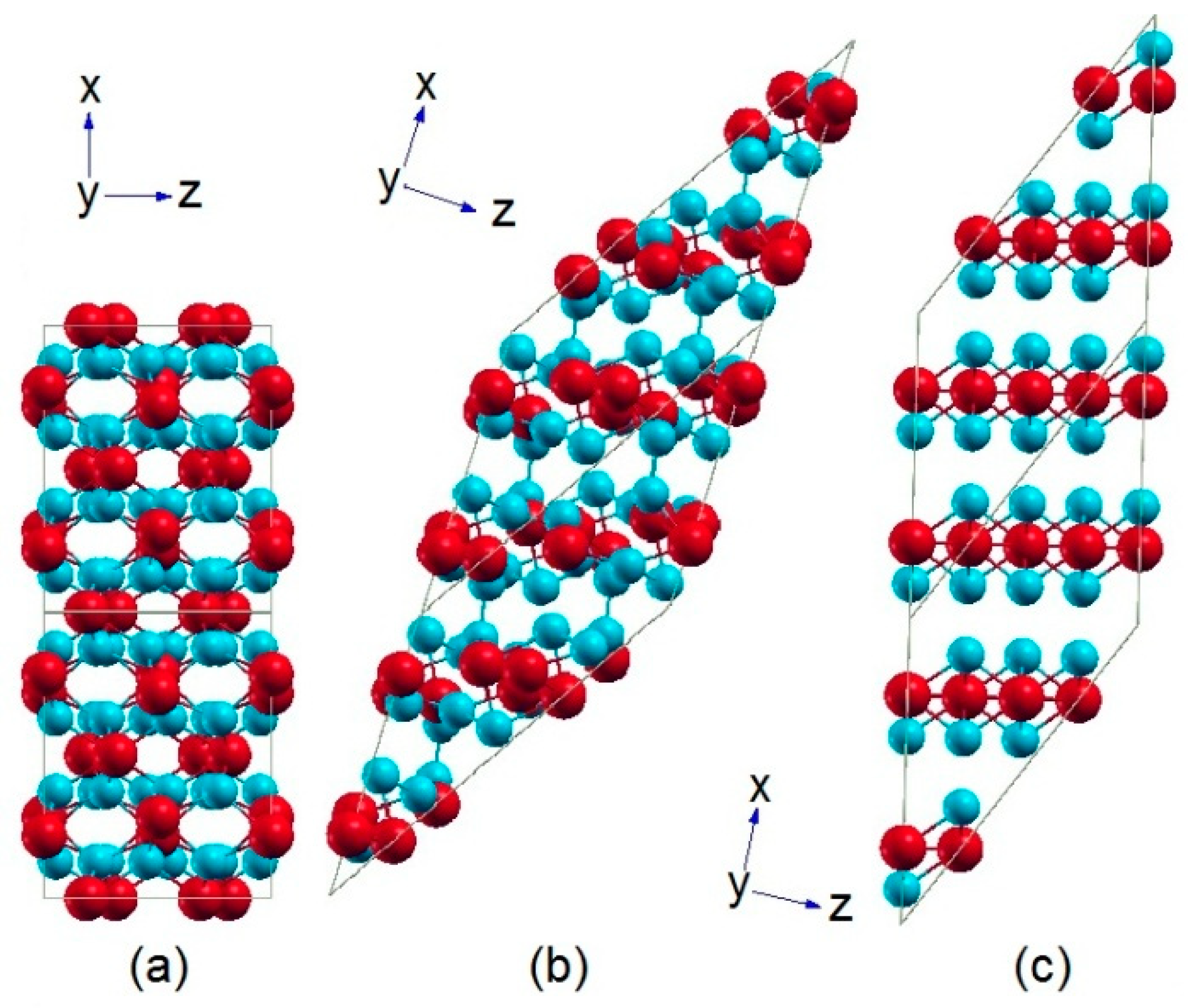
2. Crystallographic Examination and Relationships
- eutectoid: α-FeSi2 → β-FeSi2 + Si at 1233 K (960 °C)
- peritectoid: ε-FeSi + α-FeSi2 → β-FeSi2 at 1275.5 K (1002.5 °C)
- eutectic: liquid → α-FeSi2 + FeSi at 1479 K (1206 °C)
- eutectic: liquid → α-FeSi2 + Si at 1482 K (1209 °C)
- congruent melting: α-FeSi2 → liquid at 1484 K (1211 °C)
3. Computational Aspects
4. Results and Discussion
4.1. Ground State Analysis of the Ising Model
- Phase IV (bcc-based β-FeSi2 or C1) is found for J1 > 0, J2 < 0 and J1 > 0, J2 > 0, J1/J2 > 1/4;
- Phase V (bcc-based α-FeSi2) for J1 < 0, J2 > 0, 2J2/ < |J1| > 4J2 and J1 < 0, J2 > 0, |J1| > 2J2;
- Phase VI (bcc-based Θ-C16) for J1 > 0, J2 > 0, J1/J2 < 4.
