
Genome-Wide DNA Methylation Patterns of Muscle and Tail-Fat in DairyMeade Sheep and Mongolian Sheep


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling for Whole-Genome DNA Methylation Sequencing
2.2. DNA Extraction, WGBS Library Preparation and Sequencing
2.3. Reads Mapping and DNA Methylation Detection
2.4. Annotation of Genomic Regions and Methylation Level Calculation
2.5. Identification of DMR-Containing Genes
3. Results
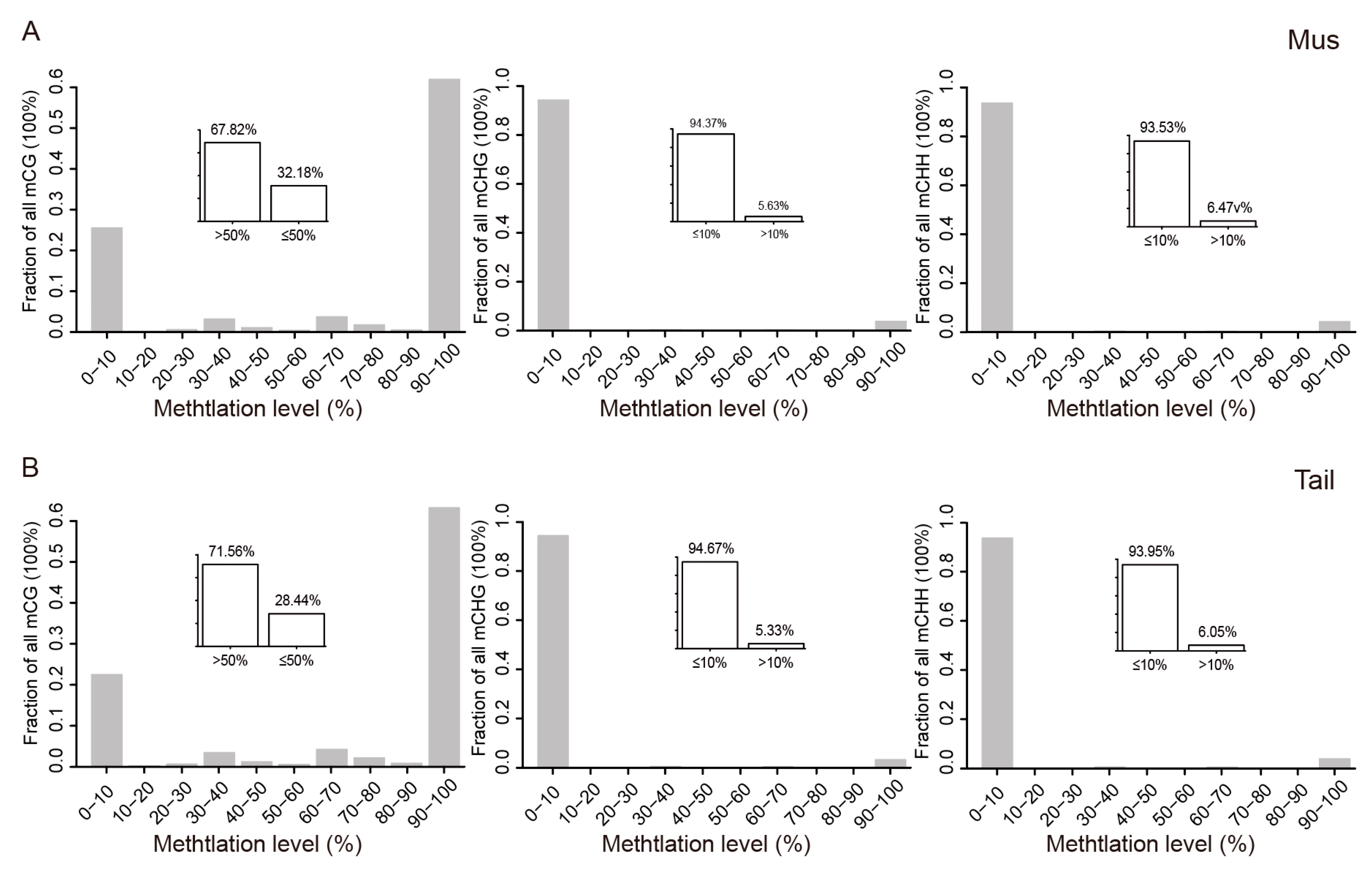
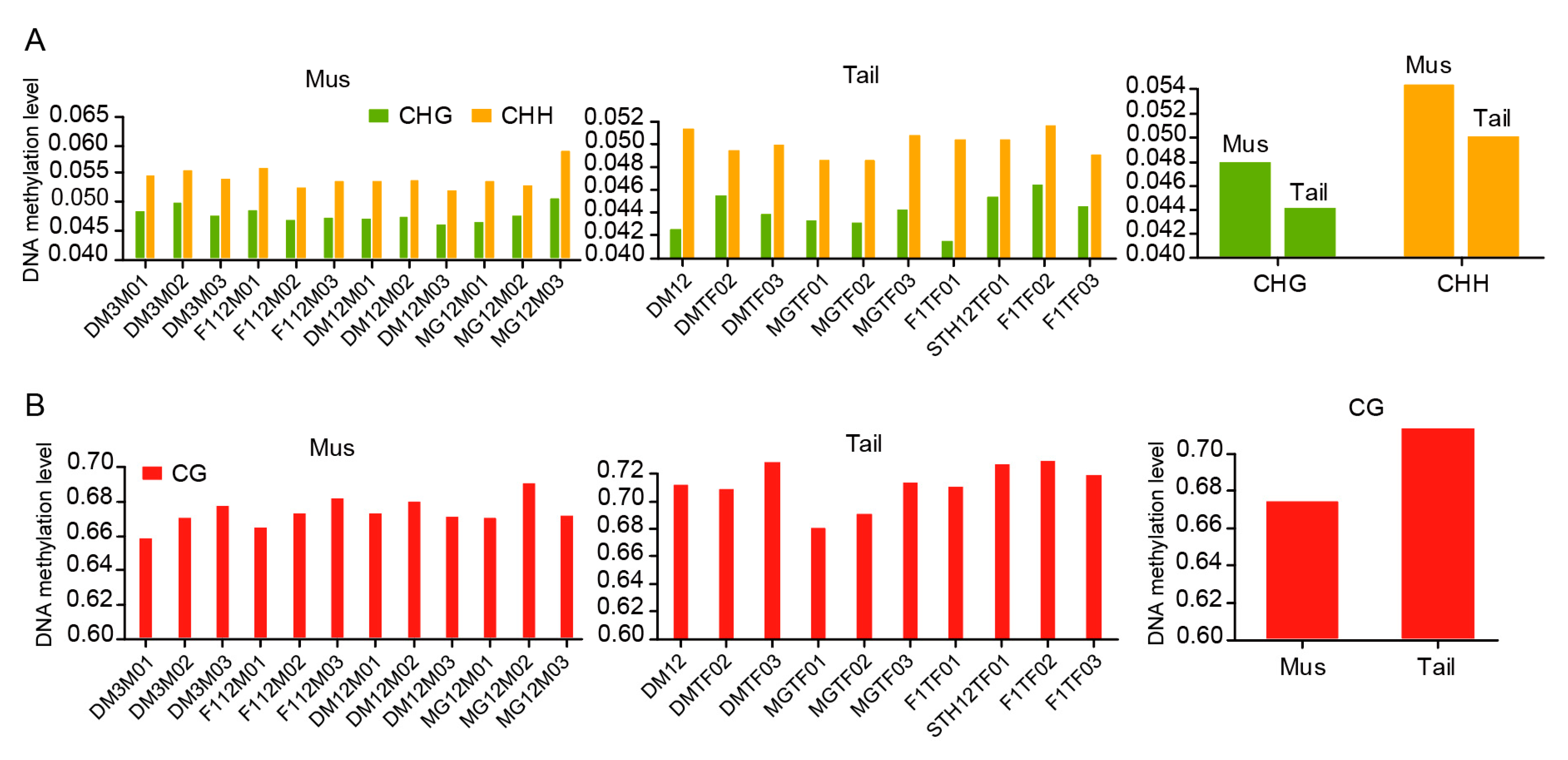
3.1. Global DNA Methylation Patterns
3.2. DNA Methylation Dynamics
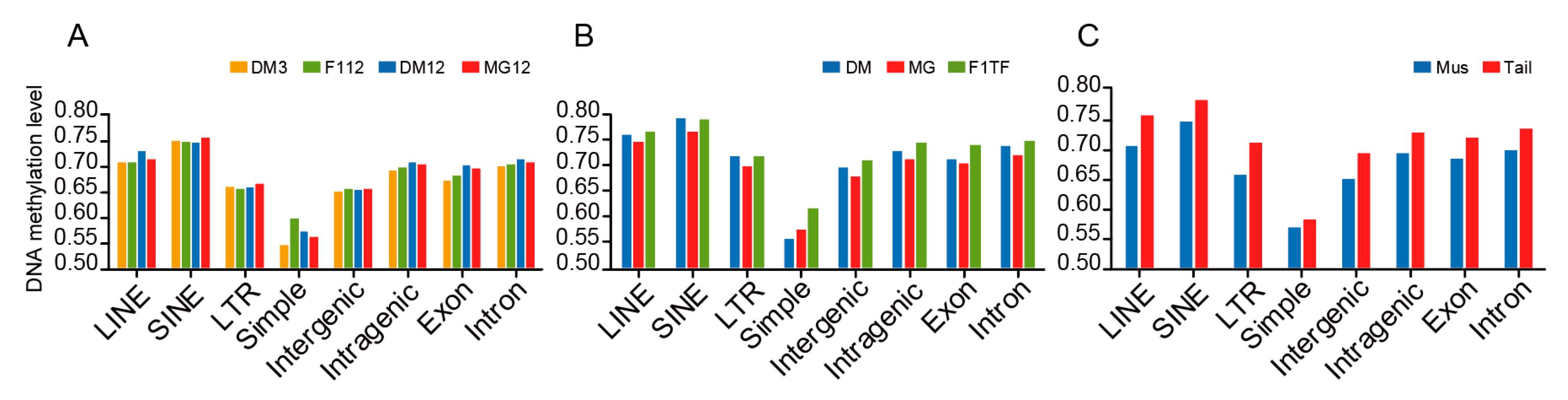
3.3. DNA Methylation of Repeat Elements and Other Genomic Regions
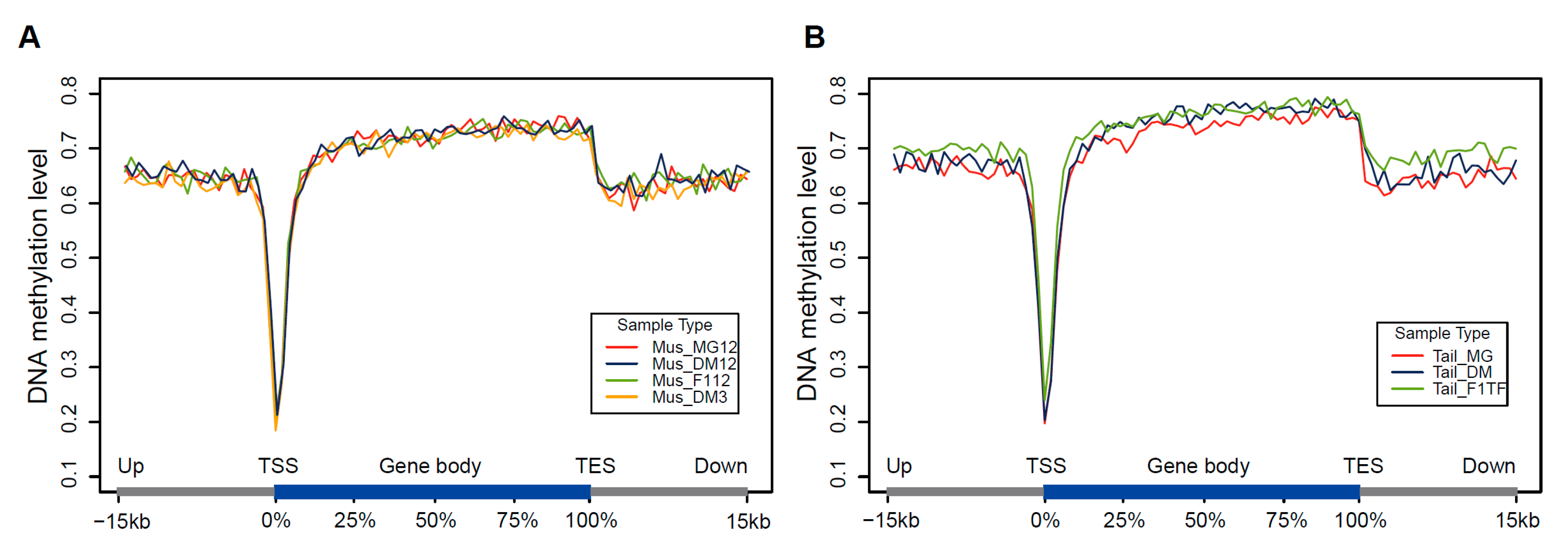
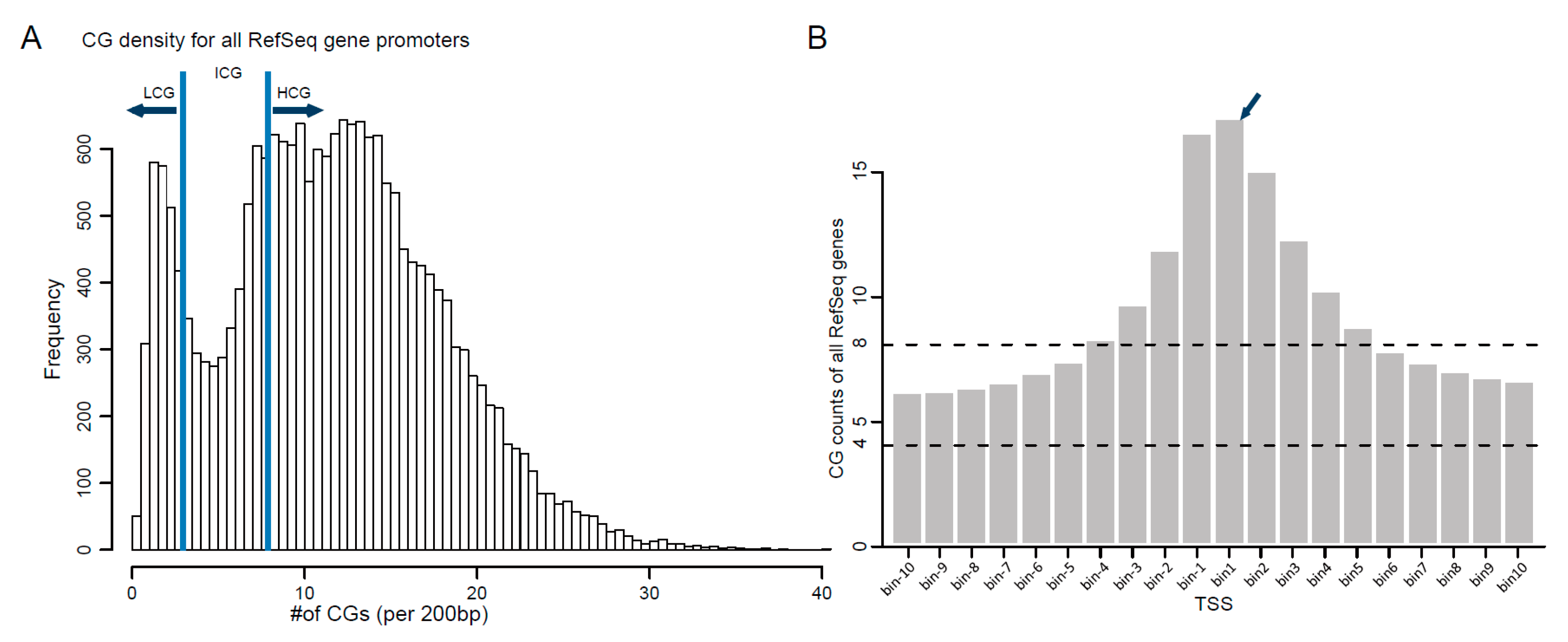
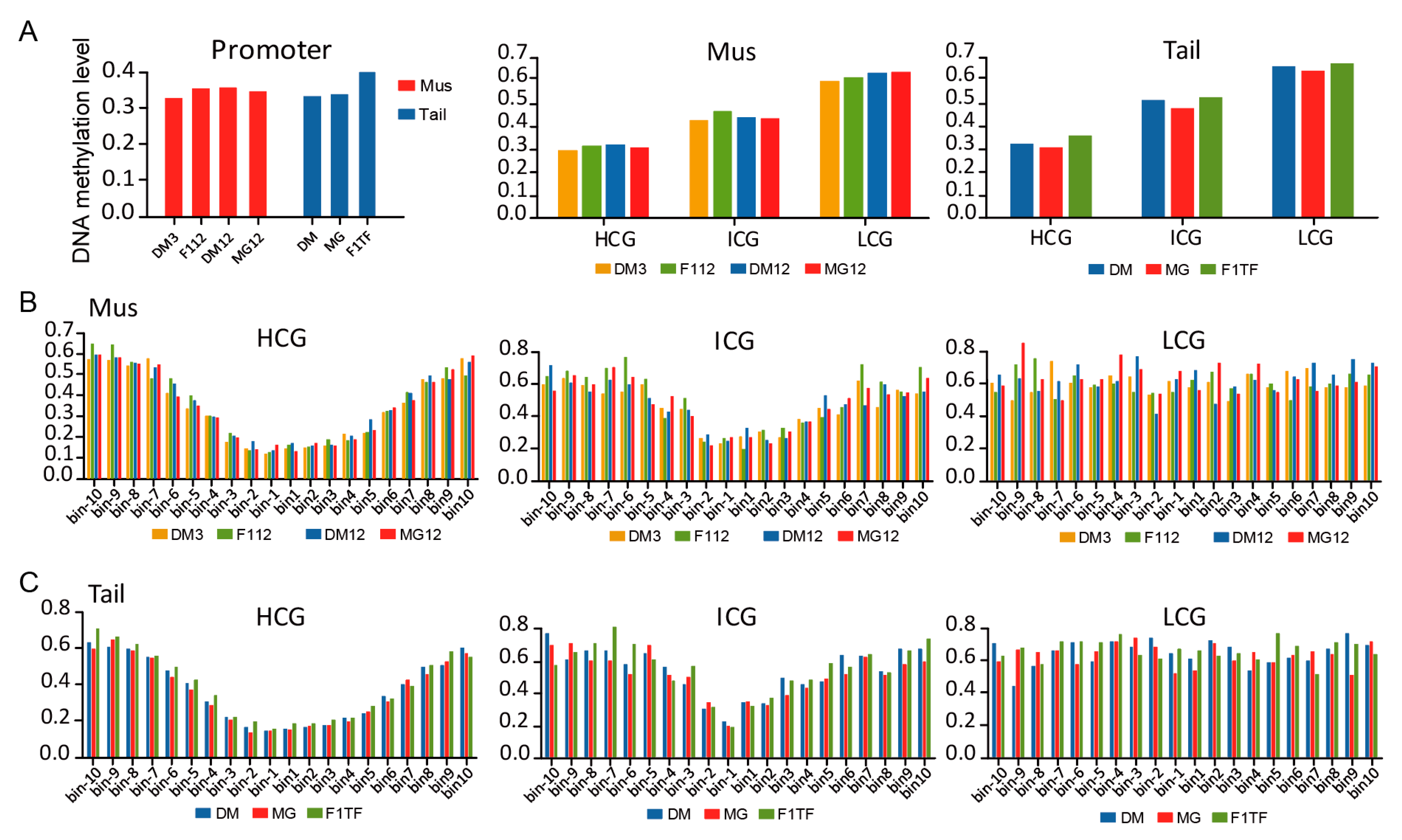
3.4. DNA Methylation of Gene Promoter Regions
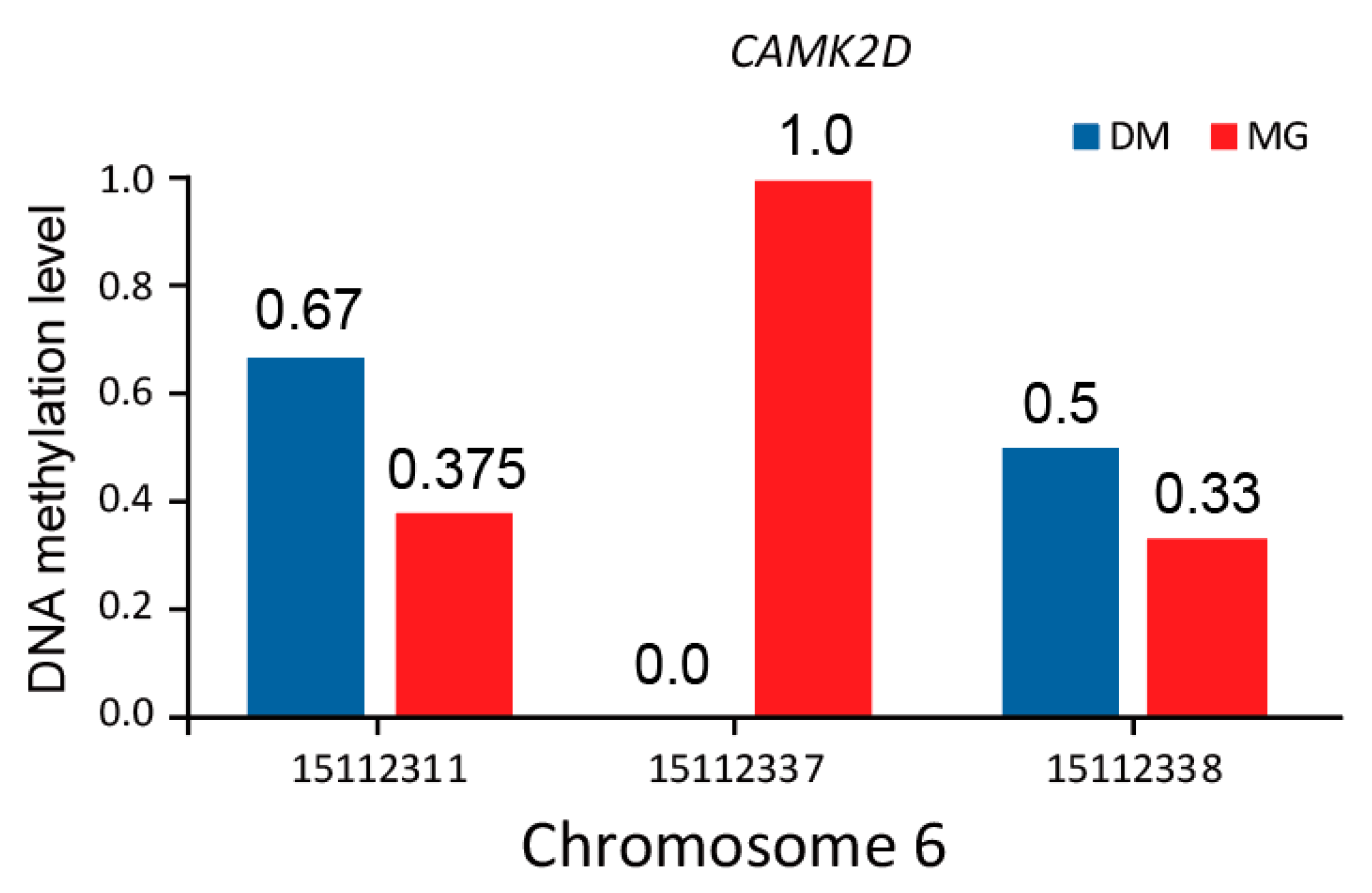
