4. Discussion
In this research, breast muscle transcriptome data were analyzed and DEGs obtained from native male turkeys individually phenotyped for feed efficiency using RNA-Seq technology. Functional annotation, investigation of the PPI network, and module analysis were conducted (see Methods). Following a key nodes analysis of the PPI network, the hub genes were found. In addition, a functional enrichment of all the expressed genes was performed between HFE and LFE turkeys.
A list of DEGs was generated using differential expression analysis which needed further analysis to create an overview of the differences between the two groups. In total, 365 significant DEGs (127 up-regulated and 238 down-regulated) were found in response to divergent FE.
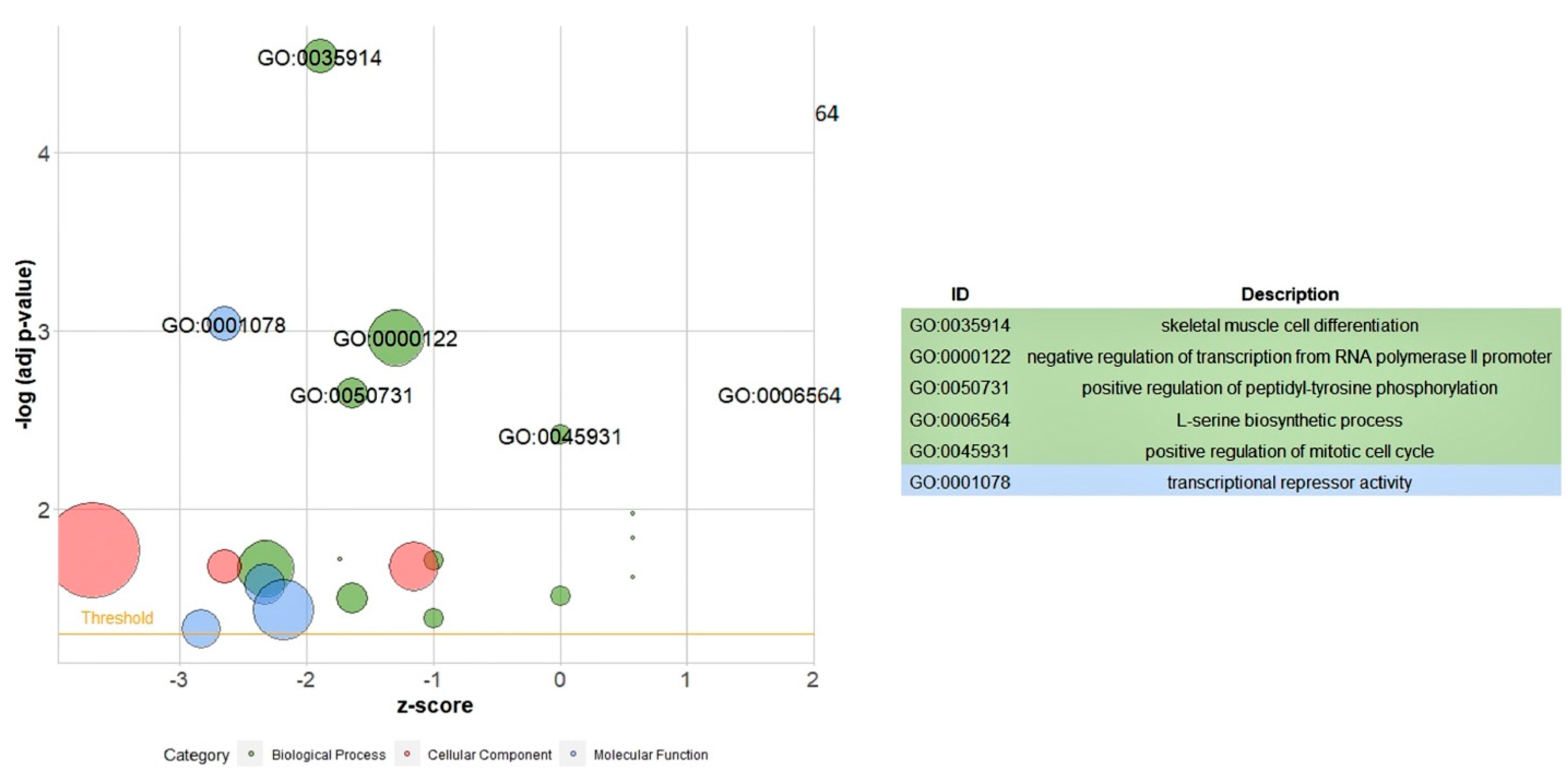
An ontology annotation of all the DEGs revealed some GO terms indicating that the most significantly enriched GOs were related to skeletal muscle cell differentiation, negative regulation of transcription from the RNA polymerase II promoter, positive regulation of peptidyl-tyrosine phosphorylation, the L-serine biosynthetic process, and positive regulation of the mitotic cell cycle in the BP term; and transcriptional repressor activity in the MF term. Therefore, most GO terms of all the DEGs were enriched in the metabolic process and cell differentiation, both of which are important in growth and feed efficiency traits. A pathway enrichment was conducted on the up-regulated DEGs. Major pathways including “glycine, serine, and threonine metabolism”, “biosynthesis of amino acids”, “adipocytokine signaling pathway”, and “metabolic pathways” were statistically significant.
Kong et al. (2011) reported that there was a down-regulation of cytoskeletal organization genes in the HFE phenotype [
5]. The glycine, serine, and threonine metabolism pathway is one of the metabolic pathways that was significant in the duodenum and adipose tissue in cattle with divergent feed efficiency on several tissues [