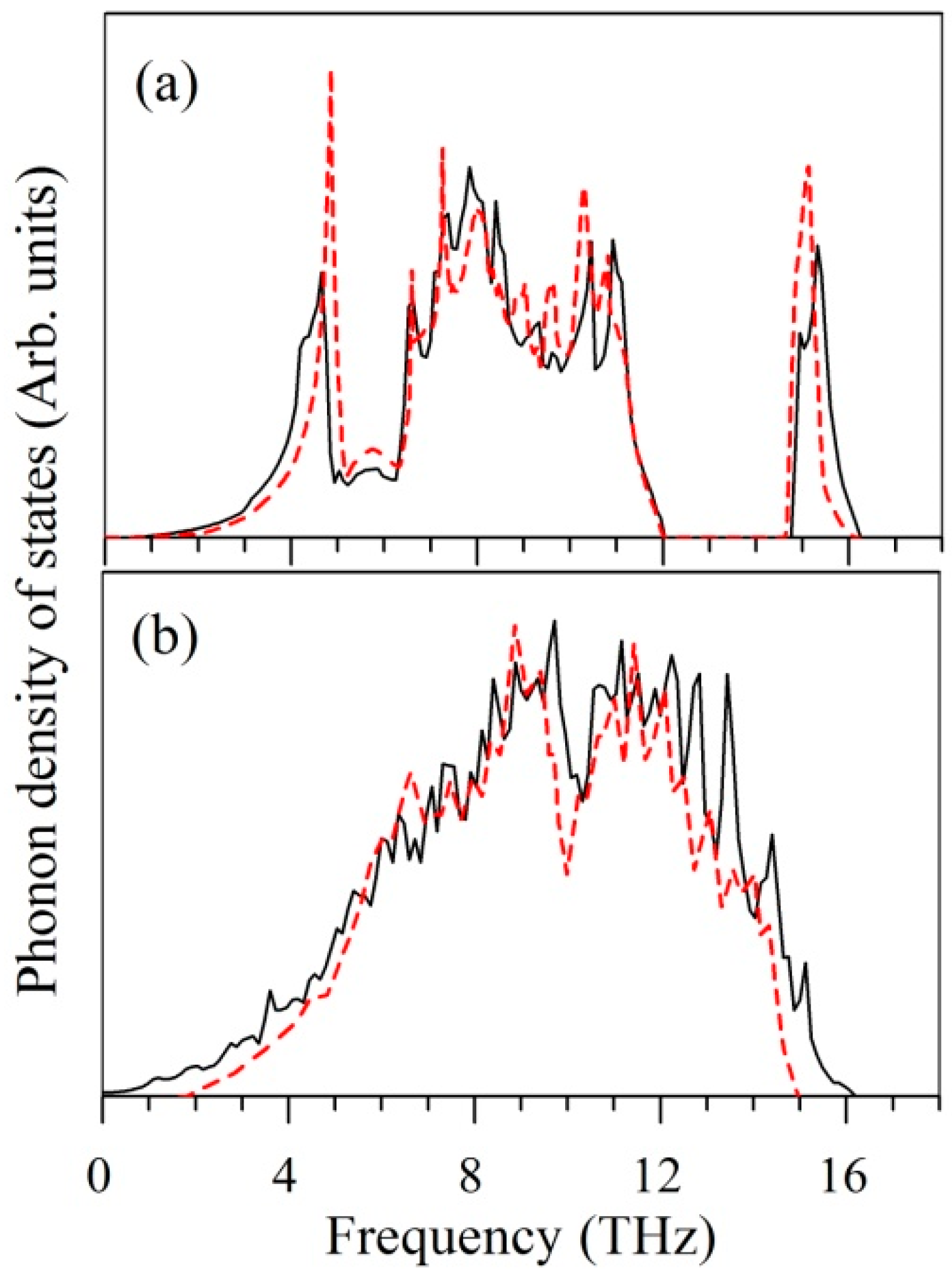
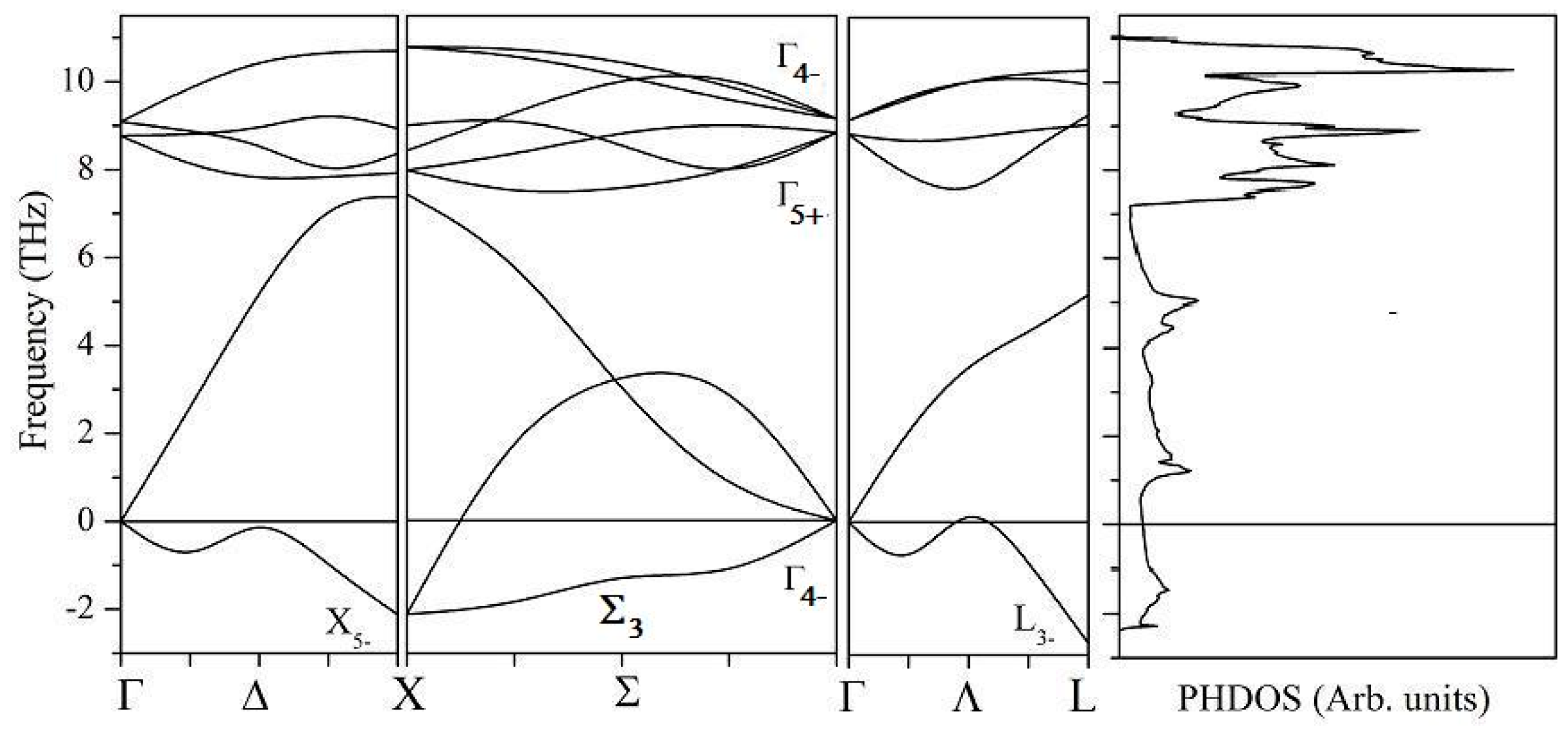
4.2. Structural Properties
4.3. Electronic Structural Properties
4.4. Mechanical Properties
4.5. Stability and Thermodynamic Properties
4.6. Temperature-Induced Phase Transformation in γ-FeSi2
4.7. Structural Transformations in β-FeSi2 and α-FeSi2
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyde, B.G.; Andersson, S. Inorganic Crystal Structures; John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Frank, F.C.; Kasper, J.S. Complex alloys structures regarded as sphere packings. I. Definitions and basic principles. Acta Cryst. 1958, 11, 184–190. [Google Scholar] [CrossRef]
- Frank, F.C.; Kasper, J.S. Complex alloys structures regarded as sphere packings. II. Classification of representative structures. Acta Cryst. 1959, 12, 483–499. [Google Scholar] [CrossRef]
- Hilbert, D. The Foundations of Geometry, 10th ed.; Open Court: La Salle, IL, USA, 1971. [Google Scholar]
- Sinha, A.K. Topologically close-packed structures of transition metal alloys. Prog. Mater. Sci. 1972, 15, 81–185. [Google Scholar] [CrossRef]
- Pearson, W.B. The Crystal Chemistry and Physics of Metals and Alloys; Wiley-Interscience: New York, NY, USA; London, UK; Sydney, Australia; Toronto, ON, Canada, 1972. [Google Scholar]
- Szeto, K.Y.; Villain, J. Filling three-dimensional space with tetrahedral: A geometric and crystallographic problem. Phys. Rev. 1987, 36, 4715–4724. [Google Scholar] [CrossRef] [PubMed]
- Damsceno, P.F.; Engel, M.; Glotzer, S.C. Predictive self-assembly of polyhedra into complex structures. Science 2012, 337, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Ansara, I.; Dupin, N.; Lukas, H.L.; Sundman, B. Thermodynamic assessment of the Al-Ni sytem. J. Alloys Compd. 1997, 247, 20–30. [Google Scholar] [CrossRef]
- Chen, H.L.; Du, Y.; Xu, H.; Xiong, W. Experimental investigation and thermodynamic modeling of the ternary Al-Cu-Fe system. J. Mater. Res. 2009, 24, 3154–3164. [Google Scholar] [CrossRef]
