

Use of Shotgun Metagenomics to Assess the Microbial Diversity and Hydrocarbons Degrading Functions of Auto-Mechanic Workshops Soils Polluted with Gasoline and Diesel Fuel
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sites Location and Soil Sampling
2.2. DNA Extraction and Next-Generation Sequencing
2.3. Annotation and Functional Profiling of the Assembled Metagenomes
2.4. Diversity Assessment
2.5. Carbon and Nitrogen Cultivable Microbial Biomass and Soil Enzymes
2.6. Determination of Soil Enzymes
2.7. Bacterial Strains’ Isolation and Assessment of Hydrocarbon-Degradation Abilities
2.8. Identification of Bacterial Strains
3. Results
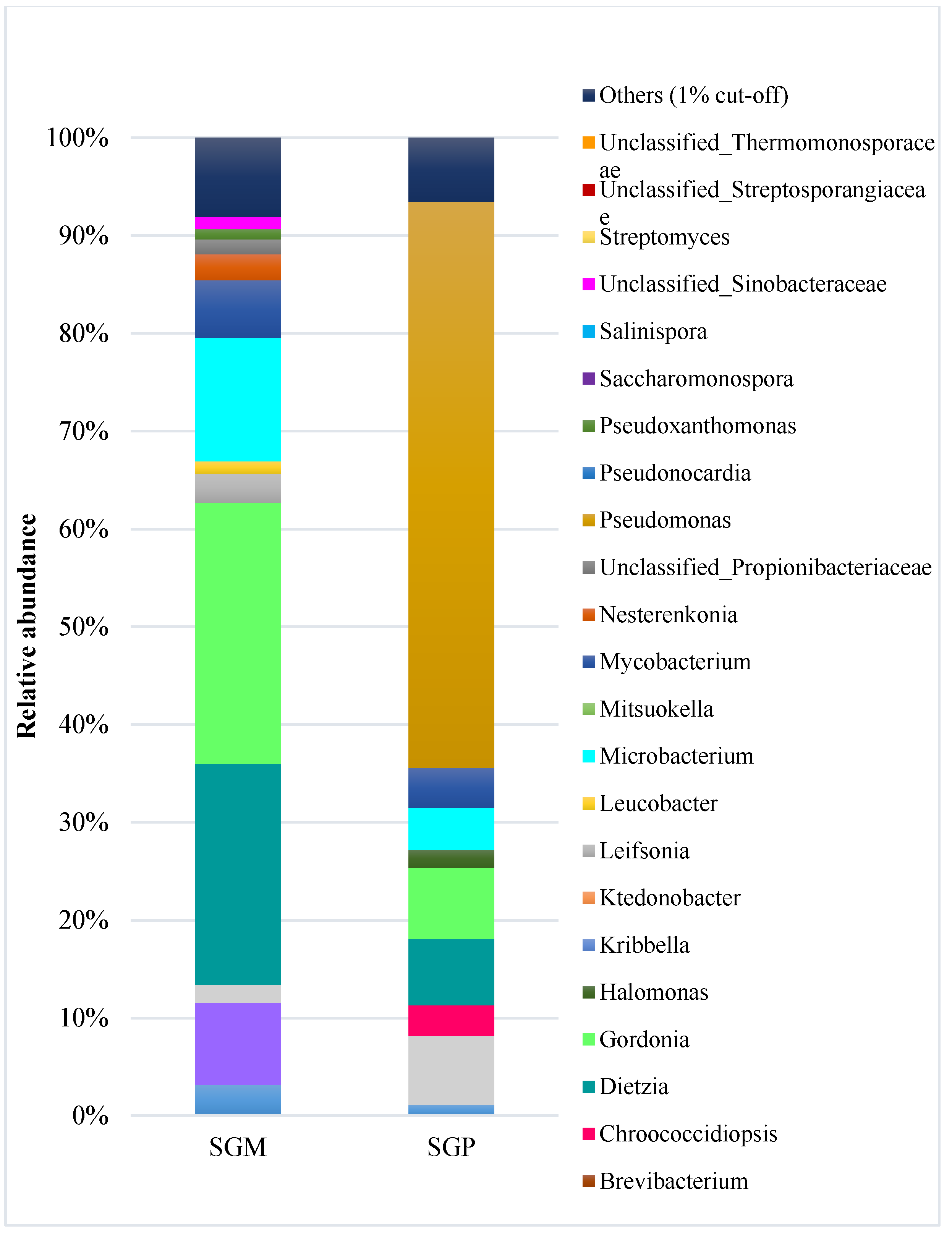
3.1. Microbial Community Composition
3.2. Functional Analysis
3.3. Soil Enzymes and Cultivable Microbial Biomass
3.4. Isolation of Bacteria and Screening for Hydrocarbon Degraders
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nkwoada, A.U.; Alisa, C.O.; Amakom, C.M. Pollution in Nigerian Auto-Mechanic Villages: A Review. IOSR J. Environ. Sci. Toxicol. Food Technol. 2018, 12, 43–54. [Google Scholar]
- Muze, N.E.; Opara, A.I.; Ibe, F.C.; Njoku, O.C. Assessment of the geo-environmental effects of activities of auto-mechanic workshops at Alaoji Aba and Elekahia Port Harcourt, Niger Delta, Nigeria. Environ. Anal. Health Toxicol. 2020, 35, e2020005. [Google Scholar] [CrossRef]
- Jolaoso, A.O.; Njoku, K.L.; Adedokun, A.H.; Adesuyi, A.A. Assessment of Automobile Mechanic Workshop Soils in Lagos and the Genotoxic Potential of the Simulated Leachate using Allium cepa L. EQA-Int. J. Environ. Qual. 2019, 34, 48–62. [Google Scholar] [CrossRef]
- Alabi, A.B.; Aiyesanmi, A.F.; Ololade, I.A. Qualitative and Quantitative Assessment of Hydrocarbons in Soil Profiles of Auto- Mechanic Workshop: A Case Study of Akure City, Nigeria. Polycycl. Aromat. Compd. 2019, 41, 1–14. [Google Scholar] [CrossRef]
- Jolaoso, A.O.; Njoku, K.L.; Adesalu, T.A. Investigation of Contaminated Soils from a Major Automobile Mechanic Workshop in Lagos Mainland, Lagos State and an Oil Spill Site in Bodo Creek, Rivers State, Nigeria. Afr. J. Phycol. 2021, 02, 013–020. [Google Scholar]
- Ferraro, A.; Massini, G.; Miritana, V.M.; Panico, A.; Pontoni, L.; Race, M.; Rosa, S.; Signorini, A.; Fabbricino, M.; Pirozzi, F. Bioaugmentation strategy to enhance polycyclic aromatic hydrocarbons anaerobic biodegradation in contaminated soils. Chemosphere 2021, 275, 130091. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.S.; Okoye, C.O.; Ezeorba, T.P.C.; Mao, G.; Chen, Y.; Xu, H.; Song, C.; Feng, W.; Wu, X. Emerging bio-dispersant and bioremediation technologies as environmentally friendly management responses toward marine oil spill: A comprehensive review. J. Environ. Manag. 2022, 322, 116123. [Google Scholar] [CrossRef] [PubMed]