16]. The glycine, serine, and threonine metabolism pathway was reported to be one of the significant metabolic pathways in Holstein and Jersey dairy cows with high and low feed efficiencies [
17]. Yang et al. (2017) reported that the glycine, serine, and threonine metabolism pathway had higher abundances in HFE pigs [
18].
Glucose is the main source of metabolic energy in the body. When glucose enters the cell, glycolysis starts in the cytoplasm. The glycolysis pathway provides an intermediate metabolite, 3-phosphoglycerate, which is catalyzed into serine by
PHGDH,
PSAT1, and
PSPH [
19]. Then, serine can be converted into glycine. Additionally, glycine can be converted to threonine. This pathway provides precursors for purine biosynthesis and the tricarboxylic acid (TCA) cycle [
20]. The TCA cycle generates ATP for protein production. It has been reported that HFE pigs accumulate more muscle mass in comparison to LFE pigs in their breast muscle [
21]. Thus, the results of the present study in turkeys suggest that the HFE animals may consume more glucose in the muscle compared to LFE birds to produce ATP for protein deposition.
On the other hand, glycine can be converted to creatine by
GATM which was over-expressed (or up-regulated) in the HFE turkey phenotype. Creatine has some anti-inflammatory and anti-oxidant characteristics, increases insulin sensitivity, and creatine metabolism and
GATM has an important role in energy expenditure and defense against diet-induced obesity [
22]. The entirety of this process is more active in HFE birds. Therefore, HFE turkeys may have better glucose absorption and have more active glycolysis or gluconeogenesis pathways. Additionally, the biosynthesis of amino acids provides a situation for producing more protein. Therefore, increasing this pathway in the breast muscle of birds results in heavier breast muscles in HFE turkeys (
Figure 11).
Fonseca et al. (2019) found metabolic pathways related to feed efficiency in beef cattle using liver proteomics. They revealed that the biosynthesis of amino acids is one of the main pathways associated with FE [
23].