- Liang, S.M.; Schmid-Fetzer, R. Thermodynamic assessment of the Al-Cu-Zn system, part II: Al–Cu binary system. CALPHAD 2015, 51, 257–260. [Google Scholar] [CrossRef]
- Liang, S.M.; Hsiao, H.M.; Schmid-Fetzer, R. Thermodynamic assessment of the Al-Cu-Zn system, part I: Cu-Zn binary system. CALPHAD 2015, 51, 224–232. [Google Scholar] [CrossRef]
- Xiong, W.; Kong, Y.; Du, Y.; Liu, Z.K.; Selleby, M.; Sun, W.H. Thermodynamic investigation of the galvanizing systems, I: Refinement of the thermodynamic description for the Fe-Zn system. CALPHAD 2009, 33, 433–440. [Google Scholar] [CrossRef]
- Lacaze, J.; Sundman, B. An assessment of the Fe-C-Si system. Met. Trans. A 1991, 22, 2211–2223. [Google Scholar] [CrossRef]
- Onda, N.; Henz, J.; Müller, E.; Mäder, K.; von Känel, H. Epitaxy of fluorite-structure silicides: Metastable cubic FeSi2 on Si(111). Appl. Surf. Sci. 1992, 56-58, 421–426. [Google Scholar] [CrossRef]
- Desimoni, J.; Bernas, H.; Behar, M.; Lin, X.W.; Washburn, J.; Liliental-Weber, Z. Ion beam synthesis of cubic FeSi2. Appl. Phys. Lett. 1993, 62, 306–308. [Google Scholar] [CrossRef]
- Lin, X.W.; Washburn, J.; Liliental-Weber, Z.; Bernas, H. Coarsening and phase transition of FeSi2 precipitates in Si. J. Appl. Phys. 1994, 75, 4686–4694. [Google Scholar] [CrossRef]
- Le Corre, C.; Genin, J.M. Transformation Mechanisms of the α ↔ β Transition in FeSi2. Phys. Stat. Sol. 1972, 51, K85–K88. [Google Scholar] [CrossRef]
- Christensen, N.E. Electronic structure of β-FeSi2. Phys. Rev. B 1990, 42, 7148–7152. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Al-Allak, H.M.; Brand, S.; Abram, R.A. Structure and electronic properties of FeSi2. Phys. Rev. B 1998, 58, 10389–10393. [Google Scholar] [CrossRef]
- Moroni, E.G.; Wolf, W.; Hafner, J.; Podloucky, R. Cohesive, structural, and electronic properties of Fe-Si compounds. Phys. Rev. B 1999, 59, 12860–12871. [Google Scholar] [CrossRef]
