



Animals 2023, 13(4), 674; https://doi.org/10.3390/ani13040674 - 15 Feb 2023
Cited by 4 | Viewed by 1953
Abstract
►
Show Figures
Tenderness is a key meat quality trait that determines the public acceptance of lamb consumption, so genetic improvement toward lamb with higher tenderness is pivotal for a sustainable sheep industry. However, unravelling the genomics controlling the tenderness is the first step. Therefore, this
[...] Read more.
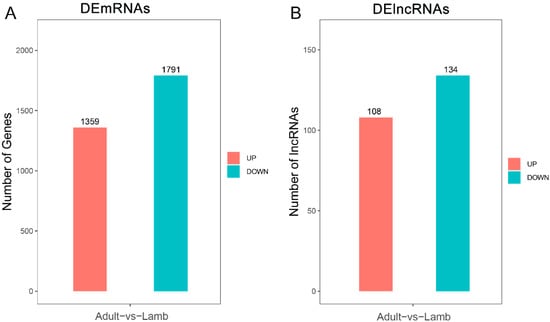
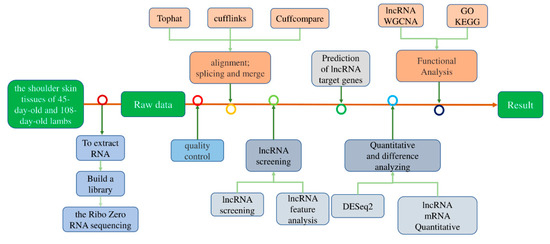
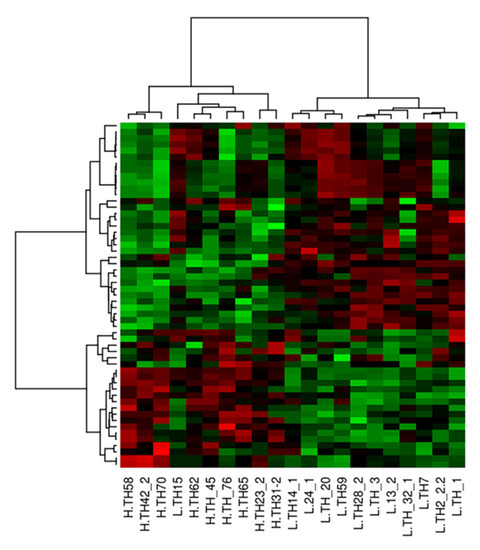
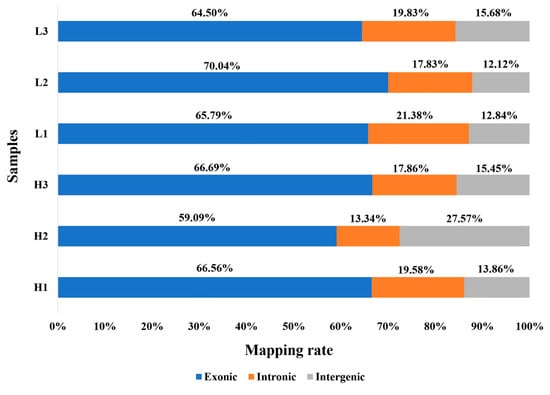
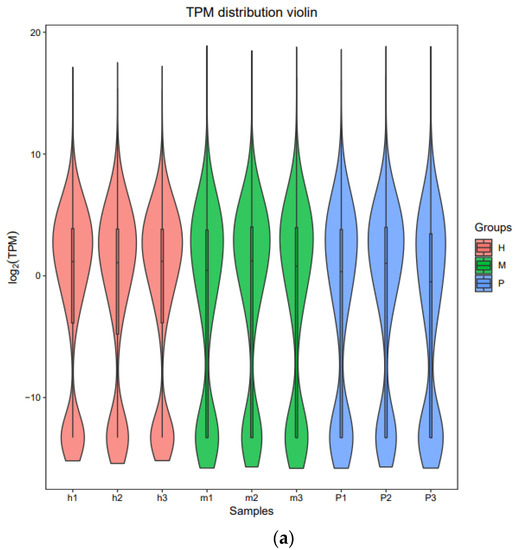
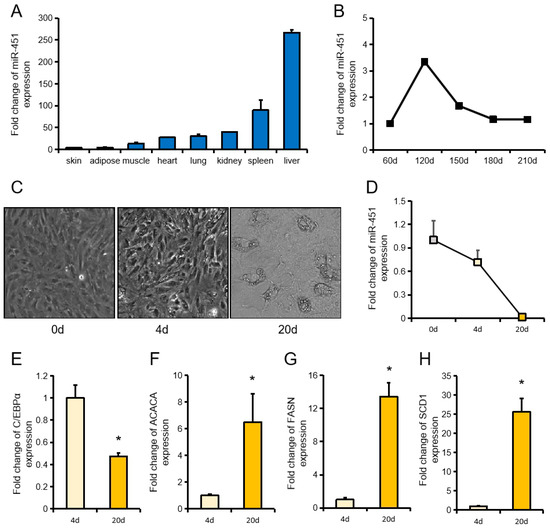
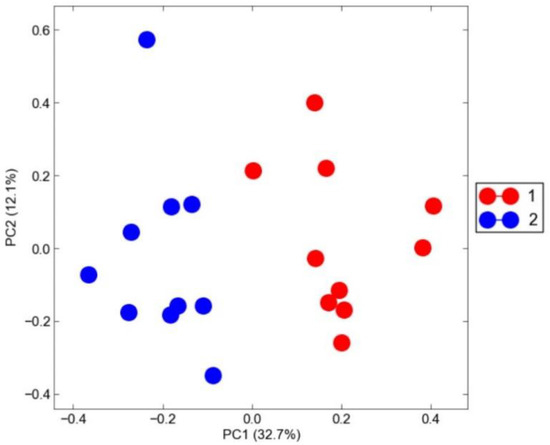
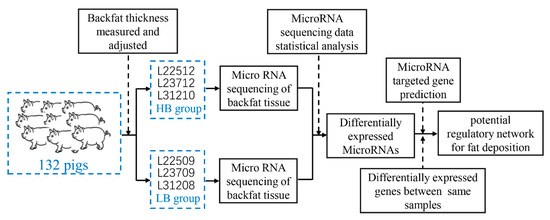
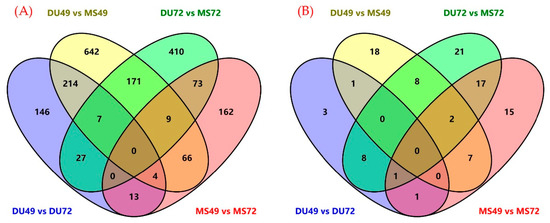
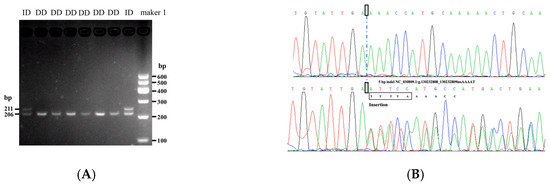
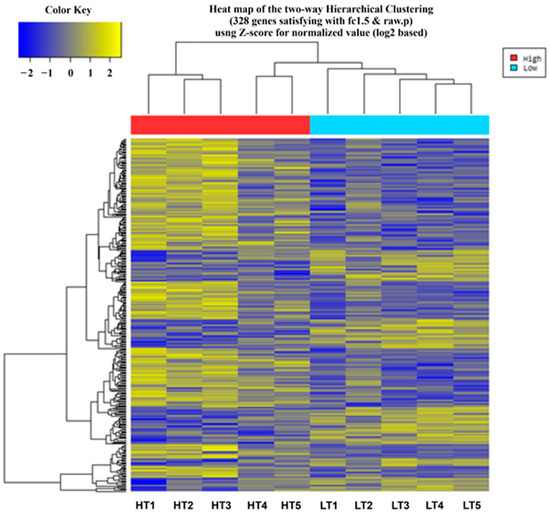
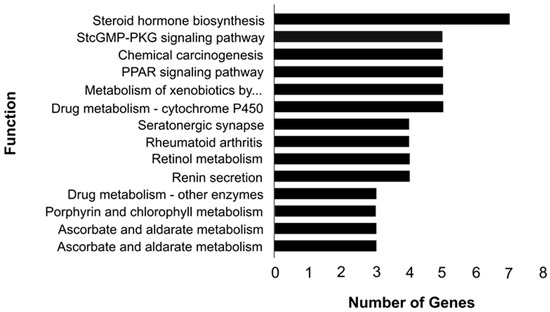
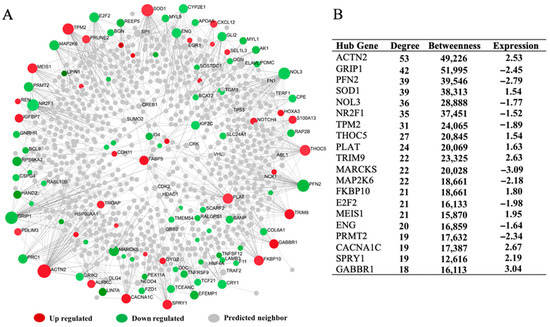