The adipocytokine signaling pathway was another of the significant pathways. Adiponectin production (a protein encoded by the
ADIPOQ gene) is negatively associated with increasing the volume and number of adipocytes. In circulation, adiponectin increases the transport of plasma fatty acids and glucose into cells. It is involved in the control of fat metabolism and has anti-inflammatory activities. Adiponectin also induces AMPK activation, which stimulates skeletal muscle glucose uptake and fatty acid oxidation. It also induces gluconeogenesis through the inhibition of the PEPCK and G6PC proteins. Therefore, these processes in HFE turkeys result in better glucose absorption and fat metabolism. The fatty acid translocase gene
CD36 is a membrane receptor that facilitates the uptake of long-chain fatty acids. It is also involved in the adipocytokine signaling pathway. Finally, it provides precursors of acetyl-CoA for the TCA cycle. Therefore, a better uptake of fatty acids, stimulating gluconeogenesis and increasing insulin sensitivity, occurs through the adipocytokine signaling pathway in HFE turkeys [
24]. The results of a study on Holstein cows showed that the adipocytokine signaling pathway is one of the significant pathways in Holstein cows with high and low feed efficiencies [
25]. In addition, Lassiter et al. (2006) reported an increased expression of AMPK in the HFE phenotype of pedigree male broilers [
26].
A pathway analysis of the down-regulated DEGs in the current study showed that the major significant pathways were “dorso-ventral axis formation” and “regulation of actin cytoskeleton”. Moreover, Yang et al. (2020) found that one of the significant modules in which the DEGs of native chickens were enriched was the regulation of the actin cytoskeleton that is involved in the regulation of cell motility [
14].
Apparent metabolizable energy is a method to assess energy utilization for feed efficiency. Pezeshkian et al. (2020) investigated the apparent metabolizable energy of chickens and showed that down-regulated genes were enriched in some pathways including regulation of the actin cytoskeleton [
27].
The ontology annotation of the DEGs revealed several biological events mostly associated with the biosynthesis of amino acids process and growth. They include the L-serine biosynthetic process, the GDP-mannose biosynthetic process, skeletal muscle tissue growth, amyloid fibril formation, the regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ions, and the negative regulation of heart rate. “Low-density lipoprotein particle” refers to a process in which a low-density lipoprotein particle is removed from the blood. “Protein homotetramerization” is the process of forming protein homotetramers, which consist of four identical subunits of proteins. Protein and lipid metabolisms are important factors in feed efficiency [
28]. Therefore, the higher expression of genes of these processes in HFE-phenotyped male native turkeys can be interpreted.
Some genes were involved in multiple pathways and GO terms. Among the DEGs,
GATM was the most highly expressed gene. The glycine aminotransferase (
GATM) gene is involved in glycine, serine, and threonine metabolism and other metabolic pathways. This enzyme converts glycine to arginine by guanidino acetate. Moreover, it was involved in the gene set of glycine, serine, and threonine metabolism in GSEA analysis. This gene was reported as a gene with different expression in a study that investigated the molecular basis of feed efficiency in broilers [
29]. Mckenna (2018) investigated the molecular control of feed efficiency in beef cattle, and the
GATM gene was introduced as a gene with different expression in muscle tissue between animals with a high and low feed efficiency [
30].
Phosphoglycerate dehydrogenase (
PHGDH), phosphoserine aminotransferase 1 (
PSAT1), and phosphoserine phosphatase (
PSPH) are genes in the serine biosynthesis process [
31]. Fonseca et al. (2019) performed liver proteomics which elucidated the metabolic pathways associated with feed efficiency in beef cattle, and the
PSAT1 gene was identified as a DEG involved in different metabolic pathways [
23]. The
PSPH gene is known as a DEG between cows with high and low residual feed intake [
32]. Keogh et al. (2016) showed that the
PSPH gene was up-regulated in HFE animals and was therefore introduced as a marker for feed efficiency [
33].
