Theoretical Evaluation of Multi-Breed Genomic Prediction in Chinese Indigenous Cattle

Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Animals and Genotype Quality Control
2.2. Simulation of Genotypes
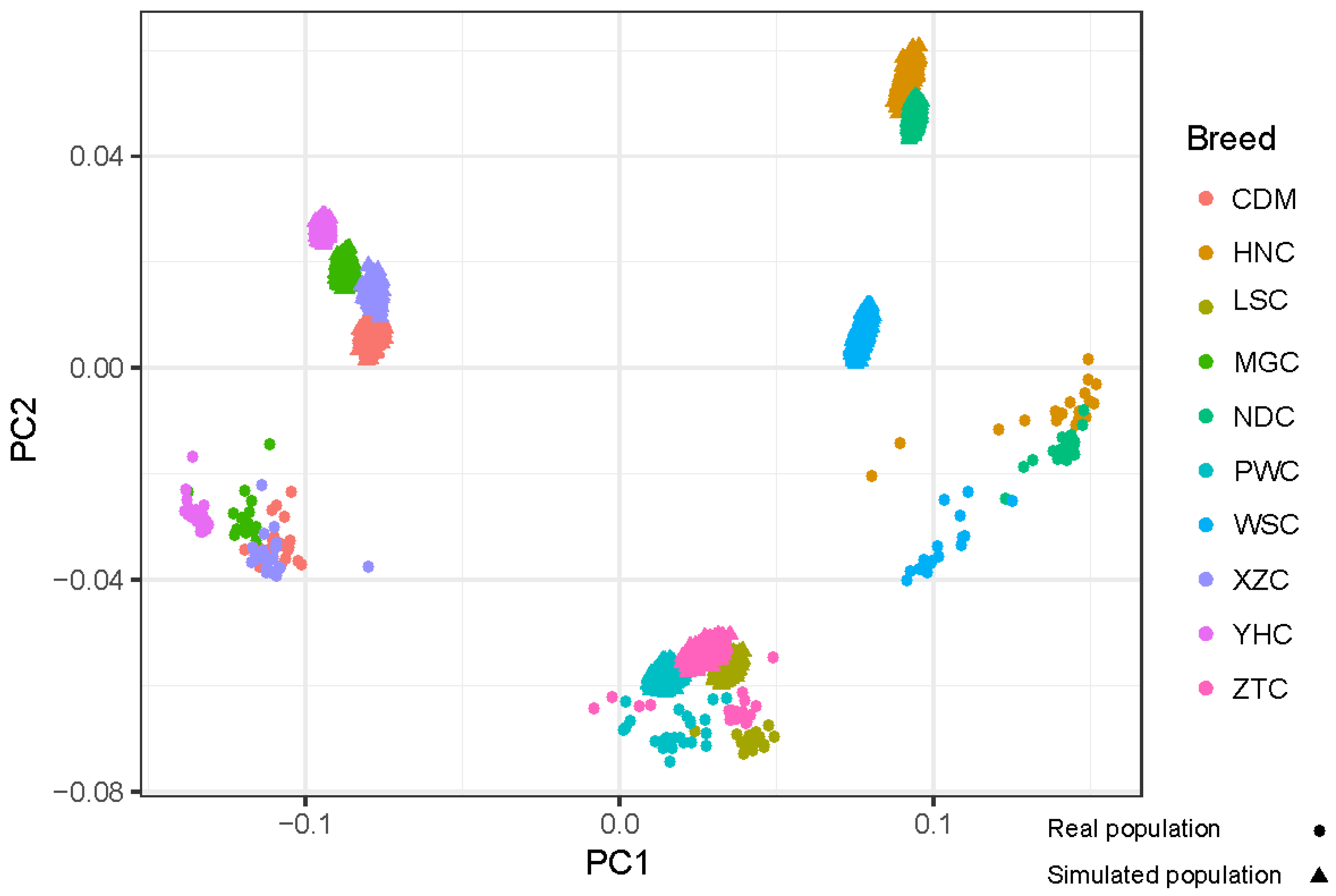
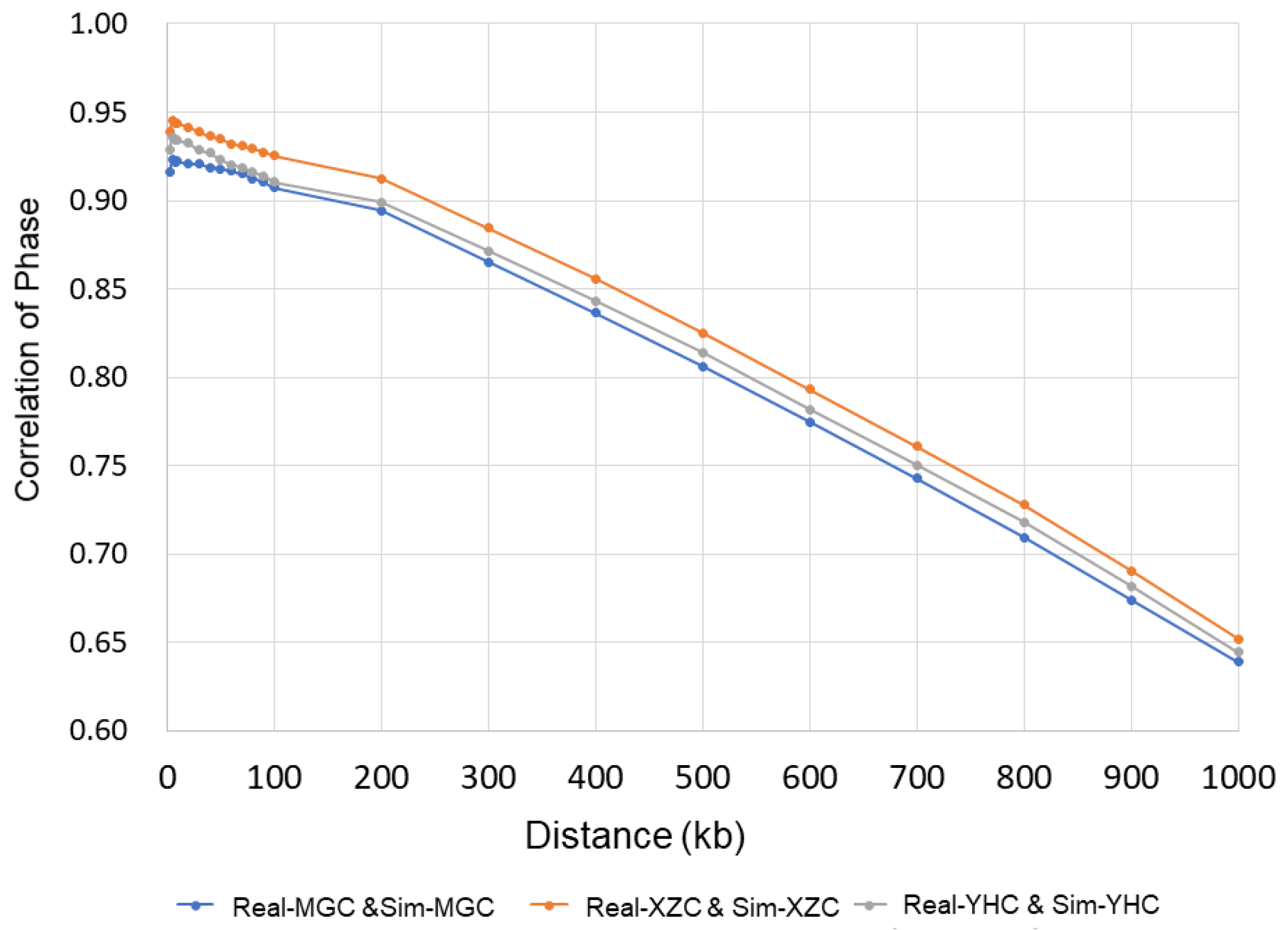
2.3. Principal Component Analysis and Persistence of Allele Phase
2.4. Simulation
2.5. Genomic Evaluation
2.6. Reference and Validation Populations
2.7. Accuracy of Genomic Prediction
3. Results
3.1. Simulation of Genotype
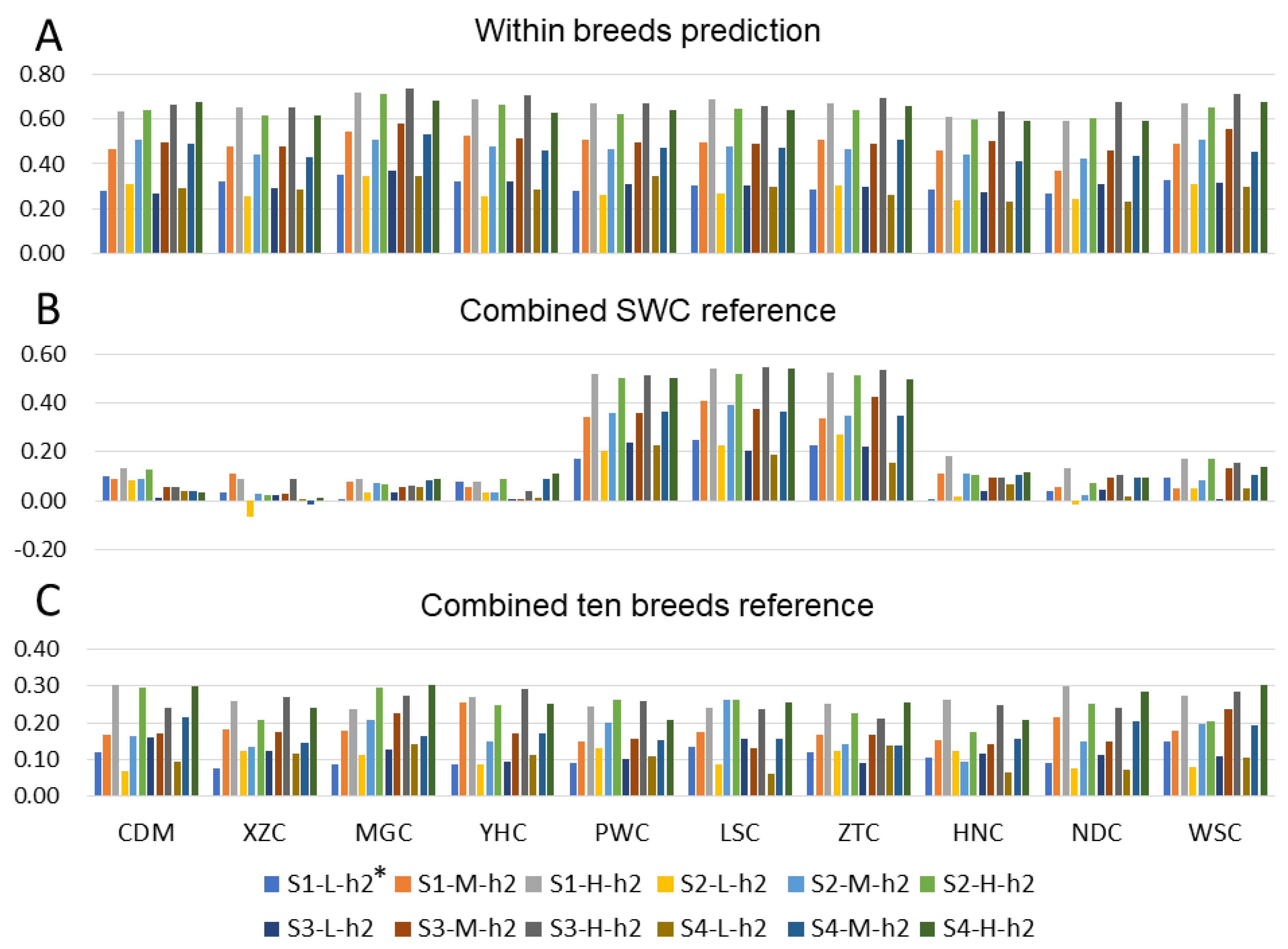
3.2. Prediction with Different Reference Population
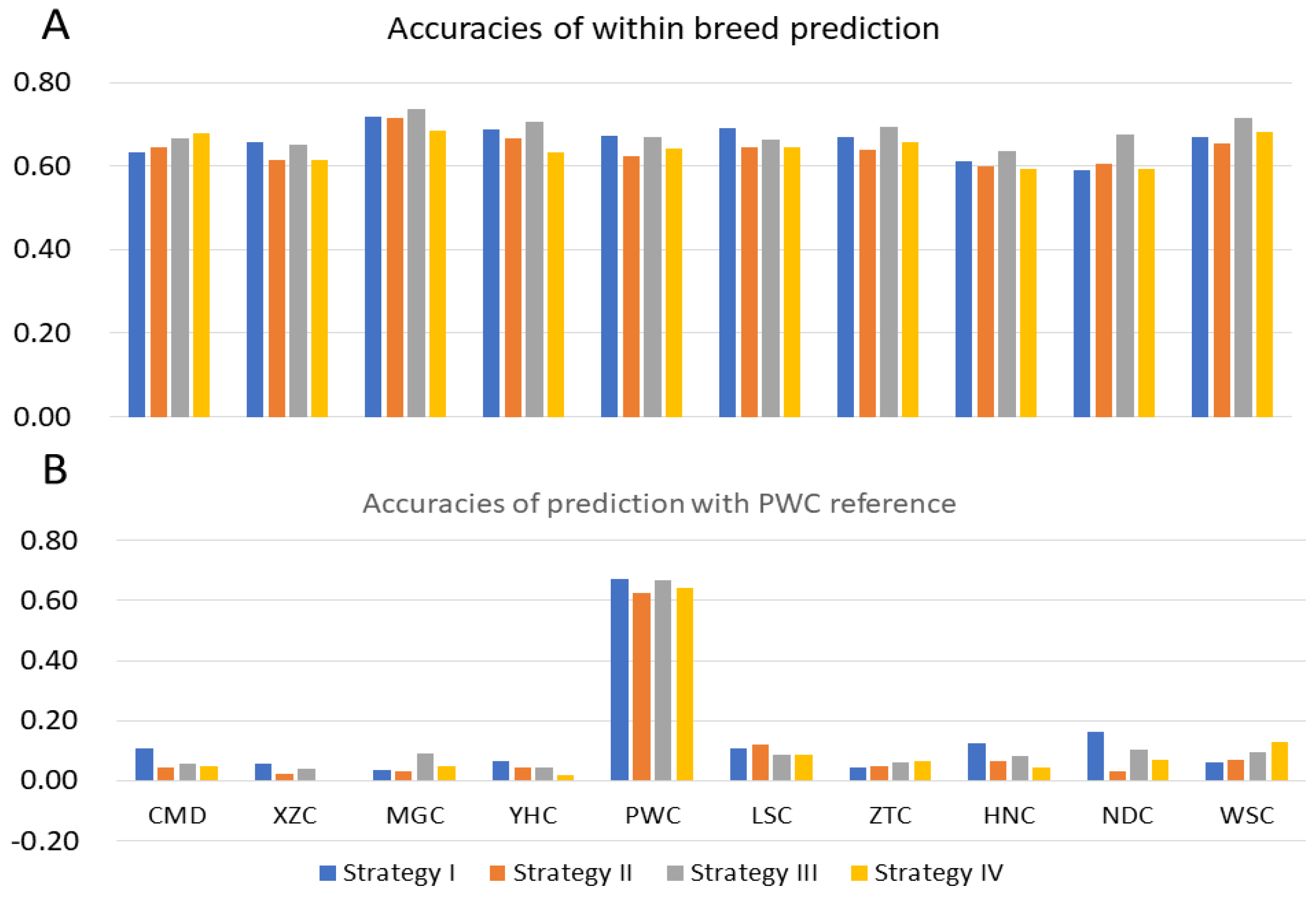
3.2.1. Prediction with Single Breed Reference
3.2.2. Admixed Breeds Reference
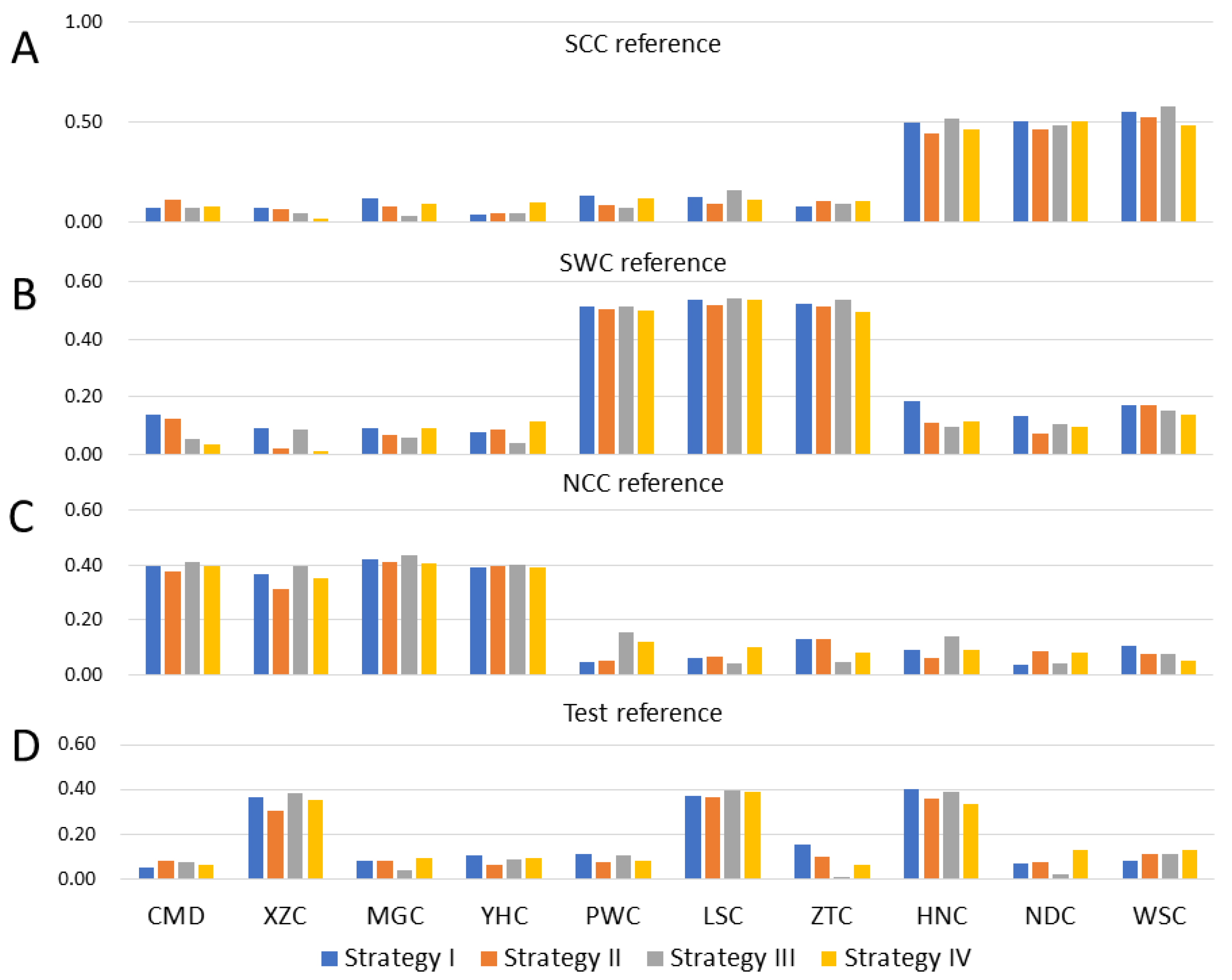
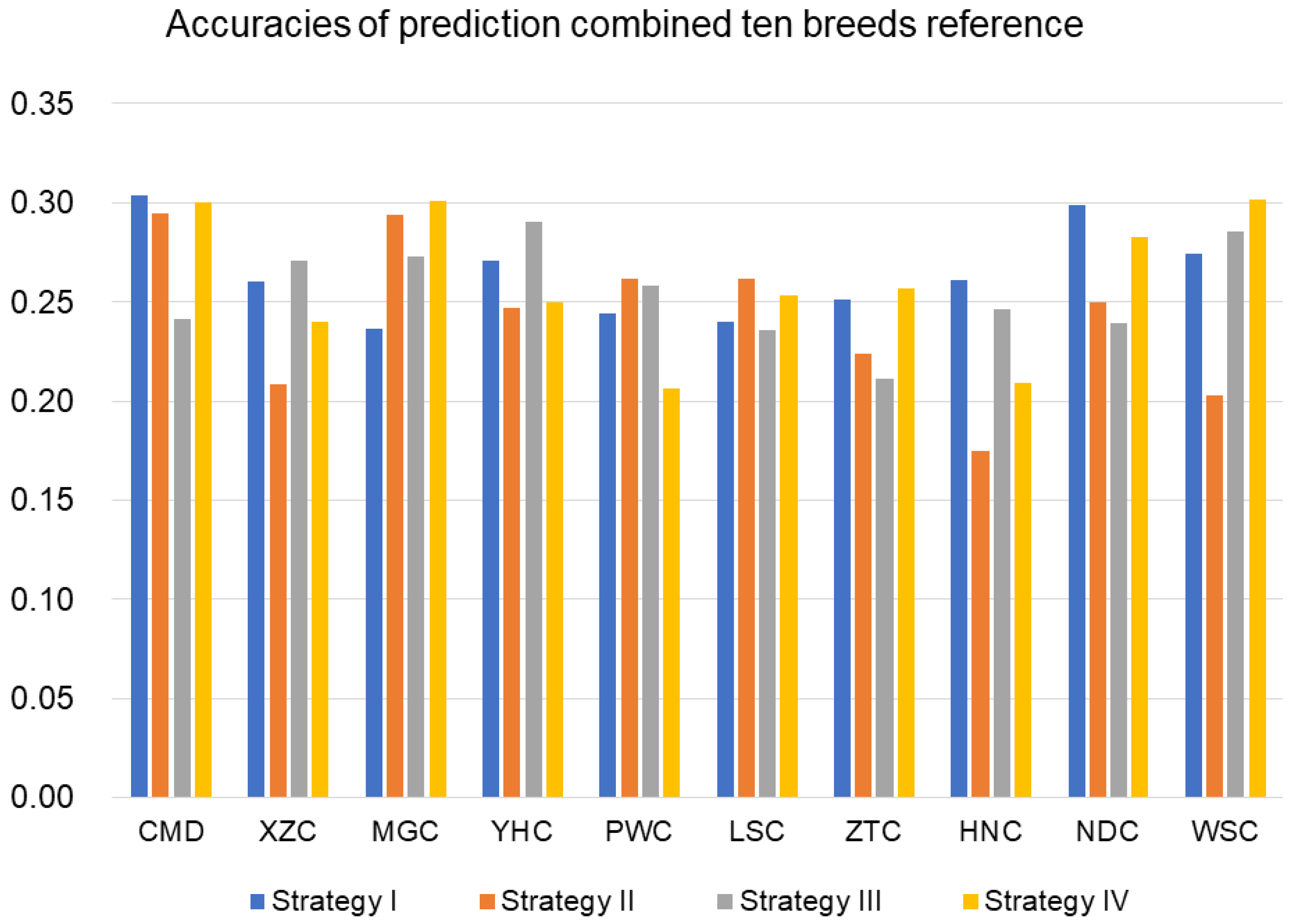
3.2.3. Prediction with Combined-10-Breeds Reference
3.3. Effect of Heritability
3.4. Effect of Genetic Architecture
4. Discussion
4.1. Simulation of Genotype and Phenotype
4.2. Predictive Accuracies from Admixed Population
4.3. Effect of Heritability and Genetic Architecture
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GEBV | Genomic estimated breeding values |
| QTL | Quantitative trait loci |
| SNP | Single nucleotide polymorphism |
| GBLUP | Genomic best linear unbiased prediction |
| GRM | Genomic relationship matrix |
| TBV | True breeding values |
| MCMC | Markov Chain Monte Carlo |
| PCA | Principal Component Analysis |
| MAF | Minor allele frequency |
| GS | Genomic selection |
| GWAS | Genome-wide association study |
| LD | Linkage disequilibrium |
| Ne | Effective population size |
| QC | Quality control |
| SD | Standard deviation |