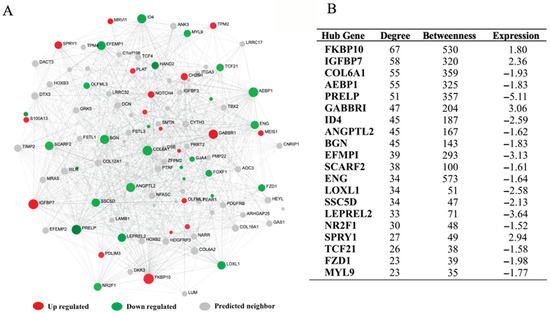
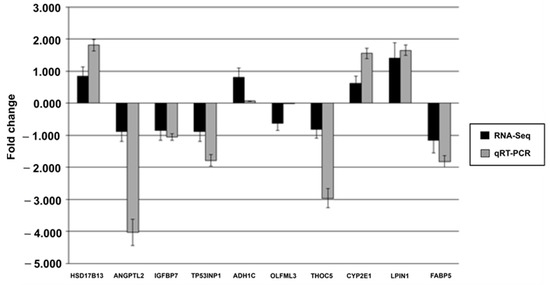
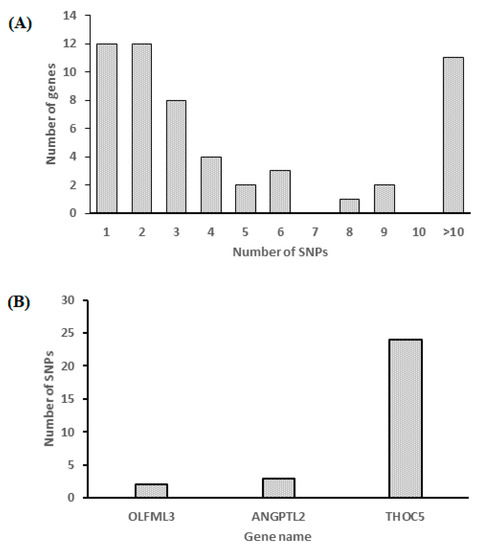
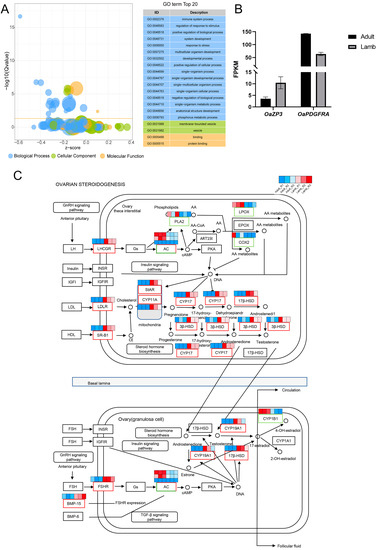
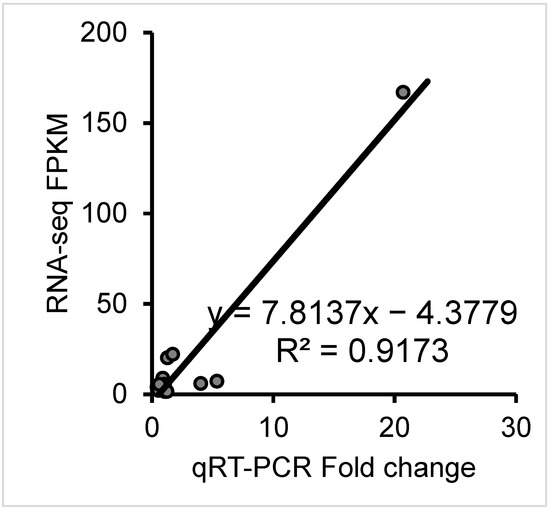
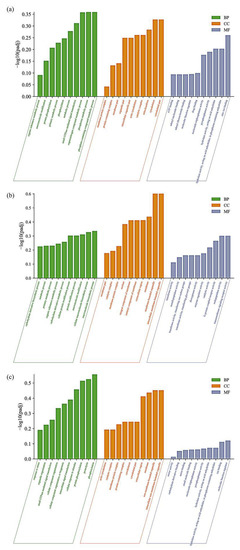
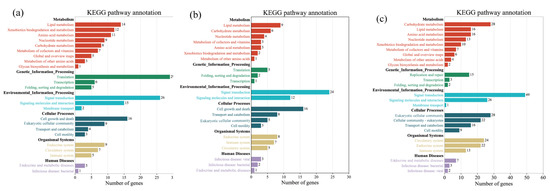
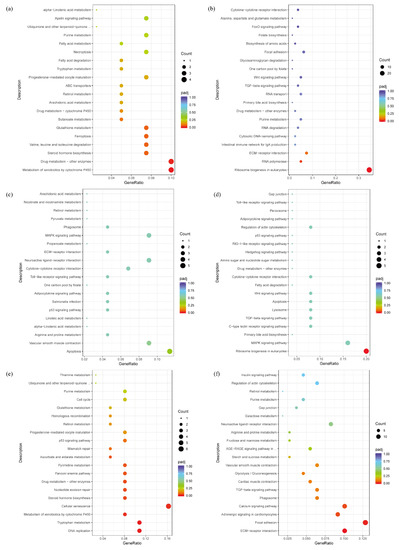
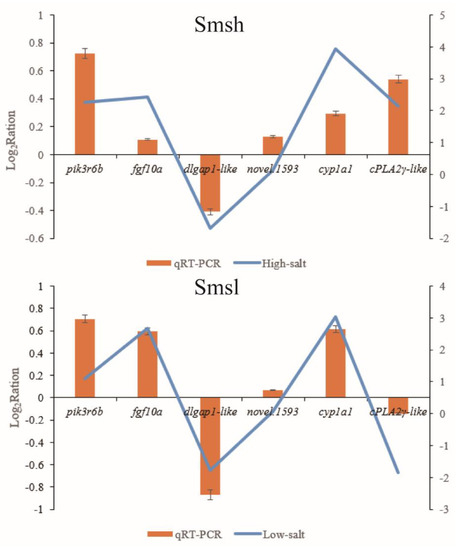
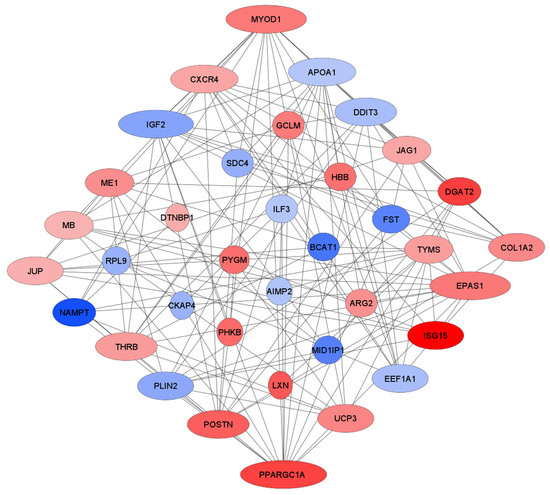
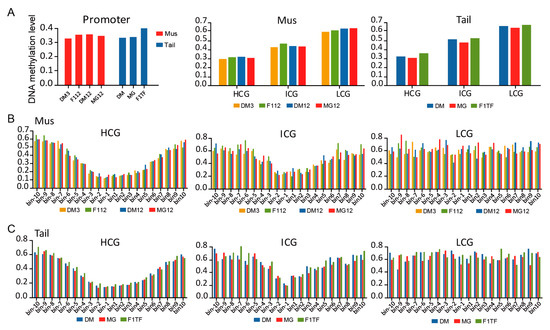
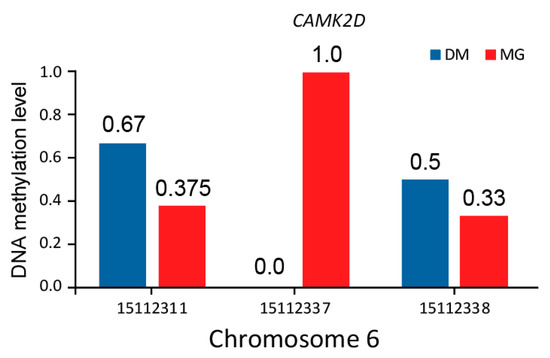
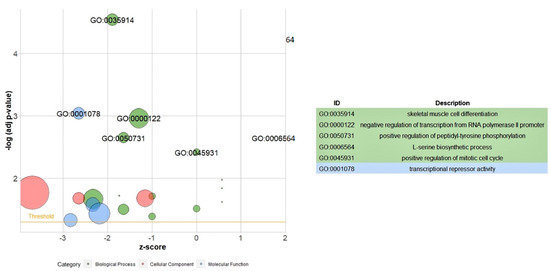
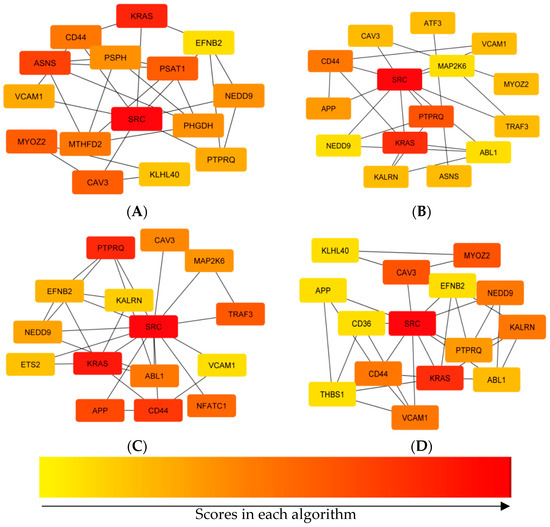
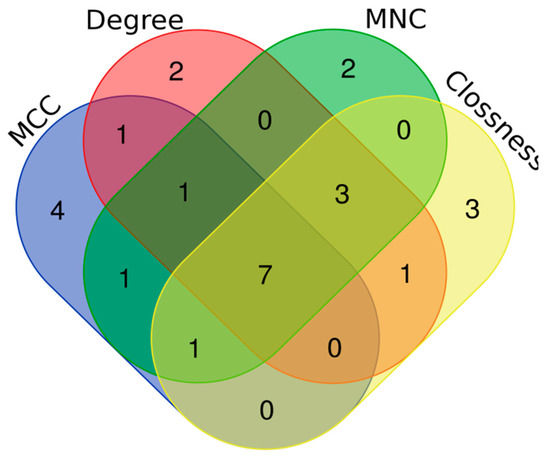
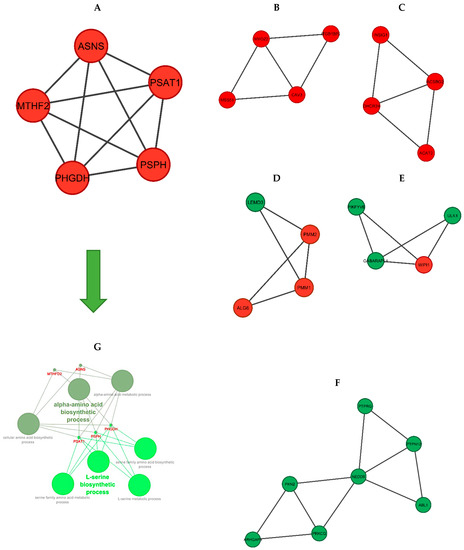
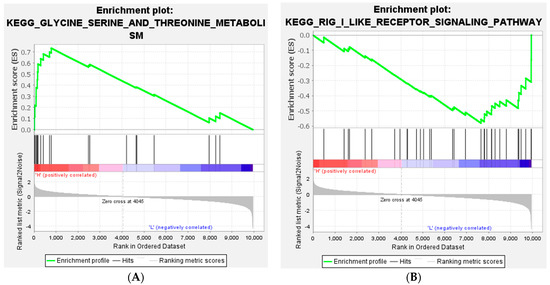
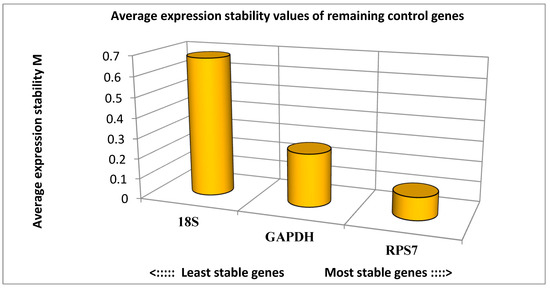
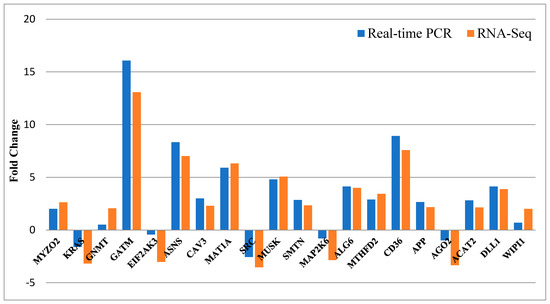
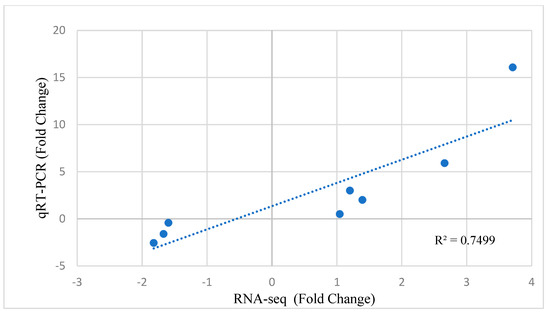
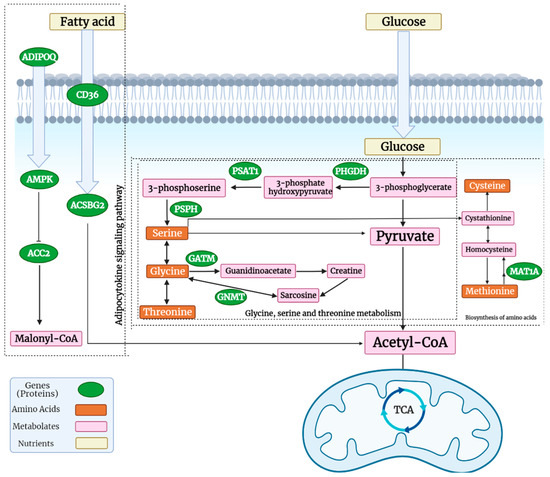
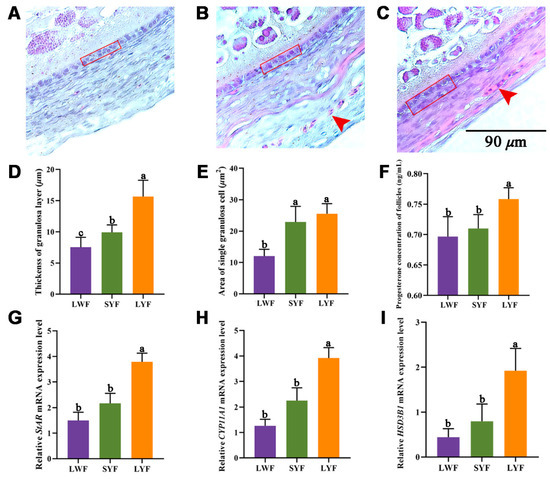
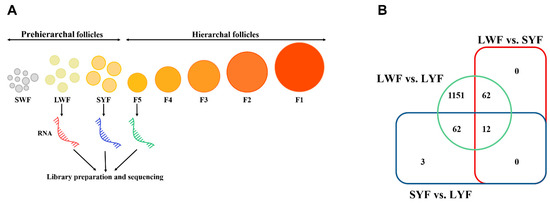