- Gagandeep, S.; Malik, D.K. Utilization of 2T engine oil by Pseudomonas sp. isolated from automobile workshop contaminated soil. Int. J. Chem. Anal. Sci. 2013, 4, 80–84. [Google Scholar] [CrossRef]
- Ruiz, O.N.; Brown, L.M.; Radwan, O.; Bowen, L.L.; Gunasekera, T.S.; Mueller, S.S.; West, Z.J.; Striebich, R.C. Metagenomic characterization reveals complex association of soil hydrocarbon-degrading bacteria. Int. Biodeterior. Biodegrad. 2021, 157, 105161. [Google Scholar] [CrossRef]
- Ebakota, O.D.; Osarueme, J.O.; Gift, O.N.; Odoligie, I.; Osazee, J.O. Isolation and Characterization of Hydrocarbon-Degrading Bacteria in Top and Subsoil of selected Mechanic Workshops in Benin City Metropolis, Nigeria. J. Applied Sci. Environ. Manag. 2017, 21, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Hassana, A.; Vincent, B.T.; Ushuji, O.D.; Yakubu, N.; Boko, U.H.; Gogo, M.F. Molecular Identification of Hydrocarbon Degrading Bacteria Isolated from Contaminated Soil of Automobile Mechanic Workshop in Lapai, Niger State. Ind. J. Pure App. Biosci. 2019, 7, 31–37. [Google Scholar] [CrossRef]
- Wu, M.; Dick, W.A.; Li, W.; Wang, X.; Yang, Q.; Wang, T.; Xu, L.; Zhang, M.; Chen, L. Bioaugmentation and biostimulation of hydrocarbon degradation and the microbial community in a petroleum-contaminated soil. Int. Biodeter. Biodegrad. 2016, 107, 158–164. [Google Scholar] [CrossRef]
- Curiel-Alegre, S.; Velasco-Arroyo, B.; Rumbo, C.; Khan, A.H.A.; Tamayo-Ramos, J.A.; Rad, C.; Gallego, J.L.R.; Barros, R. Evaluation of biostimulation, bioaugmentation, and organic amendments application on the bioremediation of recalcitrant hydrocarbons of soil. Chemosphere 2022, 307, 135638. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kadam, V.; Patil, Y. Isolation and development of a microbial consortium for the treatment of automobile service station wastewater. J. Appl. Microbiol. 2022, 132, 1048–1061. [Google Scholar] [CrossRef]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Hu, Z.; Wang, H. Metagenomic analysis exhibited the co-metabolism of polycyclic aromatic hydrocarbons by bacterial community from estuarine sediment. Environ. Int. 2019, 129, 308–319. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, M.-S.; Kim, J.-G.; Kim, S.-O. Use of Soil Enzymes as Indicators for Contaminated Soil Monitoring and Sustainable Management. Sustainability 2020, 12, 8209. [Google Scholar] [CrossRef]