| pi-hat | Proportion identity by descent |
| MGC | Mongolia cattle |
| YHC | Yanhuang cattle |
| CDM | Caidamu cattle |
| XZC | Xizang cattle |
| PWC | Pingwu cattle |
| LSC | Liangshan cattle |
| ZTC | Zhaotong cattle |
| WSC | Wenshan cattle |
| HNC | Hannan cattle |
| NDC | Nandan cattle |
| NCC | Including north Chinese cattle group (contains CDM, YHC, MGC and XZC) |
| SWC | Southwest Chinese cattle group (contains LSC, PWC and ZTC) |
| SCHC | South Chinese cattle group (contains HNC, NDC and WSC) |
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Schaeffer, L.R. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 2006, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Daetwyler, H.D.; Villanueva, B.; Woolliams, J.A. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 2008, 3, e3395. [Google Scholar] [CrossRef] [PubMed]
- de Roos, A.P.W.; Hayes, B.J.; Goddard, M.E. Reliability of Genomic Predictions Across Multiple Populations. Genetics 2009, 183, 1545–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, M.S.; De Roos, A.P.; De Vries, A.G.; Druet, T.; Ducrocq, V.; Fritz, S.; Guillaume, F.; Guldbrandtsen, B.; Liu, Z.; Reents, R.; et al. A common reference population from four European Holstein populations increases reliability of genomic predictions. Genet. Sel. Evol. 2011. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.R.; Sonstegard, T.S.; Schnabel, R.D.; Taylor, J.F.; Schenkel, F.S. Invited Review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Thomasen, J.R.; Egger-Danner, C.; Willam, A.; Guldbrandtsen, B.; Lund, M.S.; Sørensen, A.C. Genomic selection strategies in a small dairy cattle population evaluated for genetic gain and profit. J. Dairy Sci. 2014. [Google Scholar] [CrossRef]