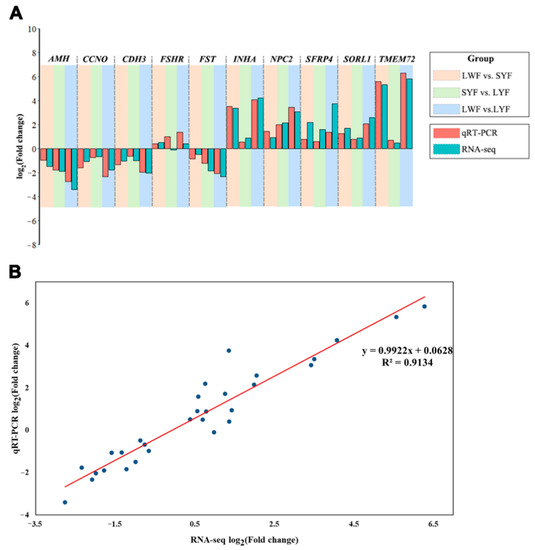
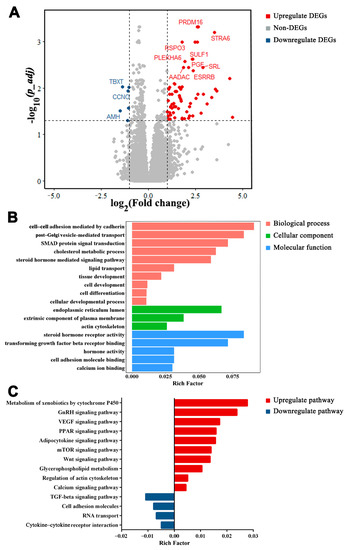
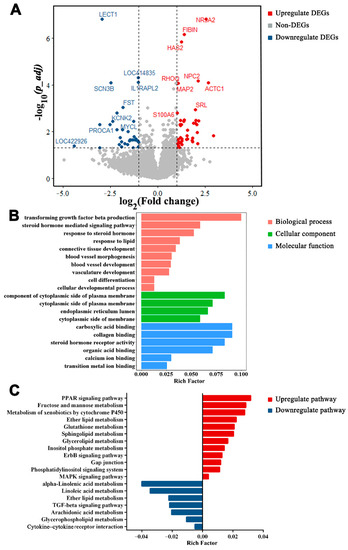
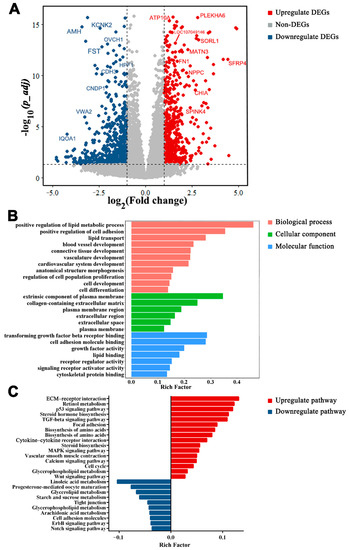
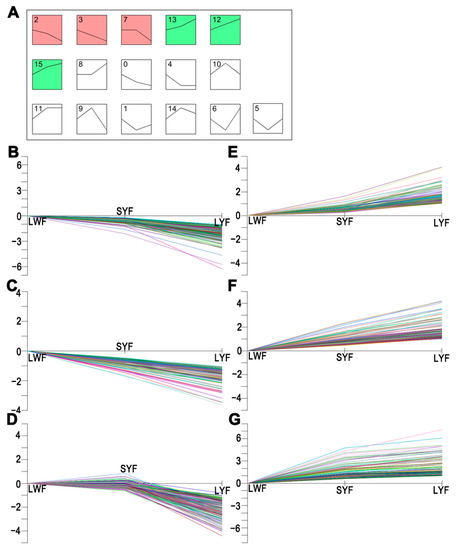
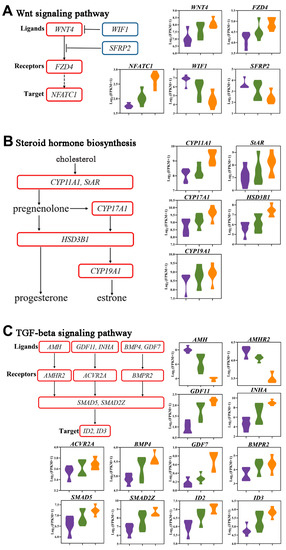
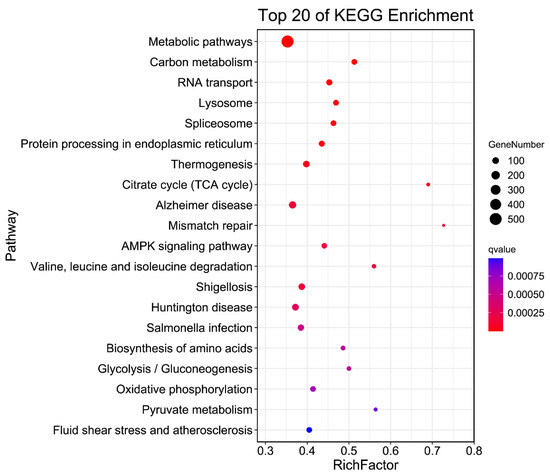
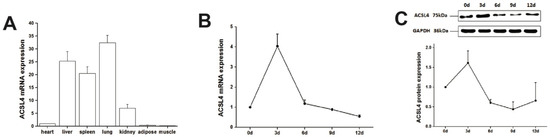
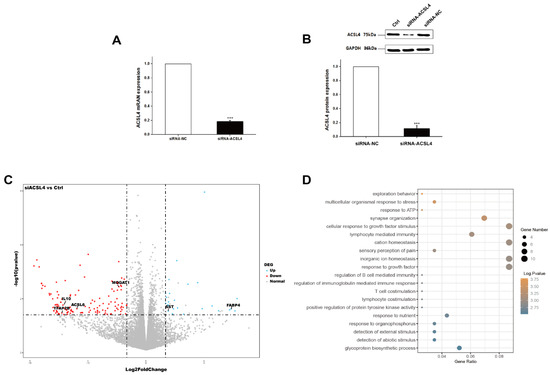
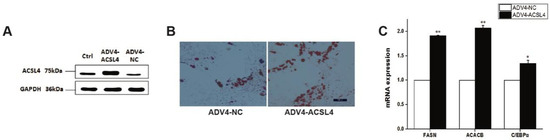
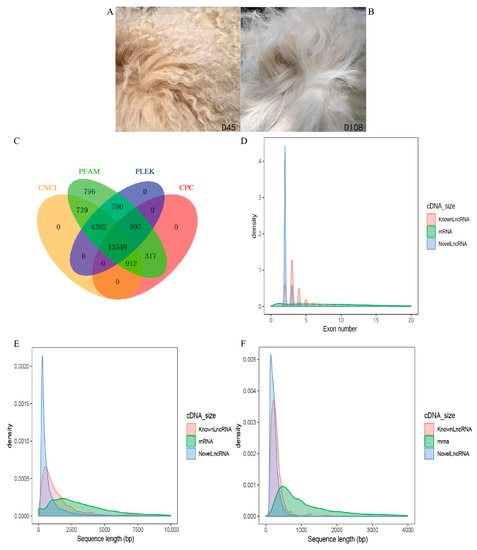
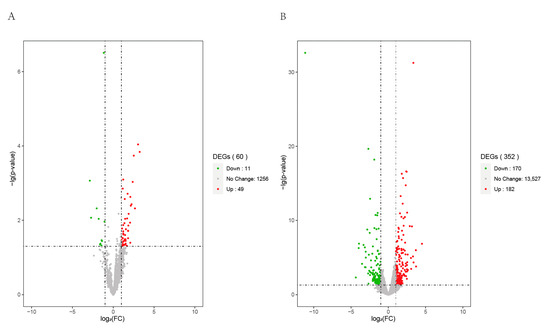
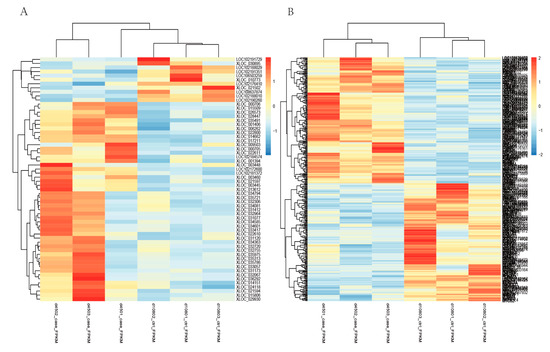
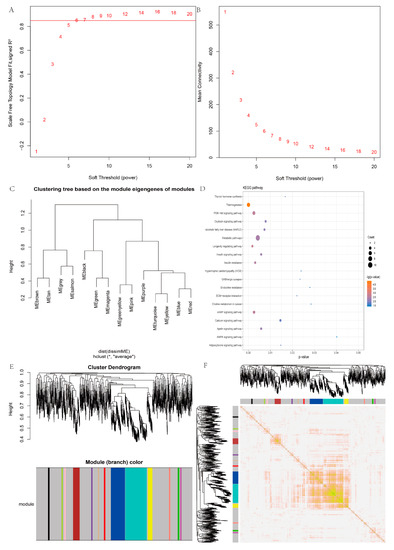
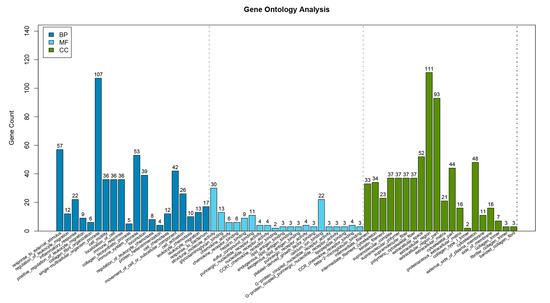
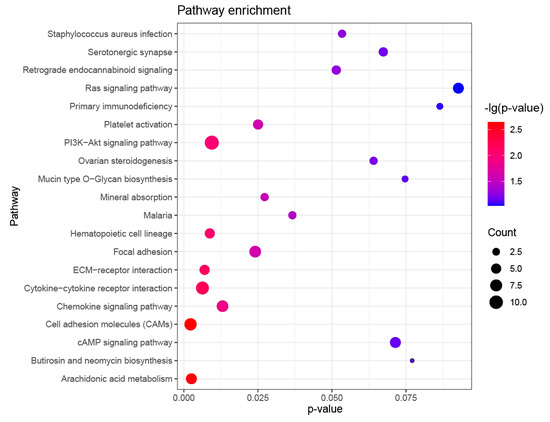
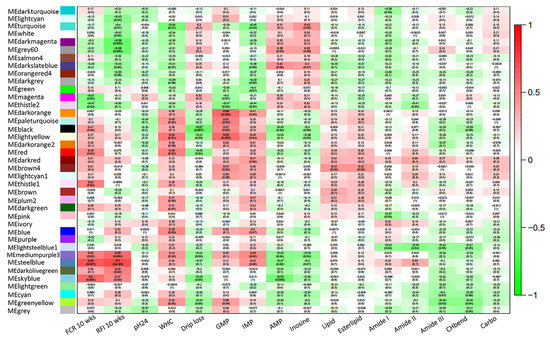
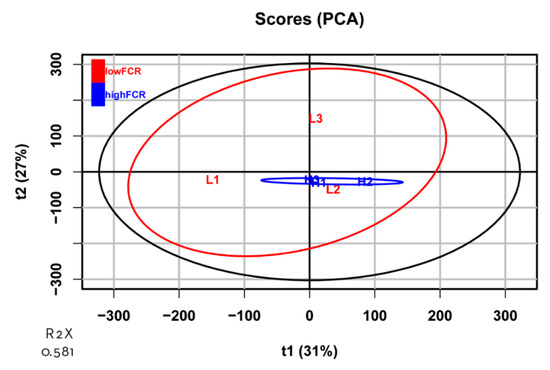
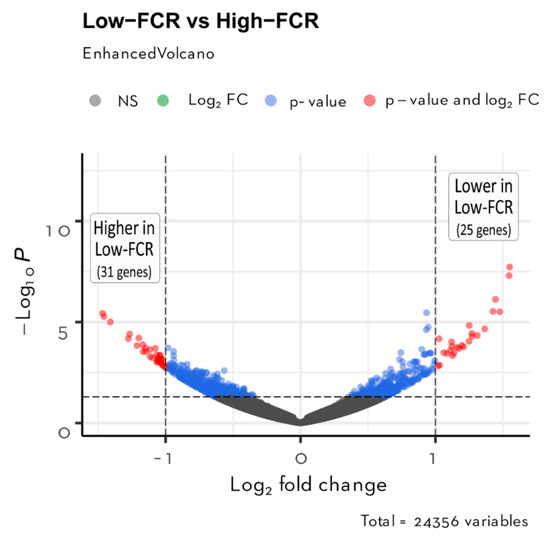
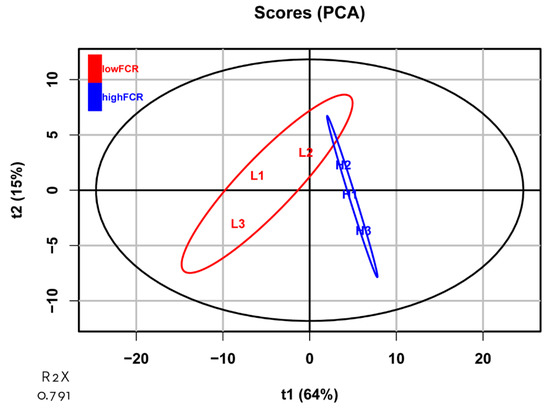
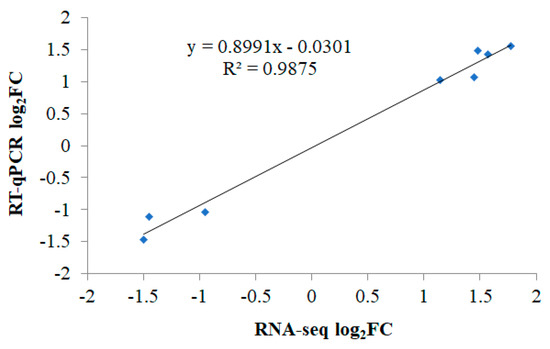
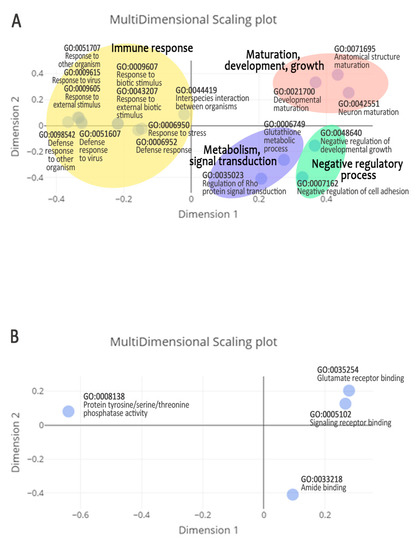
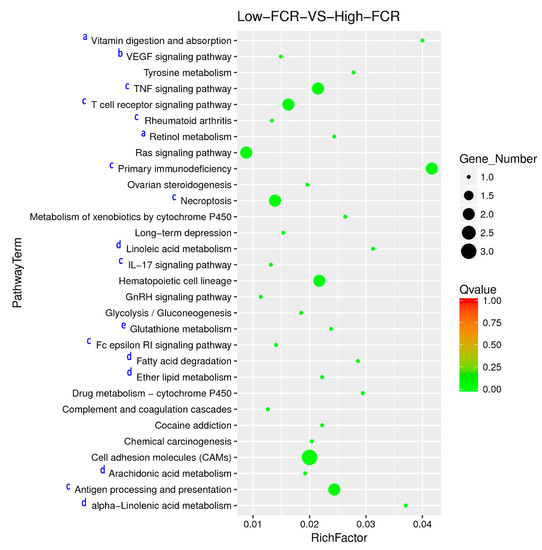