- Tani, J.I.; Takahashi, M.; Kido, H. First-principles calculations of the structural and elastic properties of β-FeSi2 at high-pressure. Intermetallics 2010, 18, 1222–1227. [Google Scholar] [CrossRef]
- Mäder, K.A.; von Känel, H.; Baldereschi, A. Electronic structure and bonding in epitaxially stabilized cubic iron silicides. Phys. Rev. B 1993, 48, 4364–4372. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.F.; Shang, S.L.; Wang, J.; Wang, Y.; Ye, F.; Lin, J.P.; Liu, Z.K. First-principles calculations of phonon and thermodynamic properties of Fe-Si Compounds. Intermetallics 2011, 19, 1374–1384. [Google Scholar] [CrossRef]
- Malegori, G.; Miglio, L. Elastic properties of NiSi2, CoSi2, and FeSi2 by tight-binding calculations. Phys. Rev. B 1993, 48, 9223–9230. [Google Scholar] [CrossRef] [PubMed]
- Miglio, L.; Malegori, G. Origin and nature of the band gap in β -FeSi2. Phys. Rev. B 1995, 52, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, S.; Calegari, C.; Velasco, V.R.; Benedek, G.; Tavazza, F.; Miglio, L. Phonon mechanism for the orthorhombic distortion in FeSi2 as compared to cubic CoSi2. Phys. Rev. B 1996, 54, 9196–9203. [Google Scholar] [CrossRef] [PubMed]
- Miglio, L.; Meregalli, V.; Tavazza, F.; Celino, M. Analysis of the metal-semiconductor structural phase transition in FeSi2 by tight-binding molecular dynamics. Europhys. Lett. 1997, 37, 415–420. [Google Scholar] [CrossRef]
- Tavazza, F.; Meregalli, V.; Miglio, L.; Celino, M. Assessing the driving force of a structural distortion by the simulated evolution of the local density of states. Phys. Rev. B 1999, 59, 3480–3488. [Google Scholar] [CrossRef]
- Dusausoy, Y.; Protas, J.; Wandji, R.; Roques, B. Structure crystalline du disiliciure de fer, FeSi2β. Acta Cryst. 1971, 27, 1209–1218. [Google Scholar] [CrossRef]
- Daams, J.L.C.; Villars, P.; van Vucht, J.H.N. Atlas of Crstal Structure Types for Intermetallic Phases; ASM International: Materials Park, OH, USA, 1991. [Google Scholar]