- Goma-Tchimbakala, E.J.C.D.; Pietrini, I.; Dal Bello, F.; Goma-Tchimbakala, J.; Lo Russo, S.; Corgnati, S.P. Great Abilities of Shinella zoogloeoides Strain from a Landfarming Soil for Crude Oil Degradation and a Synergy Model for Alginate-Bead-Entrapped Consortium Efficiency. Microorganisms 2022, 10, 1361. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Dave, B.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [Green Version]
- Dinghua, L.; Chi-Man, L.; Ruibang, L.; Kunihiko, S.; Tak-Wah, L. MEGAHIT: An ultra- fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Huttenhower, C. Metabolic Reconstruction for Metagenomic Data and Its Application to the Human Microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddle, K.; McGonigle, T.; Koiter, A. Microbe Biomass in Relation to Organic Carbon and Clay in Soil. Soil Syst. 2020, 4, 41. [Google Scholar] [CrossRef]
- Jirka, A.M.; Carter, M.J. Micro semiautomated analysis of surface and waste waters for chemical oxygen demand. Anal. Chem. 1975, 47, 1397–1402. [Google Scholar] [CrossRef]
- Bolleter, W.; Bushman, T.C.J.; Tidwell, P.W. Spectrophotometric Determination of Ammonia as Indophenol. Anal. Chem. 1961, 33, 592–594. [Google Scholar] [CrossRef]
- Joergensen, R.G.; Mueller, T. The Fumigation-Extraction Method to Estimate Soil Microbial Biomass: Calibration of the kEN Value. Soil Biol. Biochem. 1996, 28, 33–37. [Google Scholar] [CrossRef]
- Dawson, J.J.C.; Godsiffe, E.J.; Thompson, I.P.; Ralebitso-Senior, T.K.; Killham, K.S.; Paton, G.I. Application of biological indicators to assess recovery of hydrocarbon impacted soils. Soil Biol. Biochem. 2007, 39, 164–177. [Google Scholar] [CrossRef]
- Tabatabai, M.A.; Bremner, J.M. Use of p-nitrophenol phosphate for assay of soil phosphatase activity. Soil Biol. Biochem. 1969, 1, 301–307. [Google Scholar] [CrossRef]
- Parham, J.A.; Deng, S.P. Detection, quantifcation and characterization of β-glucosaminidase activity in soil. Soil Biol. Biochem. 2000, 32, 1183–1190. [Google Scholar] [CrossRef]
- Auti, A.; Narwade, N.; Deshpande, N.; Dhotre, D. Microbiome and imputed metagenome study of crude and refined petroleum-oil-contaminated soils: Potential for hydrocarbon degradation and plant-growth promotion. J. Biosci. 2019, 44, 114. [Google Scholar] [CrossRef] [PubMed]
- Sutton, N.B.; Maphosa, F.; Morillo, J.A.; Al-Soud, W.A.; Langenhoff, A.A.M.; Grotenhuis, T.; Rijnaarts, H.H.M.; Smidt, H. Impact of Long-Term Diesel Contamination on Soil Microbial Community Structure. Appl. Environ. Microbiol. 2013, 79, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Liu, Z.; Jia, X.; Lu, W. Distribution of Bacterial Communities in Petroleum-Contaminated Soils from the Dagang Oilfield, China. Trans. Tianjin Univ. 2019, 26, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Gielnik, A.; Pechaud, Y.; Huguenot, D.; C’ebron, A.; Esposito, G.; van Hullebusch, E.D. Functional potential of sewage sludge digestate microbes to degrade aliphatic hydrocarbons during bioremediation of a petroleum hydrocarbons contaminated soil. J. Environ. Manag. 