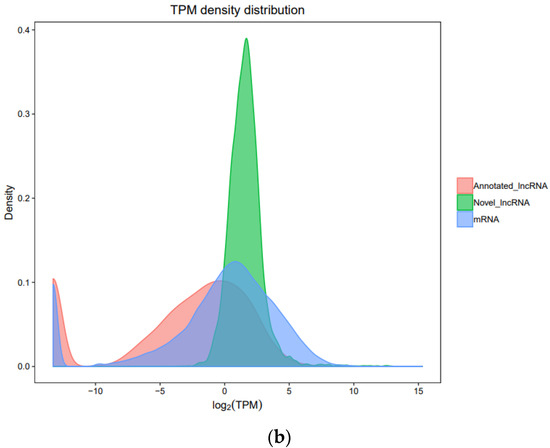
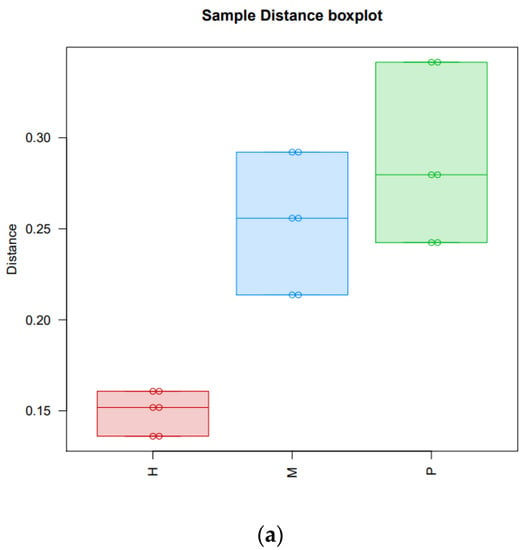
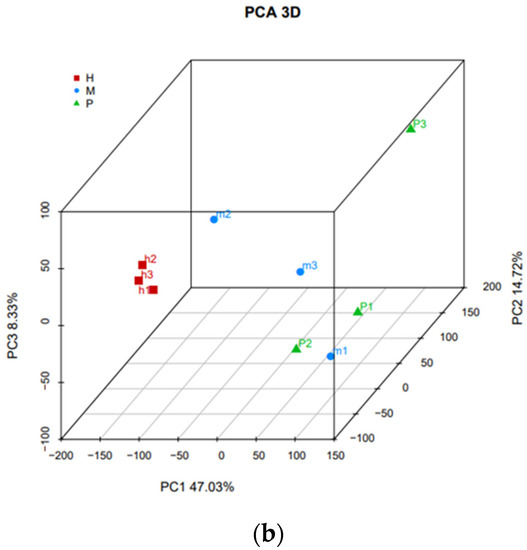
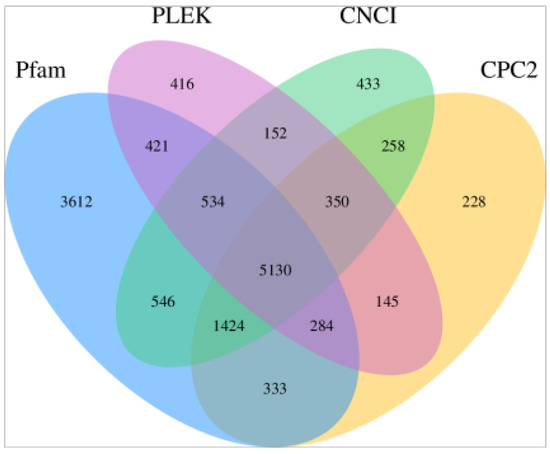
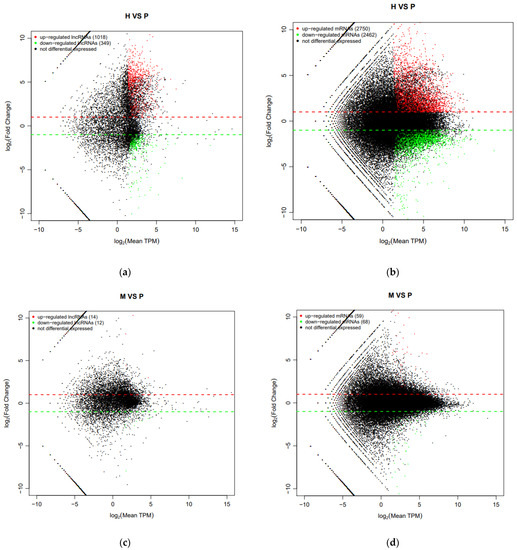
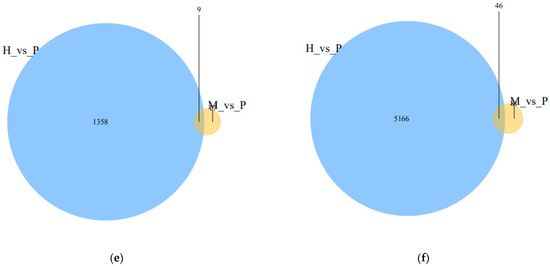
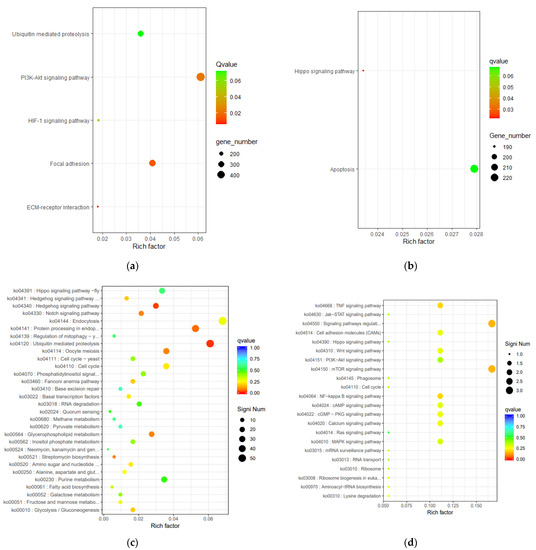
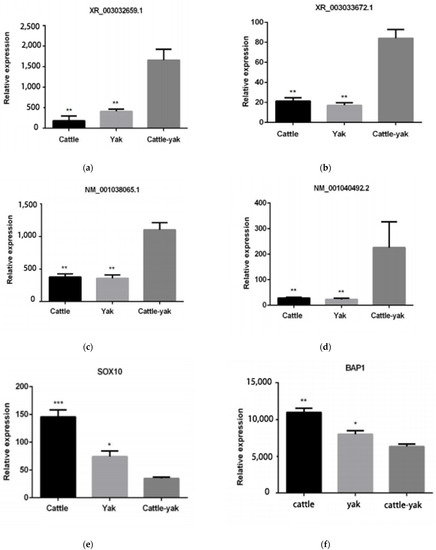
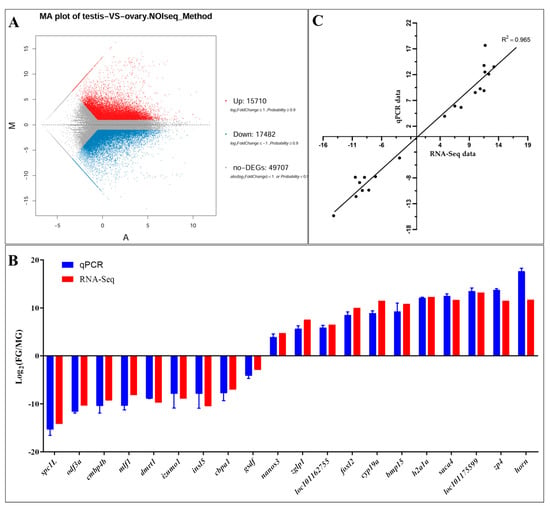
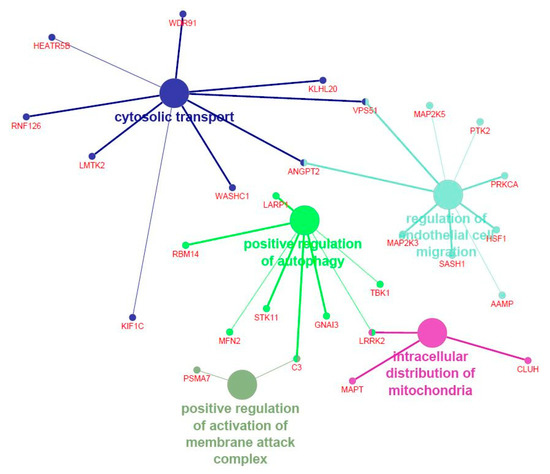
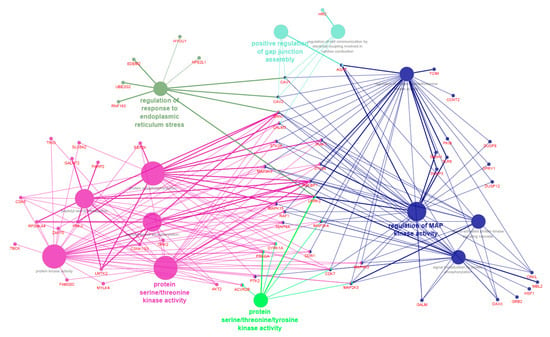
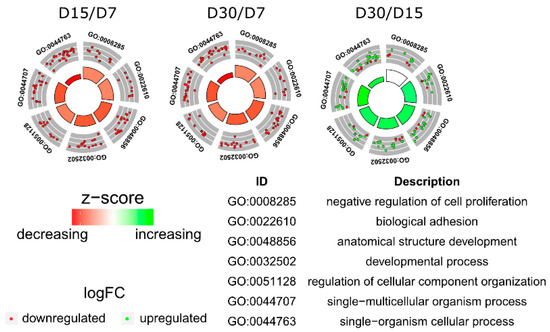
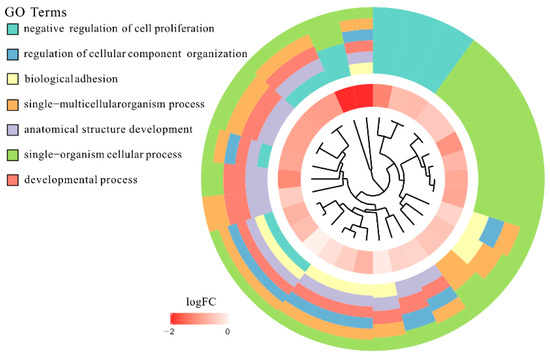
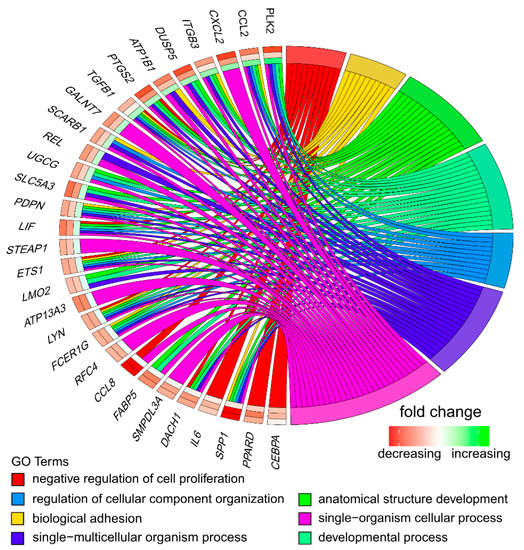
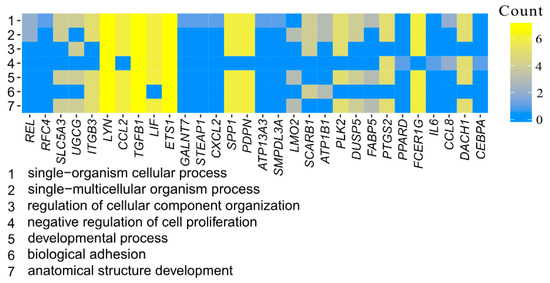
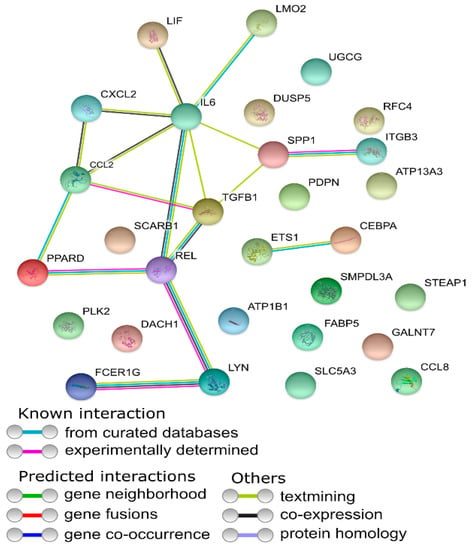
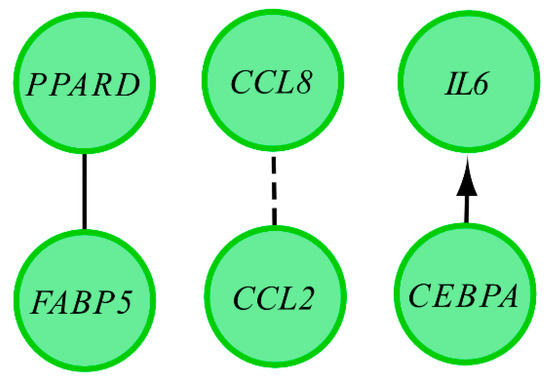
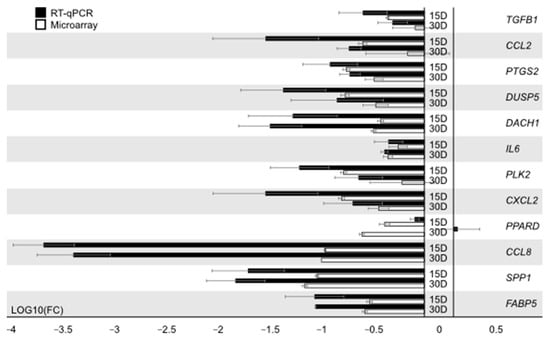
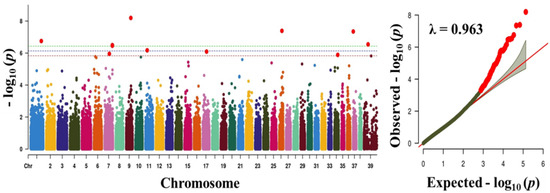
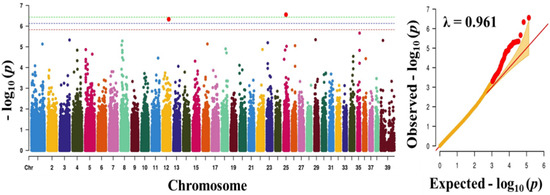
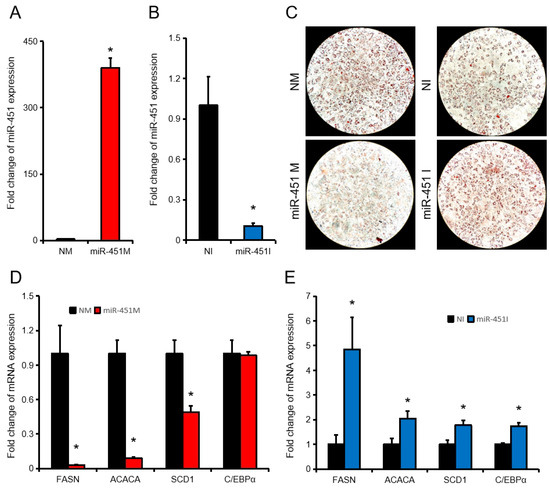
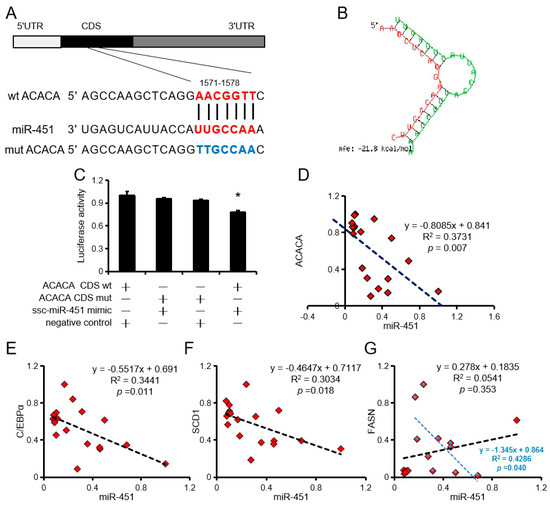
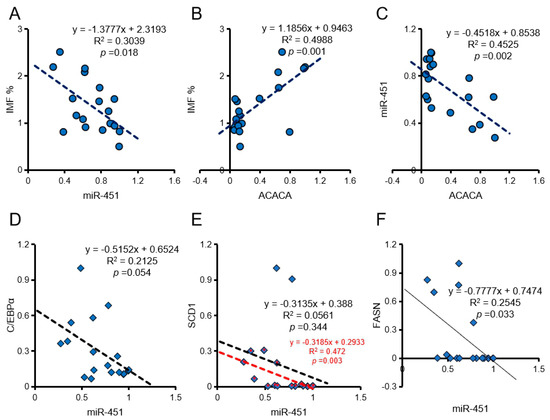
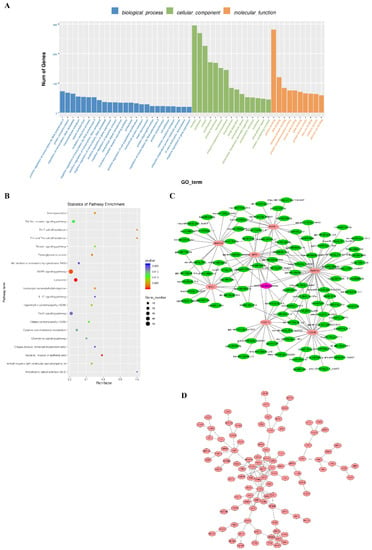