3.5. Analysis of Differentially Methylated Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walling, G.A.; Visscher, P.M.; Wilson, A.D.; McTeir, B.L.; Simm, G.; Bishop, S.C. Mapping of quantitative trait loci for growth and carcass traits in commercial sheep populations. J. Anim. Sci. 2004, 82, 2234–2245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Zhao, F.; Ren, H.; Xu, L.; Lu, J.; Zhang, S.; Zhang, X.; Wei, C.; Lu, G.; et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS ONE 2013, 8, e66569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, C.S. Animal models to study environmental epigenetics. Biol. Reprod. 2010, 82, 473–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gim, J.A.; Hong, C.P.; Kim, D.S.; Moon, J.W.; Choi, Y.; Eo, J.; Kwon, Y.J.; Lee, J.R.; Jung, Y.D.; Bae, J.H.; et al. Genome-wide analysis of DNA methylation before-and after exercise in the thoroughbred horse with MeDIP-Seq. Mol. Cells 2015, 38, 210–220. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, H.; Li, Z.; Zheng, X.; Jia, X.; Nie, Q.; Zhang, X. Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers. PLoS ONE 2013, 8, e56411. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Jiang, Z.; Xia, Y.; Lou, P.E.; Chen, L.; Wang, H.; Bai, L.; Xie, Y.; Liu, Y.; Li, W.; et al. Genome-wide DNA methylation changes in skeletal muscle between young and middle-aged pigs. BMC Genom. 2014, 15, 653. [Google Scholar]
- Cao, J.; Wei, C.; Liu, D.; Wang, H.; Wu, M.; Xie, Z.; Capellini, T.D.; Zhang, L.; Zhao, F.; Li, L.; et al. DNA methylation Landscape of body size variation in sheep. Sci. Rep. 2015, 5, 13950. [Google Scholar] [CrossRef] [Green Version]
- Lomniczi, A.; Loche, A.; Castellano, J.M.; Ronnekleiv, O.K.; Bosch, M.; Kaidar, G.; Knoll, J.G.; Wright, H.; Pfeifer, G.P.; Ojeda, S.R. Epigenetic control of female puberty. Nat. Neurosci. 2013, 16, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Abdoli, R.; Zamani, P.; Mirhoseini, S.Z.; Ghavi, H.N.; Nadri, S. A review on prolificacy genes in sheep. Reprod. Domest. Anim. 2016, 51, 631–637. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Feng, X.; Yang, H.; Zhu, A.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F. Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genom. 2017, 18, 759. [Google Scholar] [CrossRef]