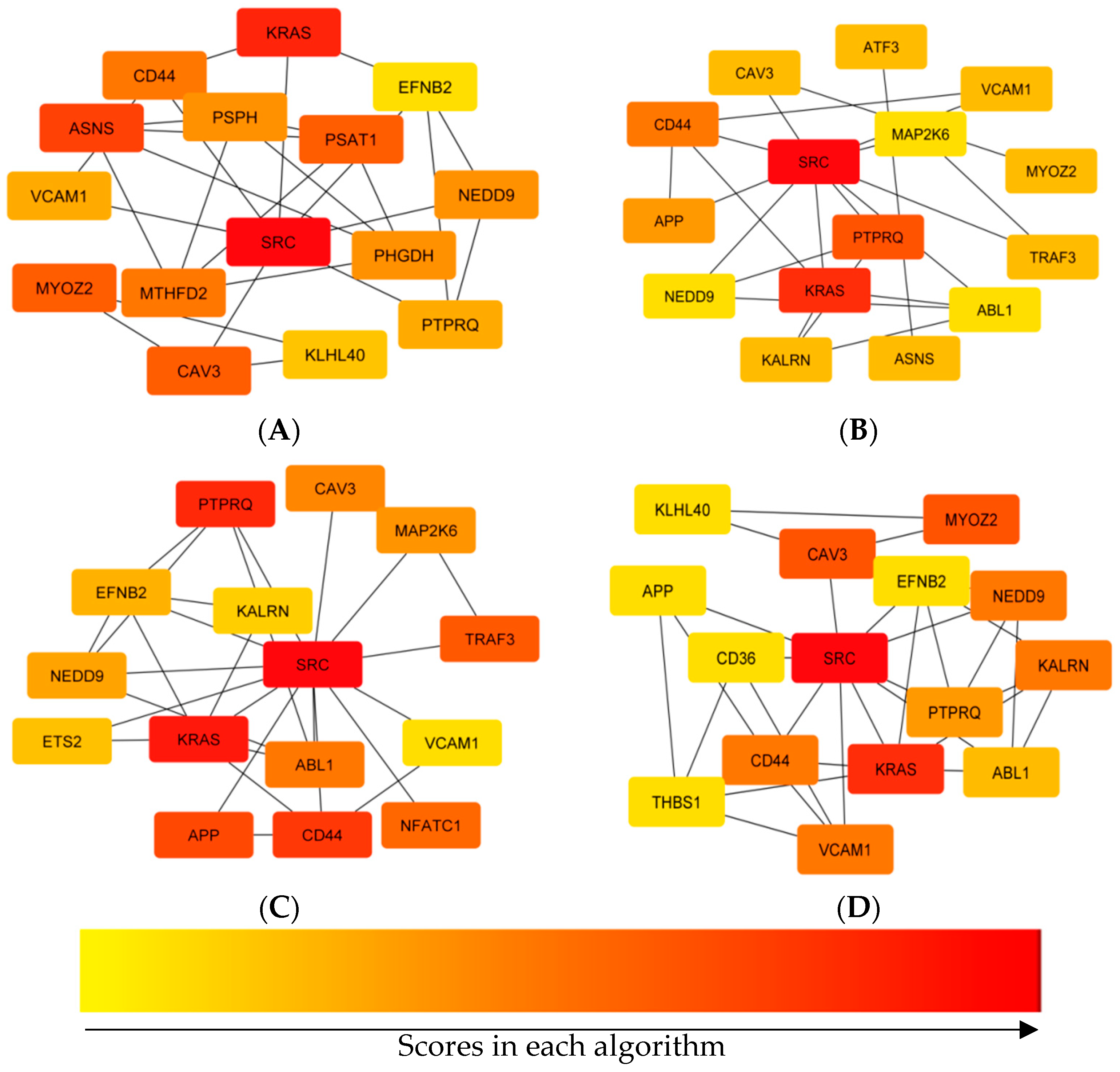
To further evaluate the intrinsic relationship between DEGs, mapping of the PPI networks was performed. Analyzing PPI networks is essential for understanding the molecular basis for complex traits. In the current study, after construction of the PPI network, the top centrality hub genes were achieved through four centrality algorithms. Finally, seven hub genes, including CD44, VCAM1, KRAS, NEDD9, SRC, PTPRQ, and CAV3 were identified.