- Sidorenko, F.A.; Gel’d, P.V.; Dubrovskaya, L.B. Nature of thee phase of the Fe-Si system. Fizika Metallo I Metalloved. 1959, 8, 735. [Google Scholar]
- Sidorenko, F.A.; Gel’d, P.V.; Shumilov, M.A. Study of the transformations of a-lebeauite. Fizika Metallo I Metalloved. 1960, 9, 861–867. [Google Scholar]
- Meetsma, A.; de Boer, J.L.; van Smaalen, S. Refinement of the crystal structure of tetragonal Al2Cu. J. Solid State Chem. 1989, 83, 370–372. [Google Scholar] [CrossRef]
- Grin, Y.; Wagner, F.R.; Armbrüster, M.; Kohout, M.; Leithe-Jasper, A.; Schwarz, U.; von Schnering, H.G. CuAl2 revisited: Composition, crystal structure, chemical bonding, compressibility and Raman spectroscopy. J. Solid State Chem. 2006, 179, 1707–1719. [Google Scholar] [CrossRef]
- Nishiyama, Z. Martensitic Transformations; Fine, M., Meshi, M., Wayman, C., Eds.; Academic Press: New York, NY, USA, 1978. [Google Scholar]
- Cuenya, B.R.; Doi, M.; Löbus, S.; Courths, R.; Keune, W. Observation of the fcc-to-bcc Bain transformation in epitaxial Fe ultrafine films on Cu3Au (001). Surf. Sci. 2001, 493, 338–360. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Billeter, S.R.; Curioni, A.; Andreoni, W. Efficient linear scaling geometry optimization and transition-state search for direct wavefunction optimization schemes in density functional theory using a plane-wave basis. Comput. Mater. Sci. 2003, 27, 437–445. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Phonopy. Available online: http://atztogo.github.io/phonopy/ (accessed on 10 July 2019).
- Golesorkhtabar, R.; Pavone, P.; Spitaler, J.; Puschnig, P.; Draxl, C. ElaStic: A tool for calculating second-order elastic constants from first-principles. Comput. Phys. Commun. 2013, 184, 1861–1873. [Google Scholar] [CrossRef]
- Niu, H.; Niu, S.; Oganov, A.R. Simple and accurate model of fracture toughness of solids. J. Appl. Phys. 2019, 125, 065105. [Google Scholar] [CrossRef]
- Stokes, H.T.; Hatch, D.M. ISOTROPY software package. Available online: http://stokes.byu.edu/isotropy.html (accessed on 10 June 2019).