2021, 280, 11648. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, W.; Tian, S.; Wang, W.; Qi, Q.; Jiang, P.; Gao, X.; Li, F.; Li, H.; Yu, H. Petroleum Hydrocarbon-Degrading Bacteria for the Remediation of Oil Pollution Under Aerobic Conditions: A Perspective Analysis. Front. Microbiol. 2018, 9, 2885. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chi, C.; Nie, Y.; Tang, Y.; Tan, Y.; Wu, G.; Wu, X. Degradation of petroleum hydrocarbons (C6–C40) and crude oil by a novel Dietzia strain. Bioresour. Technol. 2011, 102, 7755–7761. [Google Scholar] [CrossRef]
- Silva, N.M.; de Oliveira, A.M.S.A.; Pegorin, S.; Giusti, C.E.; Ferrari, V.B.; Barbosa, D.; Martins, L.F.; Morais, C.; Setubal, J.C.; Vasconcellos, S.P.; et al. Characterization of novel hydrocarbon-degrading Gordonia paraffinivorans and Gordonia sihwensis strains isolated from composting. PLoS ONE 2019, 14, e0215396. [Google Scholar] [CrossRef] [PubMed]
- Gregson, B.H.; Metodieva, G.; Metodiev, M.V.; Golyshin, P.N.; McKew, B.A. Differential Protein Expression During Growth on Medium Versus Long-Chain Alkanes in the Obligate Marine Hydrocarbon-Degrading Bacterium Thalassolituus oleivorans MIL−1. Front. Microbiol. 2018, 9, 3130. [Google Scholar] [CrossRef] [Green Version]
- Abbasian, F.; Palanisami, T.; Megharaj, M.; Naidu, R. Microbial Diversity and Hydrocarbon Degrading Gene Capacity of a Crude Oil Field Soil as Determined by Metagenomics Analysis. Biotechnol. Prog. 2016, 32, 638–648. [Google Scholar] [CrossRef]
- Baburam, C.; Feto, N.A. Mining of two novel aldehyde dehydrogenases (DHY-SC-VUT5 and DHY-G VUT7) from metagenome of hydrocarbon contaminated soils. BMC Biotechnol. 2021, 21, 18. [Google Scholar] [CrossRef]
- Frantsuzova, E.; Delegan, Y.; Bogun, A.; Sokolova, D.; Nazina, T. Comparative Genomic Analysis of the Hydrocarbon-Oxidizing Dibenzothiophene-Desulfurizing Gordonia Strains. Microorganisms 2023, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Sierra-García, I.N.; Correa Alvarez, J.; Pantaroto de Vasconcellos, S.; Pereira de Souza, A.; dos Santos Neto, E.V.; de Oliveira, V.M. New Hydrocarbon Degradation Pathways in the Microbial Metagenome from Brazilian Petroleum Reservoirs. PLoS ONE 2014, 9, e90087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenghi, F.L.G.; Berlanda, D.; Galli, E.; Sello, G.; Barbieri, P. Organization and Regulation of meta–Cleavage Pathway Genes for Toluene and o-Xylene Derivative Degradation in Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 2001, 67, 3304–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinaru, E.; Naanuri, E.; Grünbach, M.; Jõesaar, M.; Heinaru, A. Functional redundancy in phenol and toluene degradation in Pseudomonas stutzeri strains isolated from the Baltic Sea. Gene 2016, 589, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Solís-González, C.J.; Loza-Tavera, H. Alicycliphilus: Current knowledge and potential for bioremediation of xenobiotics. J. Appl. Microbiol. 2019, 126, 1643–1656. [Google Scholar] [CrossRef] [Green Version]
- Zaki, S. Detection of meta- and ortho-cleavage dioxygenases in bacterial phenol-degraders. J. Appl. Sci. Environ. Mgt. 2006, 10, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Tomás-Gallardo, L.; Gómez-Álvarez, H.; Santero, E.; Floriano, B. Combination of degradation pathways for naphthalene utilization in Rhodococcus sp. strain TFB. Microb Biotechnol. 2014, 7, 100–113. [Google Scholar] [CrossRef] [Green Version]
- Schell, M.A.; Brown, P.H.; Raju, S. Use of saturation mutagenesis to localize probable functional domains in the NahR protein, a LysR-type transcription activator. J. Biol. Chem. 1990, 265, 3844–3850. [Google Scholar] [CrossRef]
- Wells, J.T.; Ragauskas, A.T. Biotechnological opportunities with the β-ketoadipate pathway. Trends Biotechnol. 2012, 30, 627–637. [Google Scholar] [CrossRef]
- Harwood, C.S.; Parales, R.E. The beta-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 1996, 50, 553–590. [Google Scholar] [CrossRef]
- Franco, M.; Contin, G.; Bragato, M.; De Nobili, M. Microbiological resilience of soils contaminated with crude oil. Geoderma 2004, 121, 17–30. [Google Scholar] [CrossRef]
- Marschner, B.; Kalbitz, K. Controls of bioavailability and biodegradability of dissolved organic matter in soils. Geoderma 2003, 113, 211–235. [Google Scholar] [CrossRef]
- Anza, M.; Salazar, O.; Epelde, L.; Becerril, J.M.; Alkorta, I.; Garbisu, C. Remediation of Organically Contaminated Soil Through the Combination of Assisted Phytoremediation and Bioaugmentation. Appl. Sci. 2019, 9, 4757. [Google Scholar] [CrossRef] [Green Version]
- Ziervogel, K.; Arnosti, C. Enhanced protein and carbohydrate hydrolyses in plume-associated deepwaters initially sampled during the early stages of the Deepwater Horizon oil spill. Deep Sea Res. Part II Top. Stud. Oceanogr. 2016, 129, 368–373. [Google Scholar] [CrossRef]
- Muangchinda, C.; Pansri, R.; Wongwongsee, W.; Pinyakong, O. Assessment of polycyclic aromatic hydrocarbon biodegradation potential in mangrove sediment from Don Hoi Lot, Samut Songkram Province, Thailand. J. Appl. Microbiol. 2013, 114, 1311–1324. [Google Scholar] [CrossRef]