- Thomasen, J.R.; Sørensen, A.C.; Lund, M.S.; Guldbrandtsen, B. Adding cows to the reference population makes a small dairy population competitive. J. Dairy Sci. 2014, 97, 5822–5832. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.H.; Rekaya, R. A multi-compartment model for genomic selection in multi-breed populations. Livest. Sci. 2015, 177, 1–7. [Google Scholar] [CrossRef]
- Toosi, A.; Fernando, R.L.; Dekkers, J.C.M. Genomic selection in admixed and crossbred populations. J. Anim. Sci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Hozé, C.; Fritz, S.; Phocas, F.; Boichard, D.; Ducrocq, V.; Croiseau, P. Efficiency of multi-breed genomic selection for dairy cattle breeds with different sizes of reference population. J. Dairy Sci. 2014, 97, 3918–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jónás, D.; Ducrocq, V.; Fritz, S.; Baur, A.; Sanchez, M.-P.; Croiseau, P. Genomic evaluation of regional dairy cattle breeds in single-breed and multibreed contexts. J. Anim. Breed. Genet. 2017, 134, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Daetwyler, H.D.; Swan, A.A.; Van Der Werf, J.H.; Hayes, B.J. Accuracy of pedigree and genomic predictions of carcass and novel meat quality traits in multi-breed sheep data assessed by cross-validation. Genet. Sel. Evol. 2012, 44. [Google Scholar] [CrossRef] [PubMed]
- Wientjes, Y.C.J.J.; Veerkamp, R.F.; Calus, M.P.L.L. The effect of linkage disequilibrium and family relationships on the reliability of genomic prediction. Genetics 2013, 193, 621–631. [Google Scholar] [CrossRef]