- Ivashchenko, V.I.; E A Turchi, P.; Gorb, L.; Leszczynski, J.; Medukh, N.R.; Shevchenko, R. Temperature-induced phase transitions in the rock-salt type SiC: A first-principles study. J. Physics: Condens. Matter 2019, 31, 405401. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A. Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Comput. Mater. Sci. 2003, 28, 155–168. [Google Scholar] [CrossRef]
- Ivashchenko, V.I.; Turchi, P.E.A. Phonon softening and the phase transition in VN. Phys. Rev. B 2008, 78, 224113. [Google Scholar] [CrossRef]
- Landesman, J.; Treglia, G.; Turchi, P.; Ducastelle, F. Electronic structure and pairwise interactions in substoichiometric transition metal carbides and nitrides. J. Phys. 1985, 46, 1001–1015. [Google Scholar] [CrossRef]
- Ducastelle, F. Order and Stability in Alloys. In Cohesion and Structure; de Boer, F.R., Pettifor, D.G., Eds.; North-Holland Publishing Company: Amsterdam, The Netherlands; Oxford, UK; New York, NY, USA; Tokyo, Japan, 1991. [Google Scholar]
- Bieber, A.; Ducastelle, F.; Gautier, F.; Tréglia, G.; Turchi, P. Electronic structure and relative stabilities of L12 and D022 ordered structures occurring in transition metal alloys. Solid State Commun. 1983, 45, 585–590. [Google Scholar] [CrossRef]
- Kanamori, J. Magnetization process in an Ising system. Prog. Theor. Phys. 1966, 35, 16–35. [Google Scholar] [CrossRef]
- Kaburagi, M.; Kanamori, J. A Method of Determining the Ground State of the Extended-Range Classical Lattice Gas Model. Prog. Theor. Phys. 1975, 54, 30–44. [Google Scholar] [CrossRef]
- Villars, P.; Calvert, L.D. Pearson’s Handbook of Crystallographic Data for Intermetallic Phases; American Society for Metals: Materials Park, OH, USA, 1985. [Google Scholar]
- Kubaschewski, O. Phase Diagrams of Binary Iron Alloys; Okamoto, H., Ed.; ASM International: Materials Park, OH, USA, 1993; p. 380. [Google Scholar]
- Hong, S.J.; Rhee, C.K.; Chun, B.S. Phase Transition and Thermoelectric Property of Ultra-fine Structured β-FeSi2 Compounds. Solid State Phenom. 2006, 118, 591–596. [Google Scholar] [CrossRef]
- Birkholz, C.; Schelm, J. Electrical investigation of the semi-conductor-to metal transition in FeSi2. Phys. Stat. Sol. 1969, 34, K177–K180. [Google Scholar] [CrossRef]
- Milekhinea, V.; Onsøienb, M.I.; Solberga, J.K.; Skaland, T. Mechanical properties of FeSi (ε), FeSi2 (ζα) and Mg2Si. Intermetallics 2002, 10, 743–750. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Krentsis, R.P.; Gel’d, P.V.S. Nauchn. Tr., Ural’sk. Preliminary Report on the Thermodynamic Properties of Selected Light-Element and Some Related Compounds. Available online: https://books.google.rs/books?id=RTxRAAAAMAAJ&pg=PA423&lpg=PA423&dq=Krentsis,+R.+P.;+Gel%E2%80%99d,+P.+V.+Sb.+Nauchn.+Tr.,+Ural%E2%80%99sk.+Politekhn&source=bl&ots=2-ij12RiZ2&sig=ACfU3U13Ju-1CNCu7rRV5AQOhD2mGvTzRQ&hl=en&sa=X&ved=2ahUKEwjW--LZoNyCAxWbgf0HHQprBDsQ6AF6BAgIEAM#v=onepage&q=Krentsis%2C%20R.%20P.%3B%20Gel%E2%80%99d%2C%20P.%20V.%20Sb.%20Nauchn.%20Tr.%2C%20Ural%E2%80%99sk.%20Politekhn&f=false (accessed on 10 June 2019).