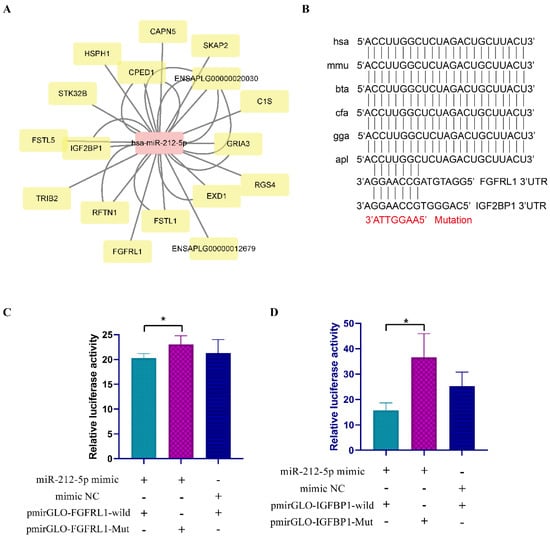
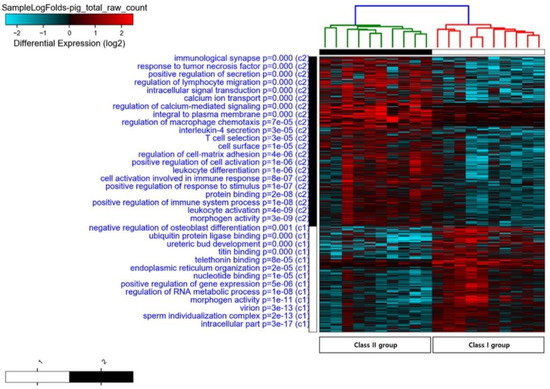
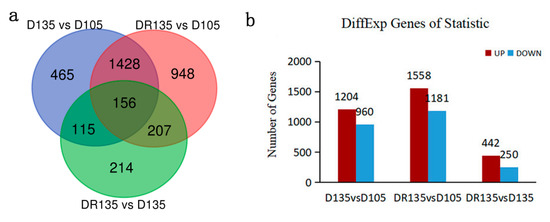
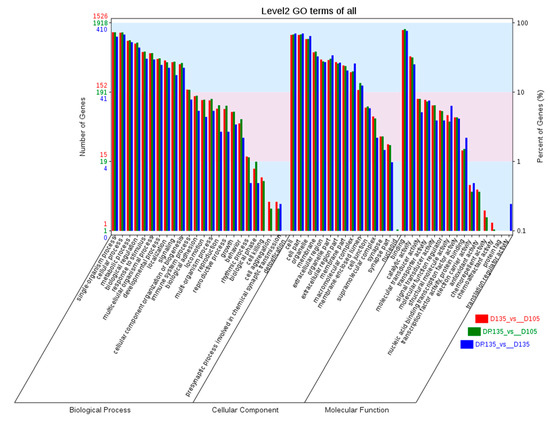
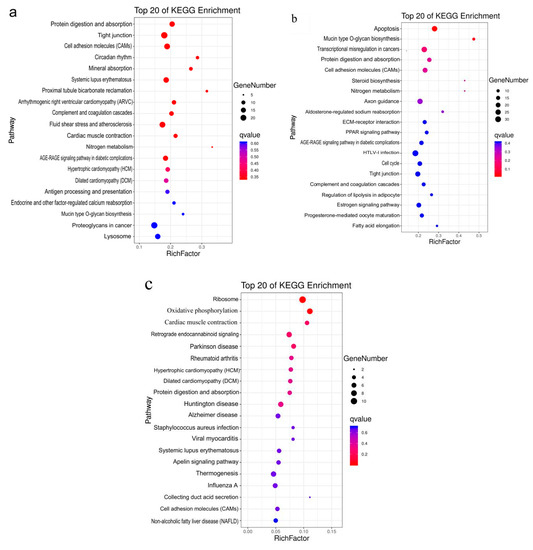
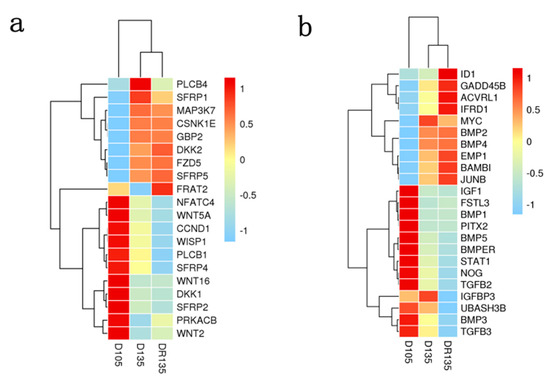
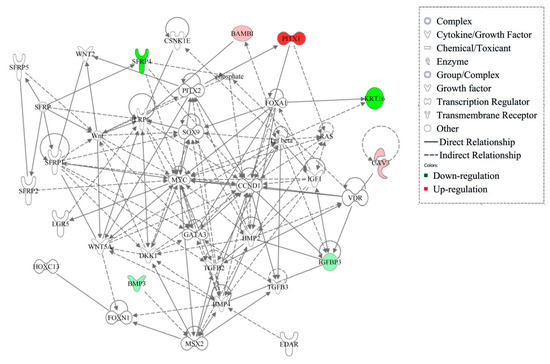
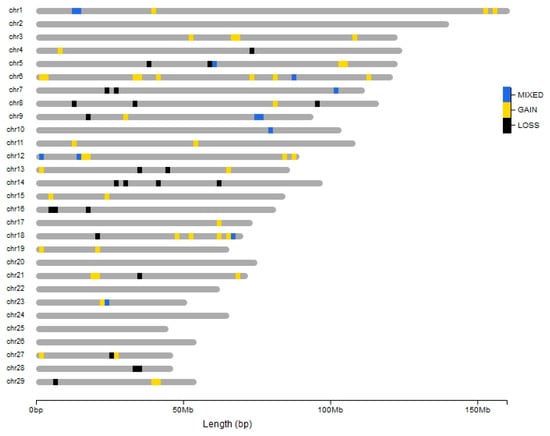
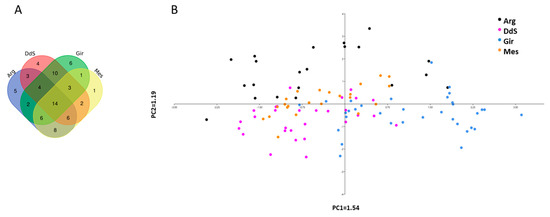
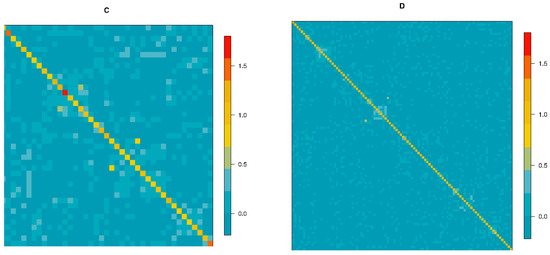
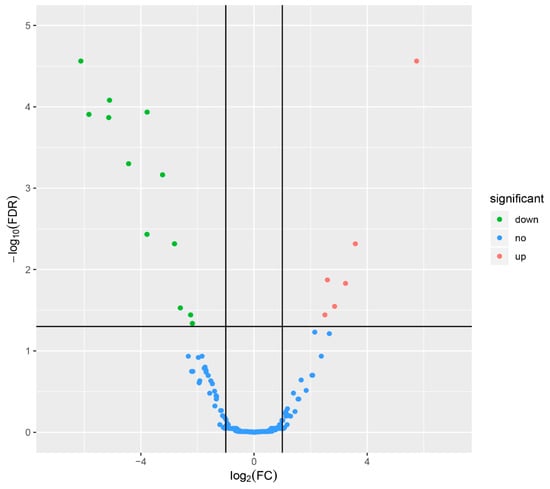
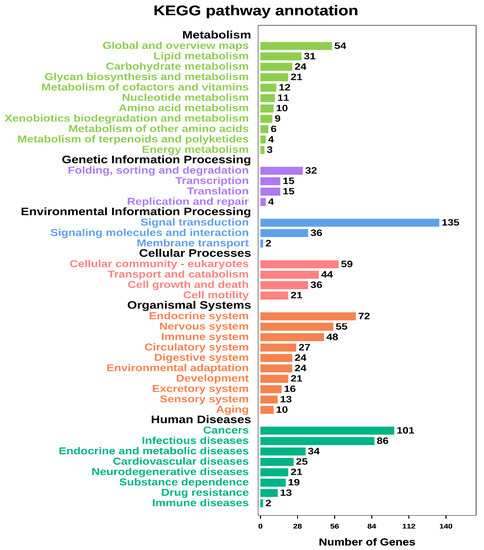
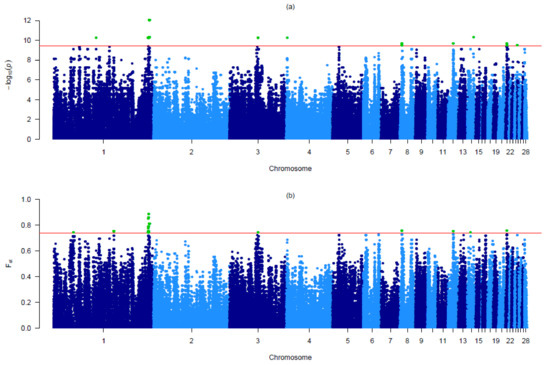
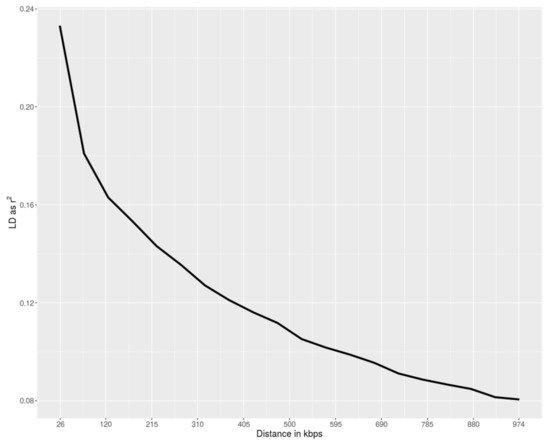
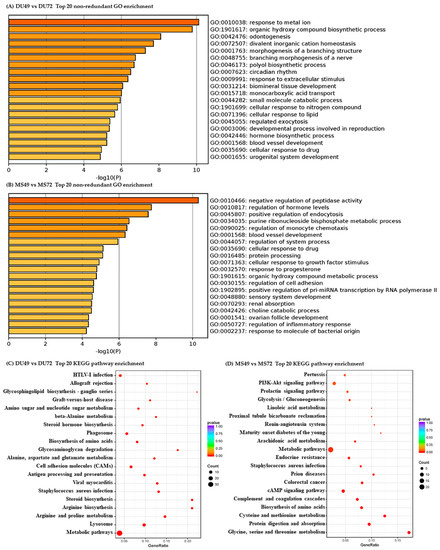
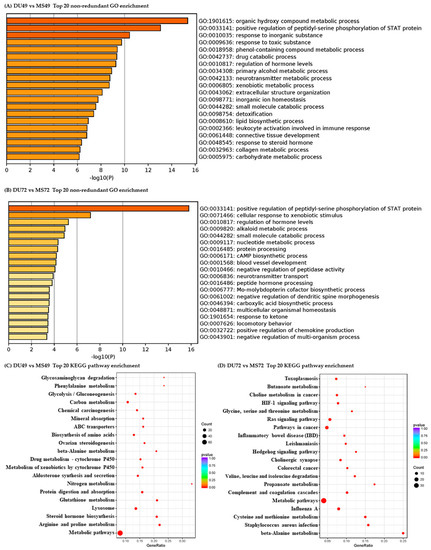
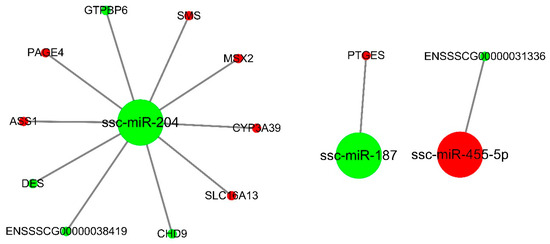
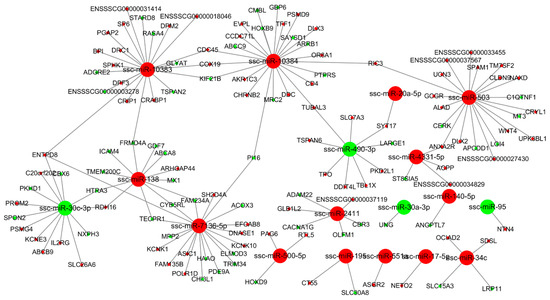
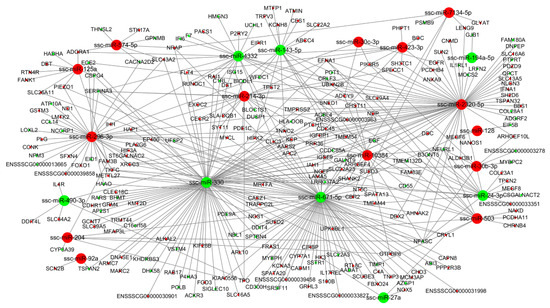
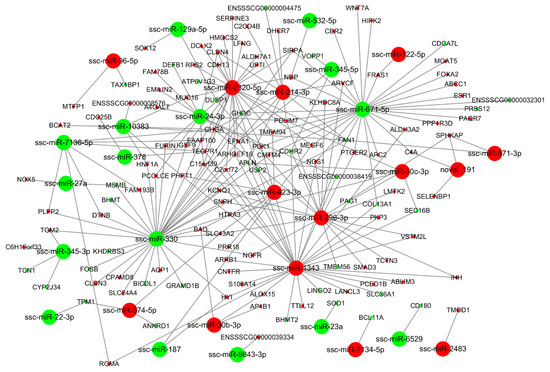
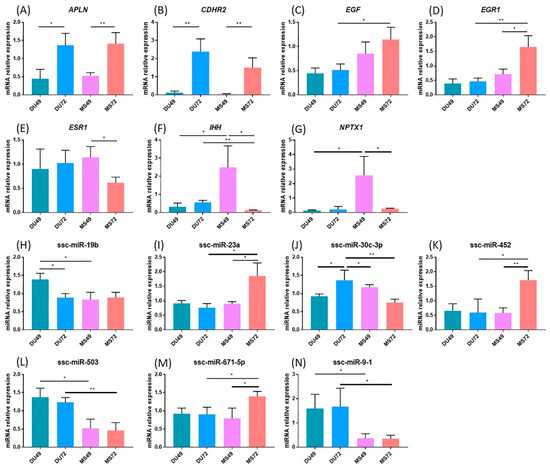
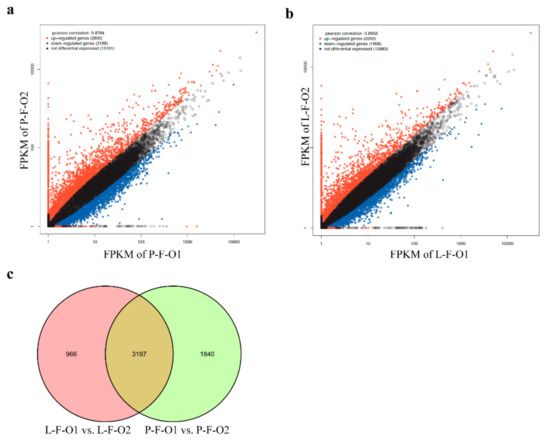
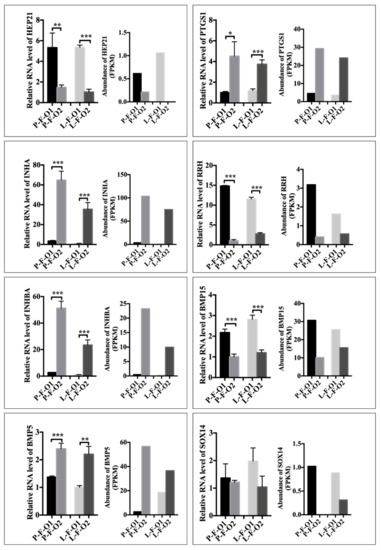