- Lambert, B.W.; Terwilliger, J.D.; Weiss, K.M. ForSim: A tool for exploring the genetic architecture of complex traits with controlled truth. Bioinformatics 2008, 24, 1821–1822. [Google Scholar] [CrossRef]
- Zhe, Z.; Xiangdong, D.; Jianfeng, L.; Guiyan, N.; Jiaqi, L.; Qin, Z.; Xiangdong, D.; Jianfeng, L.; Guiyan, N.; Jiaqi, L.; et al. Whole-Genome Genetic Data Simulation Based on Mutation-Drift Equilibrium Model. In Proceedings of the 2012 4th International Conference on Computer Modeling and Simulatio, Hong Kong, China, 17–18 February 2012; Volume 22, pp. 87–93. [Google Scholar]
- Peng, B.; Kimmel, M. simuPOP: A forward-time population genetics simulation environment. Bioinformatics 2005. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Schenkel, F.S. QMSim: A large-scale genome simulator for livestock. Bioinformatics 2009. [Google Scholar] [CrossRef]
- Hudson, R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 2002. [Google Scholar] [CrossRef]
- Liang, L.; Zöllner, S.; Abecasis, G.R. GENOME: A rapid coalescent-based whole genome simulator. Bioinformatics 2007. [Google Scholar] [CrossRef]
- Palamara, P.F. ARGON: Fast, whole-genome simulation of the discrete time Wright-fisher process. Bioinformatics 2016, 32, 3032–3034. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Umbach, D.M.; Wise, A.S.; Weinberg, C.R. Simulating autosomal genotypes with realistic linkage disequilibrium and a spiked-in genetic effect. BMC Bioinformatics 2018, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yu, G.; Langefeld, C.; Miller, D.; Guy, R.; Raghuram, J.; Yuan, X.; Herrington, D.; Wang, Y. Comparative Analysis of Methods for Detecting Interacting Loci. BMC Genomics 2011, 12, 344. [Google Scholar] [CrossRef] [PubMed]