- Maglic, K.; Parrot, J.E. Bundesministerium fur Bildung und Wissenschaft. Forschungsbericht 1970, K70-01. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | xC | yC | zC | xA | yA | zA | |
|---|---|---|---|---|---|---|---|
| Fe1 | 0.2146 | 0 | 0 | 1/4 | 0 | 0 | |
| Fe, 8d | Fe1 | 0.7854 | 0 | 0 | 3/4 | 0 | 0 |
| Fe1 | 0.7146 | 0 | 1/2 | 3/4 | 0 | 1/2 | |
| Fe1 | 0.2854 | 0 | 1/2 | 1/4 | 0 | 1/2 | |
| Fe2 | 1/2 | 0.3086 | 0.1851 | 1/2 | 1/4 | 1/4 | |
| Fe, 8f | Fe2 | 0 | 0.3086 | 0.3149 | 0 | 1/4 | 1/4 |
| Fe2 | 1/2 | 0.1914 | 0.6851 | 1/2 | 1/4 | 1/4 | |
| Fe2 | 0 | 0.1914 | 0.8149 | 0 | 1/4 | 3/4 | |
| Si1 | 0.1282 | 0.2746 | 0.0512 | 1/8 | 1/4 | 0 | |
| Si1 | 0.8718 | 0.2746 | 0.0512 | 7/8 | 1/4 | 0 | |
| Si1 | 0.6282 | 0.2746 | 0.4488 | 5/8 | 1/4 | 1/2 | |
| Si, 16g | Si1 | 0.3718 | 0.2746 | 0.4488 | 3/8 | 1/4 | 1/2 |
| Si1 | 0.1282 | 0.2254 | 0.0512 | 1/8 | 1/4 | 1/2 | |
| Si1 | 0.8718 | 0.2254 | 0.0512 | 7/8 | 1/4 | 1/2 | |
| Si1 | 0.6282 | 0.2254 | 0.9488 | 5/8 | 1/4 | 1 | |
| Si1 | 0.3718 | 0.2254 | 0.9488 | 3/8 | 1/4 | 1 | |
| Si2 | 0.3727 | 0.0450 | 0.2261 | 3/8 | 0 | 1/4 | |
| Si2 | 0.6273 | 0.0450 | 0.2261 | 3/8 | 0 | 1/4 | |
| Si2 | 0.8727 | 0.0450 | 0.2739 | 7/8 | 0 | 1/4 | |
| Si, 16g | Si2 | 0.1273 | 0.0450 | 0.2739 | 1/8 | 0 | 1/4 |
| Si2 | 0.6273 | 0.04550 | 0.7261 | 3/8 | 1/2 | 3/4 | |
| Si2 | 0.3727 | 0.04550 | 0.7261 | 3/8 | 1/2 | 3/4 | |
| Si2 | 0.1273 | 0.04550 | 0.7739 | 1/8 | 1/2 | 3/4 | |
| Si2 | 0.8727 | 0.04550 | 0.7739 | 7/8 | 1/2 | 3/4 |
| Site | xC | yC | zC | xA | yA | zA |
|---|---|---|---|---|---|---|
| Fe | 0 | 0 | 0 | 0 | 0 | 0 |
| Si | 1/2 | 1/2 | 0.280 | 1/2 | 1/2 | 1/4 |
| 1/2 | 1/2 | 0.720 | 0 | 0 | 3/4 |
| Site | xC | yC | zC | xA | yA | zA |
|---|---|---|---|---|---|---|
| Cu | 0 | 0 | 0 | 0 | 0 | 0 |
| Cu | 1/2 | 1/2 | 0 | 1/2 | 1/2 | 0 |
| Cu | 1/2 | 1/2 | 1/2 | 1/2 | 1/2 | 1/2 |
| Cu | 0 | 0 | 1/2 | 0 | 0 | 1/2 |
| Al | α | ½ − α | 1/4 | 1/4 | 1/4 | 1/4 |
| Al | ½ − α | 1/2 + α | 1/4 | 1/4 | 3/4 | 1/4 |
| Al | ½ − α | α | 1/4 | 1/4 | 1/4 | 1/4 |
| Al | α | 1/2 + α | 3/4 | 1/4 | 3/4 | 3/4 |
| Al | 1 − α | ½ − α | 3/4 | 3/4 | 1/4 | 3/4 |
| Al | 1/2 + α | 1 − α | 3/4 | 3/4 | 3/4 | 3/4 |
| - | - | - | 1/2 | 0 | 0 | |
| - | - | - | 0 | 1/2 | 0 | |
| - | - | - | 1/2 | 0 | 1/2 | |
| - | - | - | 0 | 1/2 | 1/2 |
| Structure | Na | Symmetry | a (Å) | b (Å) | c (Å) | V (Å3/atom) | ΔET (eV/atom) |
|---|---|---|---|---|---|---|---|
| 9.874 | 7.769 | 7.819 | 12.495 | ||||
| β-FeSi2 | 24 | Cmca, #64 | (9.863) a | (7.791) a | (7.833) a | (12.540) a | 0.0000 |
| (9.869) b | (7.774) b | (7.846) b | (12.540) b | ||||
| β-1400 | 12 | C2/m, #12 | 9.612 | 7.669 | 4.068 | 12.247 | 0.0357 |
| β-1800 | 6 | C2/m, #12 | 6.889 | 3.834 | 6.520 | 12.584 | 0.1064 |
| 2.705 | 2.705 | 5.143 | 12.541 | ||||
| α-FeSi2 | 3 | P4/mmm, | (2.690) c | (2.690) c | (5.130) c | 12.374 | 0.0564 |
| #123 | (2.710) d | (2.710) d | (5.140) d | 12.583 | |||
| α-Fe0.8Si2 | 45 | P1, #1 | 2.680 | 2.680 | 5.129 | 12.280 * | - |
| γ-FeSi2 | 3 | Fm3m, #225 | 5.400 | 5.400 | 5.400 | 13.122 | 0.1743 |