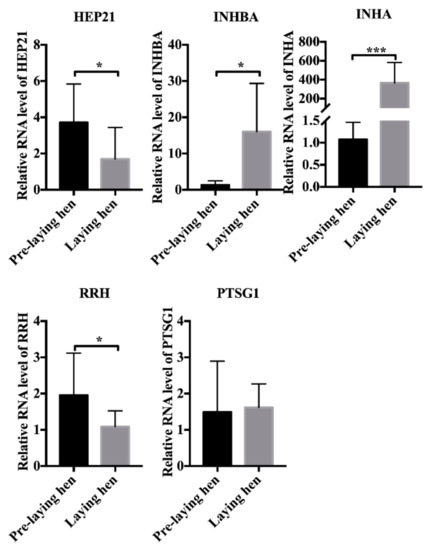
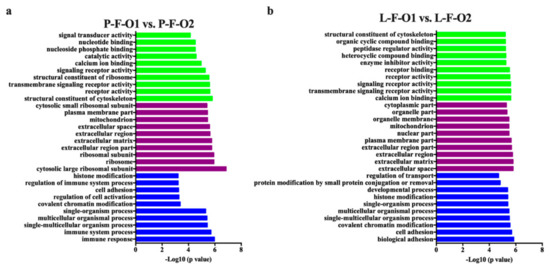
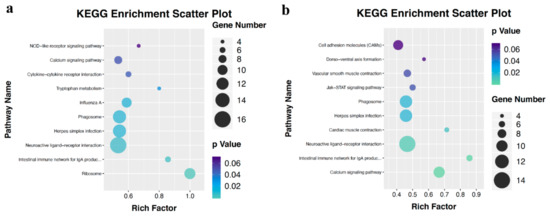
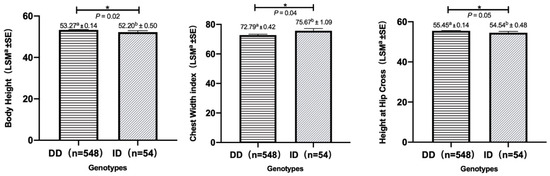
Tenderness is a key meat quality trait that determines the public acceptance of lamb consumption, so genetic improvement toward lamb with higher tenderness is pivotal for a sustainable sheep industry. However, unravelling the genomics controlling the tenderness is the first step. Therefore, this study aimed to identify the transcriptome signatures and polymorphisms related to divergent lamb tenderness using RNA deep sequencing. Since the molecules and enzymes that control muscle growth and tenderness are metabolized and synthesized in the liver, hepatic tissues of ten sheep with divergent phenotypes: five high- and five low-lamb tenderness samples were applied for deep sequencing. Sequence analysis identified the number of reads ranged from 21.37 to 25.37 million bases with a mean value of 22.90 million bases. In total, 328 genes are detected as differentially expressed (DEGs) including 110 and 218 genes that were up- and down-regulated, respectively. Pathway analysis showed steroid hormone biosynthesis as the dominant pathway behind the lamb tenderness. Gene expression analysis identified the top high (such as TP53INP1, CYP2E1, HSD17B13, ADH1C, and LPIN1) and low (such as ANGPTL2, IGFBP7, FABP5, OLFML3, and THOC5) expressed candidate genes. Polymorphism and association analysis revealed that mutation in OLFML3, ANGPTL2, and THOC5 genes could be potential candidate markers for tenderness in sheep. The genes and pathways identified in this study cause variation in tenderness, thus could be potential genetic markers to improve meat quality in sheep. However, further validation is needed to confirm the effect of these markers in different sheep populations so that these could be used in a selection program for lamb with high tenderness.