- Yuan, X.L.; Gao, N.; Xing, Y.; Zhang, H.B.; Zhang, A.L.; Liu, J.; He, J.L.; Xu, Y.; Lin, W.M.; Chen, Z.M.; et al. Profiling the genome-wide DNA methylation pattern of porcine ovaries using reduced representation bisulfite sequencing. Sci. Rep. 2016, 6, 22138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Ye, J.; Li, X.; Gao, X.; Zhang, K.; Luo, L.; Ding, J.; Zhang, Y.; Li, Y.; Cao, H.; et al. DNA Methylation Patterns in the Hypothalamus of Female Pubertal Goats. PLoS ONE 2016, 11, e0165327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relton, C.L.; Groom, A.; St, P.B.; Sayers, A.E.; Swan, D.C.; Embleton, N.D.; Pearce, M.S.; Ring, S.M.; Northstone, K.; Tobias, J.H.; et al. DNA methylation patterns in cord blood DNA and body size in childhood. PLoS ONE 2012, 7, e31821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.H.; Seale, P.; Rudnicki, M.A. Looking back to the embryo: Defining transcriptional networks in adult myogenesis. Nat. Rev. Genet. 2003, 4, 497–507. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Ma, X.; Liu, B.; Su, R.; Jiang, Y.; Wang, W.; Dong, Y. Single Base-Resolution Methylome of the Dizygotic Sheep. PLoS ONE 2015, 10, e0142034. [Google Scholar] [CrossRef]
- Fan, Y.; Liang, Y.; Deng, K.; Zhang, Z.; Zhang, G.; Zhang, Y.; Wang, F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genom. 2020, 21, 327. [Google Scholar] [CrossRef]
- Li, M.; Wu, H.; Luo, Z.; Xia, Y.; Guan, J.; Wang, T.; Gu, Y.; Chen, L.; Zhang, K.; Ma, J.; et al. An atlas of DNA methylomes in porcine adipose and muscle tissues. Nat. Commun. 2012, 3, 850. [Google Scholar] [CrossRef]
- Zykovich, A.; Hubbard, A.; Flynn, J.M.; Tarnopolsky, M.; Fraga, M.F.; Kerksick, C.; Ogborn, D.; MacNeil, L.; Mooney, S.D.; Melov, S. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 2014, 13, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Zhao, Z.; Yu, H.; Li, G.; Jiang, P.; Yang, Y.; Yang, R.; Yu, X. Comparative genome-wide methylation analysis of longissimus dorsi muscles between Japanese black (Wagyu) and Chinese Red Steppes cattle. PLoS ONE 2017, 12, e0182492. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Bai, C.; Yang, L.; Zheng, Z.; Su, G.; Gao, G.; Wei, Z.; Zuo, Y.; Li, G. DNA methylation subpatterns at distinct regulatory regions in human early embryos. Open Biol. 2018, 8, 180131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, P.J.; Gavin, R.L.; Colombo, D.; Haining, E.J.; Reyat, J.S.; Payne, H.; Thielmann, I.; Lokman, A.B.; Neag, G.; Yang, J.; et al. Tspan18 is a novel regulator of the Ca2+ channel Orai1 and von Willebrand factor release in endothelial cells. Haematologica 2019, 104, 1892–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.D.; Chen, Q.; Baye, N.L.; Huang, Y.; Healy, C.L.; Kasinathan, S.; O’Connell, T.D. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ. Res. 2008, 103, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.J.; Kiens, B.; Richter, E.A. Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. J. Physiol. 2006, 574 Pt 3, 889–903. [Google Scholar] [CrossRef]