| (5.400) d | (5.400) d | (5.400) d | (13.122) d |
| Structure | B | G | E | σ | KIC |
|---|---|---|---|---|---|
| β-FeSi2 | 180.8 | 128.1 | 310.9 | 0.21 | 2.3 |
| (172.8) a | (130.1) a | (312.0) a | (0.20) a | ||
| α-FeSi2 | 177.0 | 129.4 | 312.1 | 0.21 | 2.3 |
| (172) b | (2.5) d | ||||
| γ-FeSi2 | 173.5 | ||||
| (167) b | 29.8 | 84.6 | 0.42 | 1.1 | |
| (206) c |
| Phase, Temperature (K) | a, b, c (Å) | α β, γ (Degree) | Wyckoff Position |
|---|---|---|---|
| γ | 5.400 5.400 5.400 | 90.00 90.00 90.00 | Fe 1a 0.000 0.000 0.000 |
| T < 100 | Si 2c 0.250 0.250 0.250 | ||
| γ1 | 3.319 3.523 3.552 | 94.56 116.79 116.60 | Fe 1h 0.500 0.500 0.500 |
| 100 ≤ T < 200 | Si 1i 0.003 0.776 0.230 | ||
| γ2 | 4.731 5.268 3.234 | 90.00 129.854 90.00 | Fe 2d 0.000 0.500 0.500 |
| 200 ≤ T < 400 | Si 4g 0.000 0.226 0.000 | ||
| γ3 | Fe 4i 0.186 0.000 0.726 | ||
| 400 ≤ T ≤ 800 | 6.333 3.742 5.974 | 90.00 118.44 90.00 | Si 4i 0.598 0.000 0.875 |
| Si 4i 0.827 0.000 0.643 | |||
| Fe 4i 0.243 0.000 0.881 | |||
| γ4, γ5 | Fe 4h 0.000 0.783 0.500 | ||
| T > 800 K | 7.125 9.342 3.756 | 90.00 100.44 90.00 | Si 8j 0.278 0.126 0.414 |
| Si 4g 0.000 0.119 0.000 | |||
| Si 4g 0.000 0.634 0.000 |
| Phonon Mode | Γ5+ | L3− | |||
|---|---|---|---|---|---|
| Phase | γ1 | γ2 | γ3 | γ4 | γ5 |
| P-1, #2 | C2/m, #12 | C2/m, #12 | C2/m, #12 | C2/m, #12 | |
| Na | 1 | 1 | 6 | 12 | 12 |
| P | S1(a,b,c) | C2(a,a,b) | P2(a,a,000000) | C12(a,b,a,b,0,0,0,0) | C18(a,b,b,a,0,0,0,0) |
| Strain | e0, e1, e2, | e0, e1, | e0, e1, e2, | e0, e1, e2, εxy | e0, e1, −εxz + εyz, −εxy |
| εxy, εyz, εxz | εxz + εyz, εxy | εxz + εyz, εxy | |||
| Phase, Symmetry | a, b, c (Å) | α, β, γ (Degree) | Wyckoff Position |
|---|---|---|---|
| Fe 8d 0.716 0.000 0.000 | |||
| γ | 9.874. 7.769. 7.818 | 90.00, 90.00, 90.00 | Fe 8f 0.000 0.193 0.313 |
| CmCa | Si 16g 0.128 0.274 0.550 | ||
| Si 16g 0.373 0.448 0.727 | |||
| Fe 4g 0.000 0.243 0.000 | |||
| β-1400 | Fe 4i 0.309 0.000 0.646 | ||
| C2/m | 9.612. 7.669. 4.068 | 90.00, 101.33, 90.00 | Si 8j 0.630 0.287 0.546 |
| Si 4i 0.421 0.000 0.203 | |||
| Si 4i 0.149 0.000 0.059 | |||
| β-1800 | Fe 4i 0.188 0.000 0.721 | ||
| C2/m | 6.889. 3.834. 6.520 | 90.00, 118.70, 90.00 | Si 4i 0.405 0.000 0.126 |
| Si 4i 0.830 0.000 0.644 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turchi, P.E.A.; Ivashchenko, V.I.; Shevchenko, V.I.; Gorb, L.; Leszczynski, J.; Perron, A. Understanding the Formation of Complex Phases: The Case of FeSi2. Appl. Sci. 2023, 13, 12669. https://doi.org/10.3390/app132312669
Turchi PEA, Ivashchenko VI, Shevchenko VI, Gorb L, Leszczynski J, Perron A. Understanding the Formation of Complex Phases: The Case of FeSi2. Applied Sciences. 2023; 13(23):12669. https://doi.org/10.3390/app132312669
Chicago/Turabian StyleTurchi, Patrice E. A., Volodymyr I. Ivashchenko, V. I. Shevchenko, Leonid Gorb, Jerzy Leszczynski, and Aurélien Perron. 2023. "Understanding the Formation of Complex Phases: The Case of FeSi2" Applied Sciences 13, no. 23: 12669. https://doi.org/10.3390/app132312669