Full article
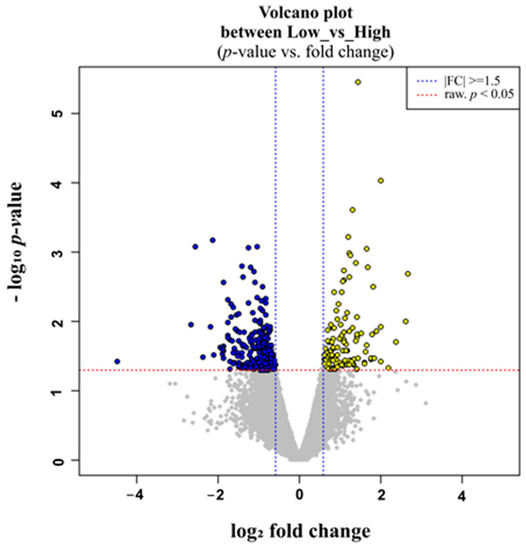
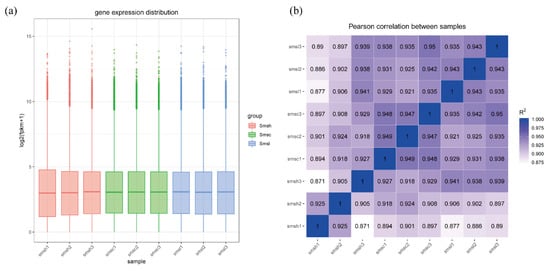
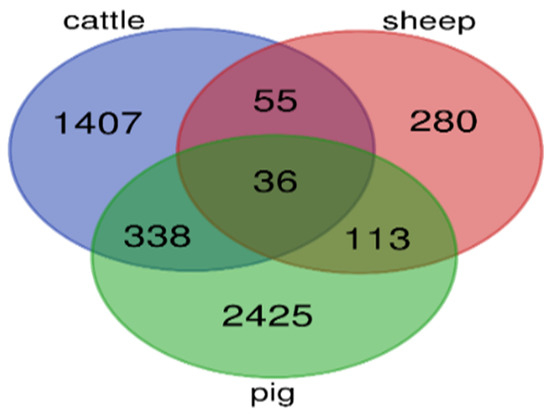
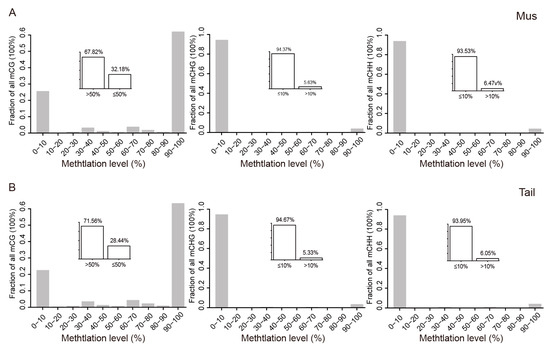
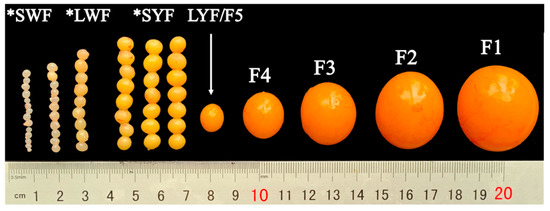
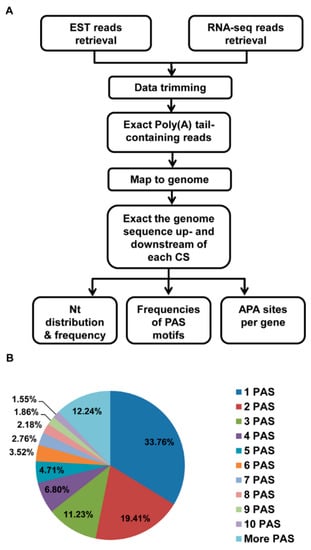
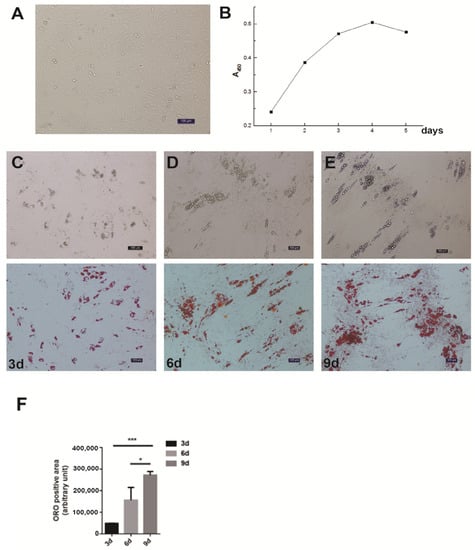
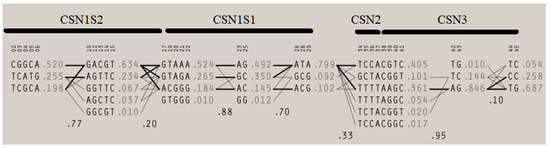
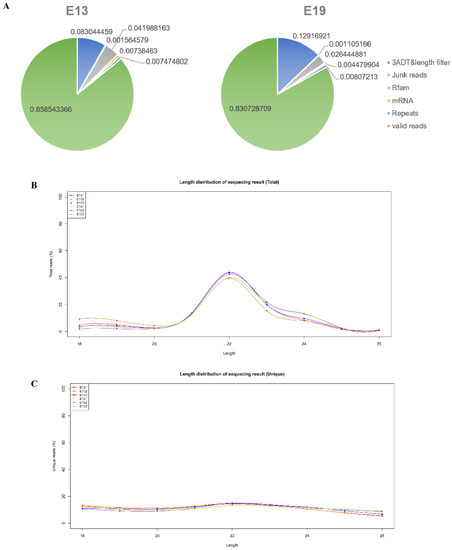
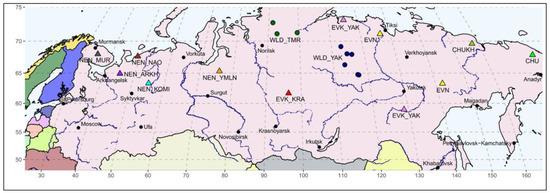
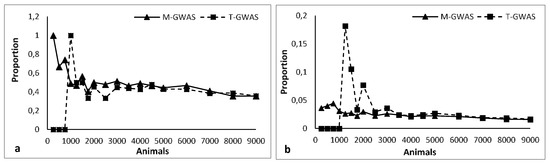
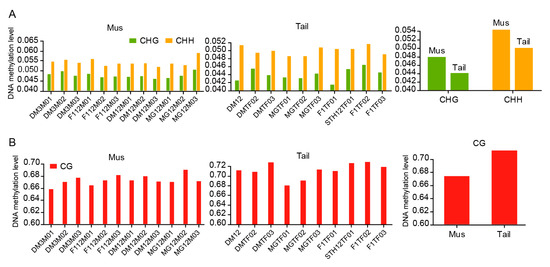
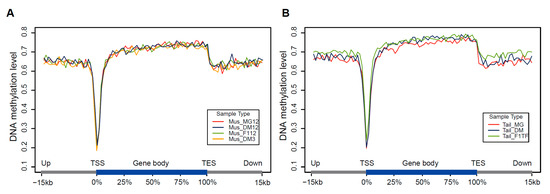
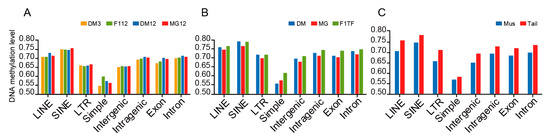
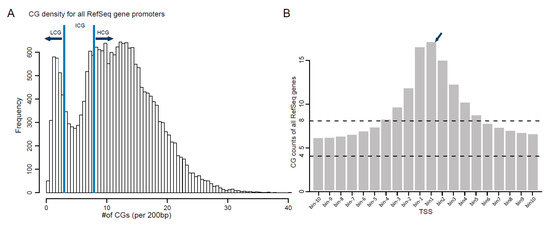
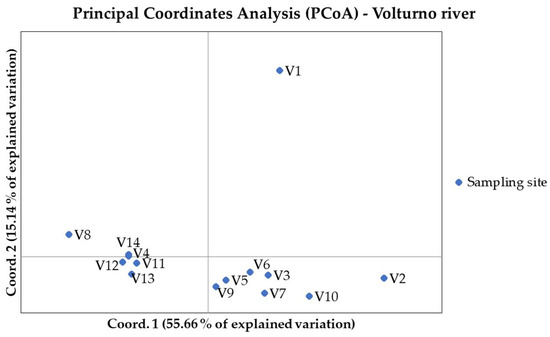
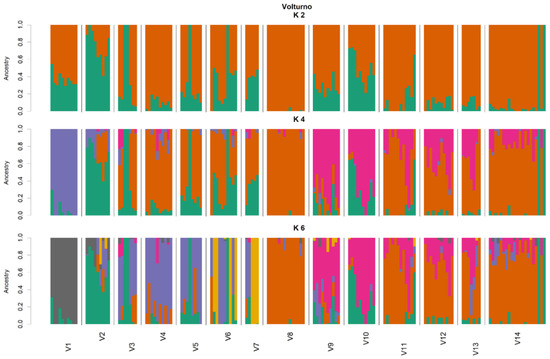
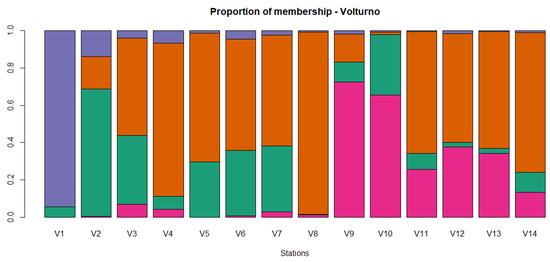
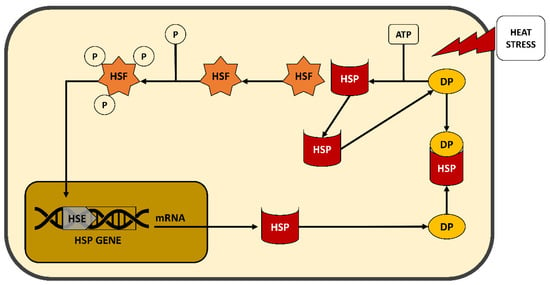
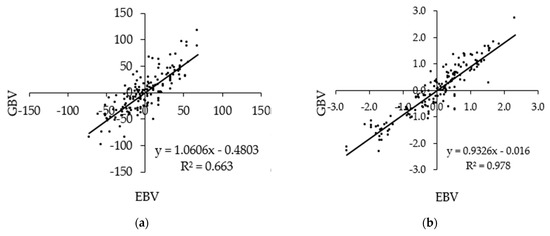
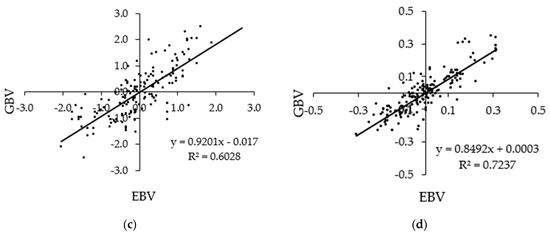
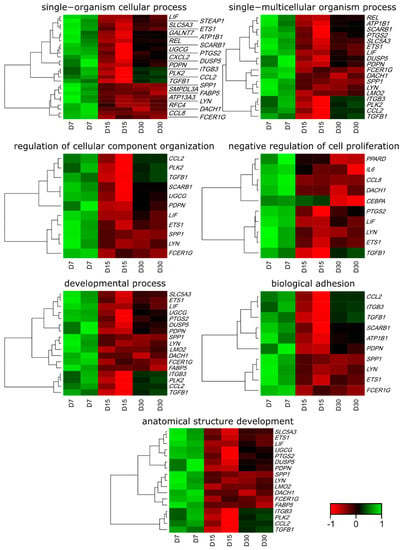
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}