- National Bureau of Statistics of China. China Statistical Yearbook; National Bureau of Statistics of China: Beijing, China, 2017; ISBN 0106279181.
- Xu, L.L.L.; Zhu, B.; Wang, Z.; Xu, L.L.L.; Liu, Y.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Zhang, S.; et al. Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals 2019, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A K-Means Clustering Algorithm. Appl. Stat. 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011. [Google Scholar] [CrossRef]
- Carvajal-Rodriguez, A. Simulation of Genomes: A Review. Curr. Genomics 2008, 9, 155–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, I.; Meuwissen, T.H.E.; MacLeod, I.M.; Goddard, M.E. Predicting the effect of reference population on the accuracy of within, across, and multibreed genomic prediction. J. Dairy Sci. 2019, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; McClure, M.C.; McKay, S.D.; Rolf, M.M.; Kim, J.; Decker, J.E.; Taxis, T.M.; Chapple, R.H.; Ramey, H.R.; Northcutt, S.L.; et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet. Sel. Evol. 2011, 43, 1–16. [Google Scholar] [CrossRef]
- Van Eenennaam, A.L.; Weigel, K.A.; Young, A.E.; Cleveland, M.A.; Dekkers, J.C.M. Applied Animal Genomics: Results from the Field. Annu. Rev. Anim. Biosci. 2013, 2, 105–139. [Google Scholar] [CrossRef] [PubMed]
- Pszczola, M.; Strabel, T.; Mulder, H.A.; Calus, M.P.L. Reliability of direct genomic values for animals with different relationships within and to the reference population. J. Dairy Sci. 2012, 95, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Wientjes, Y.C.C.J.J.C.; Calus, M.P.P.L.L.; Goddard, M.E.; Hayes, B.J. Impact of QTL properties on the accuracy of multi-breed genomic prediction. Genet. Sel. Evol. 2015, 47, 42. [Google Scholar] [CrossRef]