- Little, G.H.; Bai, Y.; Williams, T.; Poizat, C. Nuclear calcium/calmodulin-dependent protein kinase IIdelta preferentially transmits signals to histone deacetylase 4 in cardiac cells. J. Biol. Chem. 2007, 282, 7219–7231. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Guo, F.; Yan, L.; Guo, H.; Li, L.; Hu, B.; Zhao, Y.; Yong, J.; Hu, Y.; Wang, X.; Wei, Y.; et al. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell 2015, 161, 1437–1452. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wu, K.; Liu, Z.; Yao, X.; Yuan, S.; Tao, W.; Yi, L.; Yu, G.; Hou, Z.; Fan, D.; et al. Chromatin Accessibility Landscape in Human Early Embryos and Its Association with Evolution. Cell 2018, 173, 248–259.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Wang, Y.; Zhang, F.; Yao, F.; Sun, C. Calcium Signaling Pathways: Key Pathways in the Regulation of Obesity. Int. J. Mol. Sci. 2019, 20, 2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Shin, D.; Song, K.D. Dominance effects of ion transport and ion transport regulator genes on the final weight and backfat thickness of Landrace pigs by dominance deviation analysis. Genes Genom. 2018, 40, 1331–1338. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, R.; Dai, X.; Zhang, L.; Li, G.; Zheng, Z. Genome-Wide DNA Methylation Patterns of Muscle and Tail-Fat in DairyMeade Sheep and Mongolian Sheep. Animals 2022, 12, 1399. https://doi.org/10.3390/ani12111399
Luo R, Dai X, Zhang L, Li G, Zheng Z. Genome-Wide DNA Methylation Patterns of Muscle and Tail-Fat in DairyMeade Sheep and Mongolian Sheep. Animals. 2022; 12(11):1399. https://doi.org/10.3390/ani12111399
Chicago/Turabian StyleLuo, Rongsong, Xuelei Dai, Li Zhang, Guangpeng Li, and Zhong Zheng. 2022. "Genome-Wide DNA Methylation Patterns of Muscle and Tail-Fat in DairyMeade Sheep and Mongolian Sheep" Animals 12, no. 11: 1399. https://doi.org/10.3390/ani12111399