- Sarma, P.M.; Bhattacharya, D.; Krishnan, S.; Banwari, L. Degradation of Polycyclic Aromatic Hydrocarbons by a Newly Discovered Enteric Bacterium, Leclercia adecarboxylata. Appl. Environ. Microbiol. 2004, 70, 3163–3166. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Soils | Clay (%) | Silt (%) | Sand (%) | Fe (%) | NH4 (%) | Mg (%) | C (‰) | N (‰) | P (‰) |
|---|---|---|---|---|---|---|---|---|---|
| SGM | 9.5 | 19.94 | 70.52 | 0.25 | 0.17 | 0.09 | 12.5 | 1.0 | 0.03 |
| SGP | 7.03 | 15.33 | 77.64 | 0.33 | 0.15 | 0.07 | 14.2 | 1.2 | 0.02 |
| EAR | 9.77 | 19.14 | 71.10 | 0.37 | 0.18 | 0.10 | 16.2 | 1.7 | 0.04 |
| Alpha Diversity Index | SGM | SGP |
|---|---|---|
| S (number of genera) | 111 | 81 |
| Individuals | 85 | 90 |
| Simpson 1-D | 0.84 | 0.64 |
| Shannon H′ | 2.42 | 1.76 |
| Evenness_e^H′/S | 0.10 | 0.07 |
| Equitability_J | 0.51 | 0.40 |
| Chao−1 | 111 | 81 |
| Gene Family/Enzymes | Genus | Relative Abundance |
|---|---|---|
| ABC transporter | Mycobacterium, Gordonia, Agromyces, Modestobacter, Agrococcus, Isoptericola, Cellulomonas, Janibacter, Microbacterium, Dietzia, Modestobacter, Rhodococcus | 0.02% |
| Alcohol dehydrogenase | Gordonia, Mycobacterium, Rhodococcus, Dietzia, Paracoccus, Pseudomonas, Sphingobium, Geodermatophilus, Stenotrophomonas | 0.03% |
| Aldehyde dehydrogenase | Dietzia, Stenotrophomonas, Rhodococcus, Mycobacterium, Isoptericola, Gordonia, Cellulomonas, Paracoccus, Janibacter, Xanthomonas, Sciscionella, Dechloromonas, Shingobium, Agromyces, Geodermatophilus, Agrococcus, Blastococcus, Modestobacter, Pseudomonas, Ornithinimicrobium | 0.03% |
| Alkane 1-monooxygenase | Gordonia, Mycobacterium, Dietzia, Rhodococcus, Nevskia, Paracoccus | 0.004% |
| Alkane monooxygenase | Gordonia, Rhodococcus, Mycobacterium | 0.00004% |
| Catechol 1,2-dioxygenase | Gordonia, Dietzia, Geodermatophilus | 0.005% |
| Catechol-O-methyltransferase | Mycobacterium | 0.0001% |
| Cytochrome P450 | Gordonia, Rhodococcus, Mycobacterium, Dietzia, Modestobacter, Blastococcus, Nevskia | 0.02% |
| Cytochrome P450 alkane hydroxylase | Gordonia, Mycobacterium, Dietzia | 0.02% |
| Cytochrome P450 monooxygenase | Gordonia | 0.0005% |
| Toluene efflux pump membrane transporter TtgB | Stenotrophomonas, Pseudomonas | 0.0005% |
| Toluene tolerance protein | Xanthomonas, Stenotrophomonas | 0.003% |
| Pathways | Genus | Relative abundance |
| Acetylene degradation | Gordonia, Rhodococcus, Pseudomonas, Mycobacterium | 0.02% |
| Alkane degradation | Pseudomonas, Nevskia | 0.00005% |
| Benzene degradation | Rhodococcus | 0.0001% |
| Beta-ketoadipate pathway (aromatic compounds degradation via 3-oxoadipate) | Gordonia, Rhodococcus, Pseudomonas, Dietzia, Agrococcus, Janibacter, Blastococcus, Xanthomonas | 0.02% |