During inflammation, vascular cell adhesion molecule 1 (
VCAM1) is mainly expressed in endothelial cells and causes vascular inflammation [
34].
VCAM1 was reported as a key gene related to the feed efficiency of native chickens [
14].
KRAS plays an important role in regulating cell proliferation and was down-regulated in HFE turkeys in the current study. In a RNA-Seq transcriptomics and pathway analysis study on dairy cattle with a high and low residual feed intake, one of the genes with a significantly different expression was
KRAS gene [
35]. Zarek et al. (2017) investigated gene expression related to weight gain and feed intake in liver tissue and showed that the
KRAS gene was one of the DEGs between groups with a different phenotype of weight gain and feed intake [
36]. Mukherjee et al. (2020) found that the phosphatidylinositol phosphatase (
PTPRQ) gene was up-regulated in low growth bovines. This gene can regulate cell survival and proliferation [
37]. The caveolin 3 gene (
CAV3) is a member of the caveolin family and acts as a specific muscle isoform of the caveolin protein. Lassiter et al. (2019) showed that the
CAV3 and
MYOZ2 genes were among the DEGs that were involved in muscle development and differentiation in broilers with a high and low feed efficiency [
38]. The
SRC gene encodes protein tyrosine kinases. Lam et al. (2021) used RNA-Seq to identify that the
SRC gene is among the most important genes with different expression in a group of animals with low feed efficiency [
39]. Bottje et al. (2017) investigated broilers with divergent feed efficiency and one of the top enrichment down-regulated genes in the HFE group was
CD44 [
40]. Onteru et al. (2013) found that
NEDD9 (neural precursor cell expressed, developmentally down-regulated 9) was one of the significant single nucleotide polymorphisms between pigs with a low and high residual feed intake (RFI) [
41].