- Kachman, S.D.; Spanger, M.L.; Bennett, G.L.; Hanford, K.J.; Kuehn, L.A.; Snelling, W.M.; Thallman, R.M.; Saatchi, M.; Garrick, D.J.; Schnabel, R.D.; et al. Comparison of molecular breeding values based on within- and across-breed training in beef cattle. Genet. Sel. Evol. 2013, 45, 30. [Google Scholar] [CrossRef]
- Weber, K.L.; Thallman, R.M.; Keele, J.W.; Snelling, W.M.; Bennett, G.L.; Smith, T.P.L.; McDaneld, T.G.; Allan, M.F.; Van Eenennaam, A.L.; Kuehn, L.A. Accuracy of genomic breeding values in multibreed beef cattle populations derived from deregressed breeding values and phenotypes. J. Anim. Sci. 2012. [Google Scholar] [CrossRef]
- Mouresan, E.F.; Cañas-Álvarez, J.J.; González-Rodríguez, A.; Munilla, S.; Altarriba, J.; Díaz, C.; Baró, J.A.; Molina, A.; Piedrafita, J.; Varona, L. Evaluation of the potential use of a meta-population for genomic selection in autochthonous beef cattle populations. Animal 2018, 12, 1350–1357. [Google Scholar] [CrossRef]
- Erbe, M.; Hayes, B.J.J.; Matukumalli, L.K.K.; Goswami, S.; Bowman, P.J.J.; Reich, C.M.M.; Mason, B.A.A.; Goddard, M.E.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 97, 6622. [Google Scholar] [CrossRef]
- Karoui, S.; Carabaño, M.J.; Díaz, C.; Legarra, A. Joint genomic evaluation of French dairy cattle breeds using multiple-trait models. Genet. Sel. Evol. 2012, 44. [Google Scholar] [CrossRef]