| Biphenyl degradation | Gordonia, Rhodococcus, Pseudomonas, Dechloromonas | 0.003% |
| Catechol degradation (ortho-cleavage pathway & beta; -ketoadipate) | Not assigned | 0.003% |
| Naphthalene degradation | Gordonia, Rhodococcus, Pseudomonas, Dechloromonas | 0.0003% |
| Octane oxidation | Gordonia, Mycobacterium | 0.03% |
| P-cumate degradation | Gordonia, Rhodococcus, Pseudomonas, Dechloromonas | 0.008% |
| Polychlorinated biphenyl degradation | Gordonia, Rhodococcus, Pseudomonas, Dechloromonas, Nevskia | 0.02% |
| Protocatechuate degradation II (ortho-cleavage pathway) | Not assigned | 0.0001% |
| Toluene degradation | Dietzia, Gordonia, Rhodococcus, Pseudomonas, Dechloromonas, Paracoccus, Mycobacterium, Geodermatophilus | 0.002% |
| Superpathway of salicylate degradation | Not assigned | 0.0001% |
| Gene Family/Enzymes | Genus | Relative Abundance |
|---|---|---|
| ABC transporter | Gordonia, Mycobacterium, Dietzia, Pseudomonas, Agrococcus, Rhodococcus, Blastococcus, Microbacterium, Geodermatophilus | 0.01% |
| Alcohol dehydrogenase | Mycobacterium, Pseudomonas, Paracoccus, Dietzia, Gordonia, Geodermatophilus, Rhodococcus, Agrococcus, Blastococcus, Chroococcidiopsis | 0.05% |
| Aldehyde dehydrogenase | Pseudomonas, Thauera, Paracoccus, Mycobacterium, Dietzia, Rhodococcus, Xanthomonas, Acidiphilium, Gordonia, Blastococcus, Geodermatophilus, Chroococcidiopsis, Nevskia, Agrococcus | 0.03% |
| Alkane 1-monooxygenase | Gordonia, Dietzia, Pseudomonas, Mycobacterium, Paracoccus, Nevskia | 0.002% |
| Alkane monooxygenase | Gordonia | 0.0001% |
| Aromatic hydrocarbon degradation protein | Pseudomonas | 0.0006% |
| Biphenyl-2,3-diol 1,2-dioxygenase protein | Pseudomonas | 0.0003% |
| Catechol 1,2-dioxygenase | Pseudomonas, Gordonia, Dietzia, Rhodococcus | 0.005% |
| Catechol oxidase | Chroococcidiopsis | 0.0002% |
| Catechol-2,3-dioxygenase | Pseudomonas | 0.0002% |
| Catechol-O-methyltransferase | Mycobacterium | 0.0003% |
| Cytochrome P450 | Dietzia, Mycobacterium, Gordonia, Rhodococcus, Chroococcidiopsis, Blastococcus, Geodermatophilus, Oceanicola | 0.007% |
| Cytochrome P450 alkane hydroxylase | Gordonia, Dietzia, Mycobacterium | 0.005% |
| Cytochrome P450 monooxygenase | Gordonia | 0.0001% |
| Naphthalene 1,2-dioxygenase | Pseudomonas | 0.002% |
| Protocatechuate 3,4-dioxygenase | Pseudomonas, Gordonia | 0.002% |
| Salicylate hydroxylase | Pseudomonas | 0.001% |
| Toluene efflux pump membrane transporter TtgB | Pseudomonas | 0.0002% |
| Toluene tolerance protein | Pseudomonas | 0.007% |
| Pathways | Genus | Relative abundance |
| Acetylene degradation | Pseudomonas, Gordonia | 0.01% |