To further analyze the PPI network, we investigated the significant modules and found six modules with an MCODE score of higher than three. Both KEGG pathway and GO analyses were performed using the ClueGO plugin, and only the first module (including PSPH, PHGDH, PSAT1, MTHFD2, and ASNS) showed significant enrichment terms. The most important of these enrichment analyses were the alpha-amino acid biosynthetic process and the L-serine biosynthetic process. As mentioned before, the amino acid and protein biosynthetic process has a significant effect on the feed efficiency of animals.
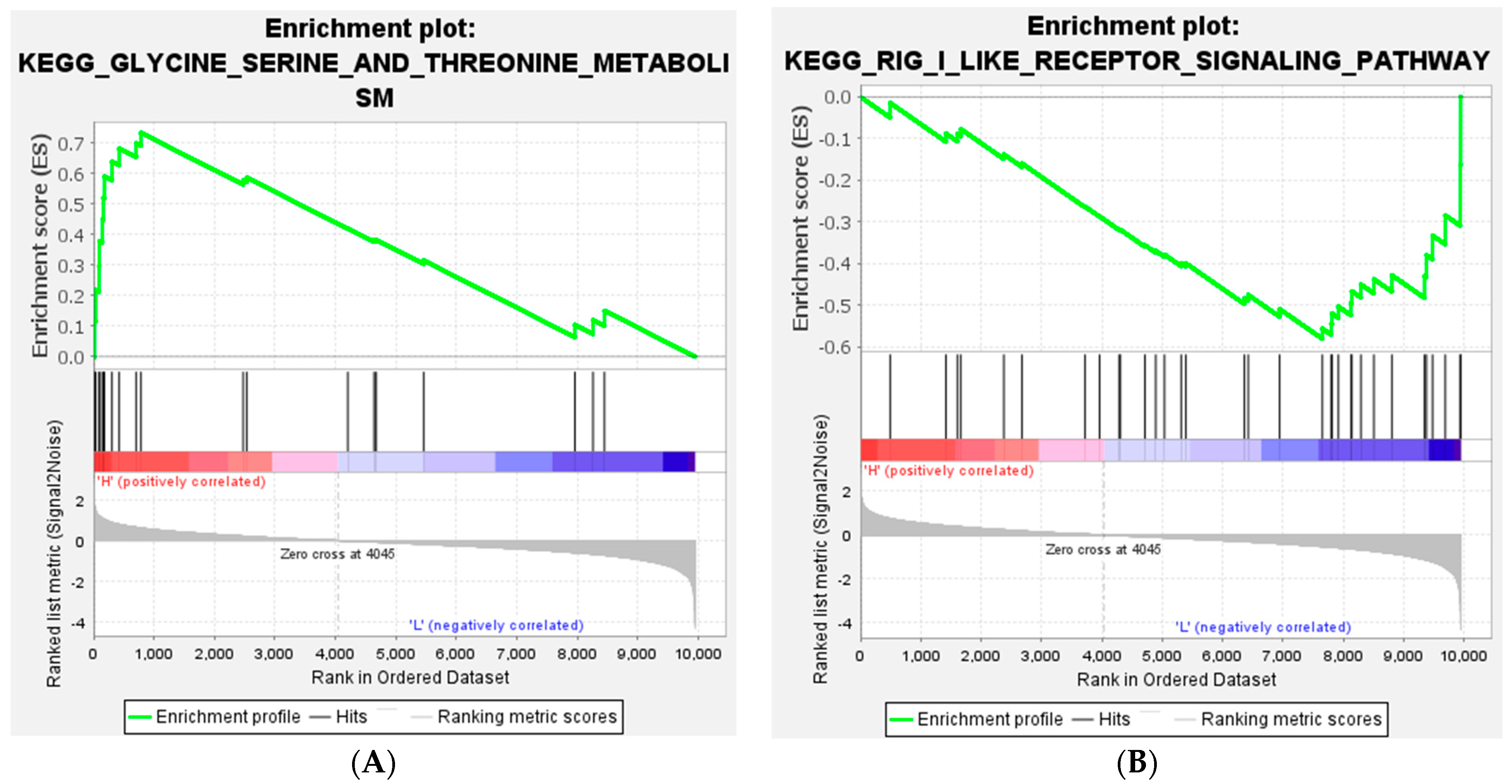
In the current study, we applied the GSEA method to change the RNA-Seq data into biological interpretations. Therefore, there are not any limitations and thresholds for the expressed genes to perform function analysis. In addition, GSEA can reliably identify gene sets with biological significance [
42]. As a result, the most significant gene set that was highly expressed in HFE was glycine, serine, and threonine metabolism. As mentioned before, amino acid metabolism has a key effect on feed efficiency. On the other hand, the most significant gene lists in the LFE group were the RIG-I-like receptor signaling pathway and the Jak-STAT signaling pathway. Both of these pathways are related to the inflammatory response and the immune response. This might suggest increased oxidative stress in the LFE phenotype which agrees with findings in the muscle [
43], intestines [
44], liver [
45], and heart [
46] and those of the study of Lassiter et al. (2006) on high and low FE broilers [
24]. It has been reported that the high production of pro-inflammatory agents may cause damage to the intestinal integrity and decrease feed efficiency [
47]. These results showed that pathways associated with the inflammatory and immune responses are related to feed efficiency. The study of Yang et al. (2020) reported an overexpression of genes related to the Jak-STAT signaling pathway in native chickens with a high RFI [
14].
To confirm the putative results from RNA-Seq, we randomly selected a subset of DEGs for qRT-PCR assays. Overall, there was an excellent agreement and high concordance between the computational and experimental results, which revealed a good detection sensitivity and accuracy.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}