- Zhou, L.; Heringstad, B.; Su, G.; Guldbrandtsen, B.; Meuwissen, T.H.E.H.E.; Svendsen, M.; Grove, H.; Nielsen, U.S.S.; Lund, M.S.S. Genomic predictions based on a joint reference population for the Nordic Red cattle breeds. J. Dairy Sci. 2014, 97, 4485–4496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de los Campos, G.; Vazquez, A.I.; Fernando, R.; Klimentidis, Y.C.; Sorensen, D. Prediction of Complex Human Traits Using the Genomic Best Linear Unbiased Predictor. PLoS Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Calus, M.P.L.; Meuwissen, T.H.E.; de Roos, A.P.W.; Veerkamp, R.F. Accuracy of Genomic Selection Using Different Methods to Define Haplotypes. Genetics 2008, 178, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgante, F.; Huang, W.; Maltecca, C.; Mackay, T.F.C. Effect of genetic architecture on the prediction accuracy of quantitative traits in samples of unrelated individuals. Heredity 2018, 120, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef]
- Wiggans, G.R.; VanRaden, P.M.; Cooper, T.A. The genomic evaluation system in the United States: Past, present, future. J. Dairy Sci. 2011. [Google Scholar] [CrossRef]
- Chen, L.; Li, C.; Miller, S.; Schenkel, F. Multi-population genomic prediction using a multi-task Bayesian learning model. BMC Genet. 2014, 15, 53. [Google Scholar] [CrossRef]
- Raymond, B.; Bouwman, A.C.; Wientjes, Y.C.J.; Schrooten, C.; Houwing-Duistermaat, J.; Veerkamp, R.F. Genomic prediction for numerically small breeds, using models with pre-selected and differentially weighted markers. Genet. Sel. Evol. 2018, 50, 49. [Google Scholar] [CrossRef]