| Benzene degradation | Pseudomonas | 0.001% |
| Beta-ketoadipate pathway (aromatic compounds degradation via 3-oxoadipate) | Pseudomonas | 0.05% |
| Biphenyl degradation | Pseudomonas, Gordonia, Rhodococcus, Thauera | 0.006% |
| Catechol degradation | Pseudomonas | 0.01% |
| Chlorosalicylate degradation | Not assigned | 0.0001% |
| 4-methylcatechol degradation (ortho cleavage) | Not assigned | 0.002% |
| Meta cleavage pathway of aromatic compounds | Not assigned | 0.0005% |
| Naphthalene degradation | Pseudomonas, Gordonia, Rhodococcus, Thauera | 0.003% |
| Octane oxidation | Pseudomonas, Gordonia, Mycobacterium | 0.02% |
| P-cumate degradation | Pseudomonas, Gordonia, Rhodococcus, Thauera | 0.007% |
| Polychlorinated biphenyl degradation | Pseudomonas, Gordonia, Thauera, Nevskia, Rhodococcus, Mycobacterium | 0.01% |
| Protocatechuate degradation II (ortho-cleavage pathway) | Pseudomonas | 0.007% |
| Superpathway of salicylate degradation | Pseudomonas | 0.003% |
| Toluene degradation | Pseudomonas, Dietzia, Paracoccus, Gordonia, Rhodococcus, Thauera, Mycobacterium, Geodermatophilus, | 0.01% |
| Xylene degradation | Pseudomonas | 0.001% |
| Parameter | Unit Parameter | SGM | SGP |
|---|---|---|---|
| Microbial biomass | MBC (mgC/kg soil) | 688 ± 21.4 | 621 ± 17.4 |
| MBN (mgN/kg soil) | 186.83 ± 8.8 | 138.57 ± 10.5 | |
| Soil enzymes (µg pN/g sol/h) | β glucosidase | 397.13 ± 7.6 | 351.3 ± 9 |
| β glucosaminidase | 101.05 ± 1.6 | 90.27 ± 5.3 | |
| Acid phosphatase | 804.17 ± 20.5 | 677.33 ± 32.1 | |
| Hydrocarbons | TPH (g/kg soil) | 327.67 ± 14.2 | 248.33 ± 20.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goma-Tchimbakala, E.J.C.D.; Pietrini, I.; Goma-Tchimbakala, J.; Corgnati, S.P. Use of Shotgun Metagenomics to Assess the Microbial Diversity and Hydrocarbons Degrading Functions of Auto-Mechanic Workshops Soils Polluted with Gasoline and Diesel Fuel. Microorganisms 2023, 11, 722. https://doi.org/10.3390/microorganisms11030722
Goma-Tchimbakala EJCD, Pietrini I, Goma-Tchimbakala J, Corgnati SP. Use of Shotgun Metagenomics to Assess the Microbial Diversity and Hydrocarbons Degrading Functions of Auto-Mechanic Workshops Soils Polluted with Gasoline and Diesel Fuel. Microorganisms. 2023; 11(3):722. https://doi.org/10.3390/microorganisms11030722
Chicago/Turabian StyleGoma-Tchimbakala, Emerance Jessica Claire D’Assise, Ilaria Pietrini, Joseph Goma-Tchimbakala, and Stefano Paolo Corgnati. 2023. "Use of Shotgun Metagenomics to Assess the Microbial Diversity and Hydrocarbons Degrading Functions of Auto-Mechanic Workshops Soils Polluted with Gasoline and Diesel Fuel" Microorganisms 11, no. 3: 722. https://doi.org/10.3390/microorganisms11030722