- Lund, M.S.; Su, G.; Janss, L.; Guldbrandtsen, B.; Brøndum, R.F.; Lund Mogens Sandøand Su, G.; Janss, L.; Guldbrandtsen, B.; Brøndum, R.F.; Lund, M.S.; et al. Invited review: Genomic evaluation of cattle in a multi-breed context. Livest. Sci. 2014, 166, 101–110. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulation Strategy | nQTL 1 | nS 2 | nM 3 | nL 4 | Heritability |
|---|---|---|---|---|---|
| I | 100 | 0 | 0 | 100 | 0.1/0.3/0.6 |
| II | 2000 | 1361 | 614 | 25 | 0.1/0.3/0.6 |
| III | 5000 | 4595 | 390 | 15 | 0.1/0.3/0.6 |
| IV | 10,000 | 10,000 | 0 | 0 | 0.1/0.3/0.6 |
| Within Breed | Combined Breeds | |||||||
|---|---|---|---|---|---|---|---|---|
| Strategy * | I | II | III | IV | I | II | III | IV |
| L-h2 * | 0.30 | 0.28 | 0.31 | 0.29 | 0.11 | 0.10 | 0.12 | 0.10 |
| M-h2 * | 0.48 | 0.47 | 0.51 | 0.47 | 0.18 | 0.17 | 0.17 | 0.17 |
| H-h2 * | 0.66 | 0.64 | 0.68 | 0.64 | 0.26 | 0.24 | 0.26 | 0.26 |
| Average | 0.48 | 0.46 | 0.50 | 0.47 | 0.18 | 0.17 | 0.18 | 0.18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Wang, Z.; Zhu, B.; Liu, Y.; Li, H.; Bordbar, F.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; et al. Theoretical Evaluation of Multi-Breed Genomic Prediction in Chinese Indigenous Cattle. Animals 2019, 9, 789. https://doi.org/10.3390/ani9100789
Xu L, Wang Z, Zhu B, Liu Y, Li H, Bordbar F, Chen Y, Zhang L, Gao X, Gao H, et al. Theoretical Evaluation of Multi-Breed Genomic Prediction in Chinese Indigenous Cattle. Animals. 2019; 9(10):789. https://doi.org/10.3390/ani9100789
Chicago/Turabian StyleXu, Lei, Zezhao Wang, Bo Zhu, Ying Liu, Hongwei Li, Farhad Bordbar, Yan Chen, Lupei Zhang, Xue Gao, Huijiang Gao, and et al. 2019. "Theoretical Evaluation of Multi-Breed Genomic Prediction in Chinese Indigenous Cattle" Animals 9, no. 10: 789. https://doi.org/10.3390/ani9100789