

Genetic Diversity among Rose Rosette Virus Isolates: A Roadmap towards Studies of Gene Function and Pathogenicity



Abstract
:1. Introduction
2. Materials and Methods
2.1. RRV Sequence Database Construction
2.2. Performance of ML Analysis and MAFTT Alignments and Analysis of Sequence Variability and Mutations
3. Results
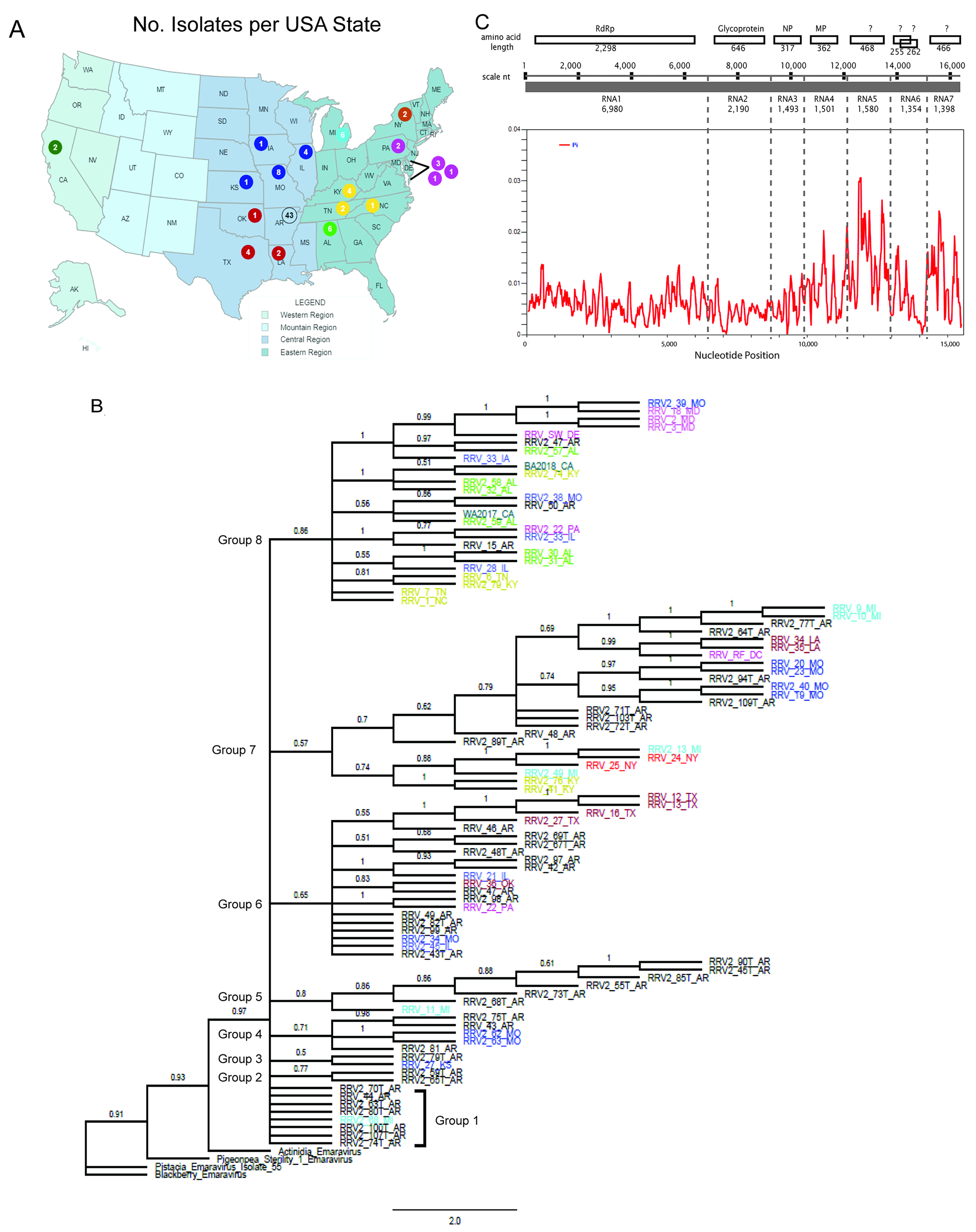
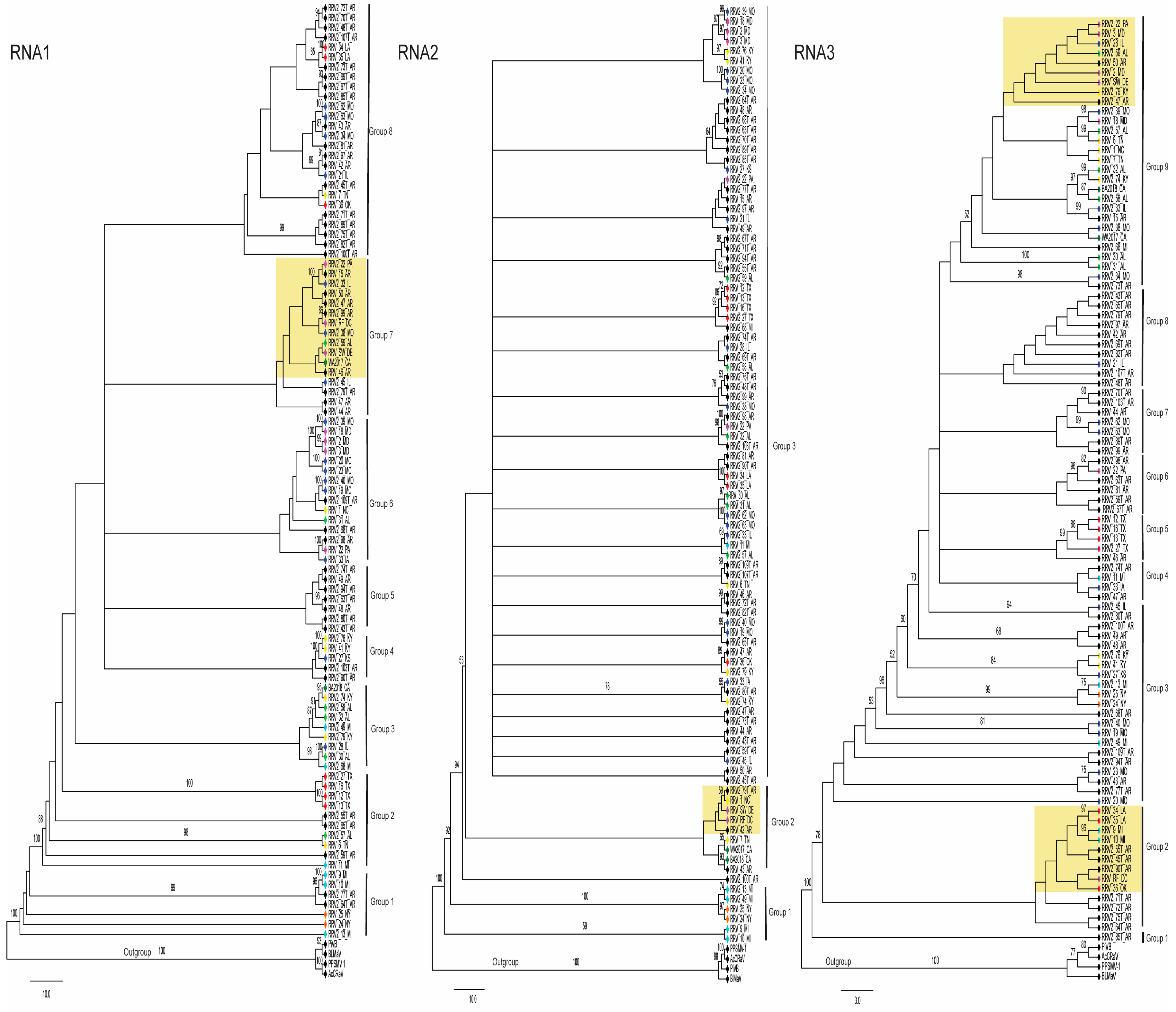
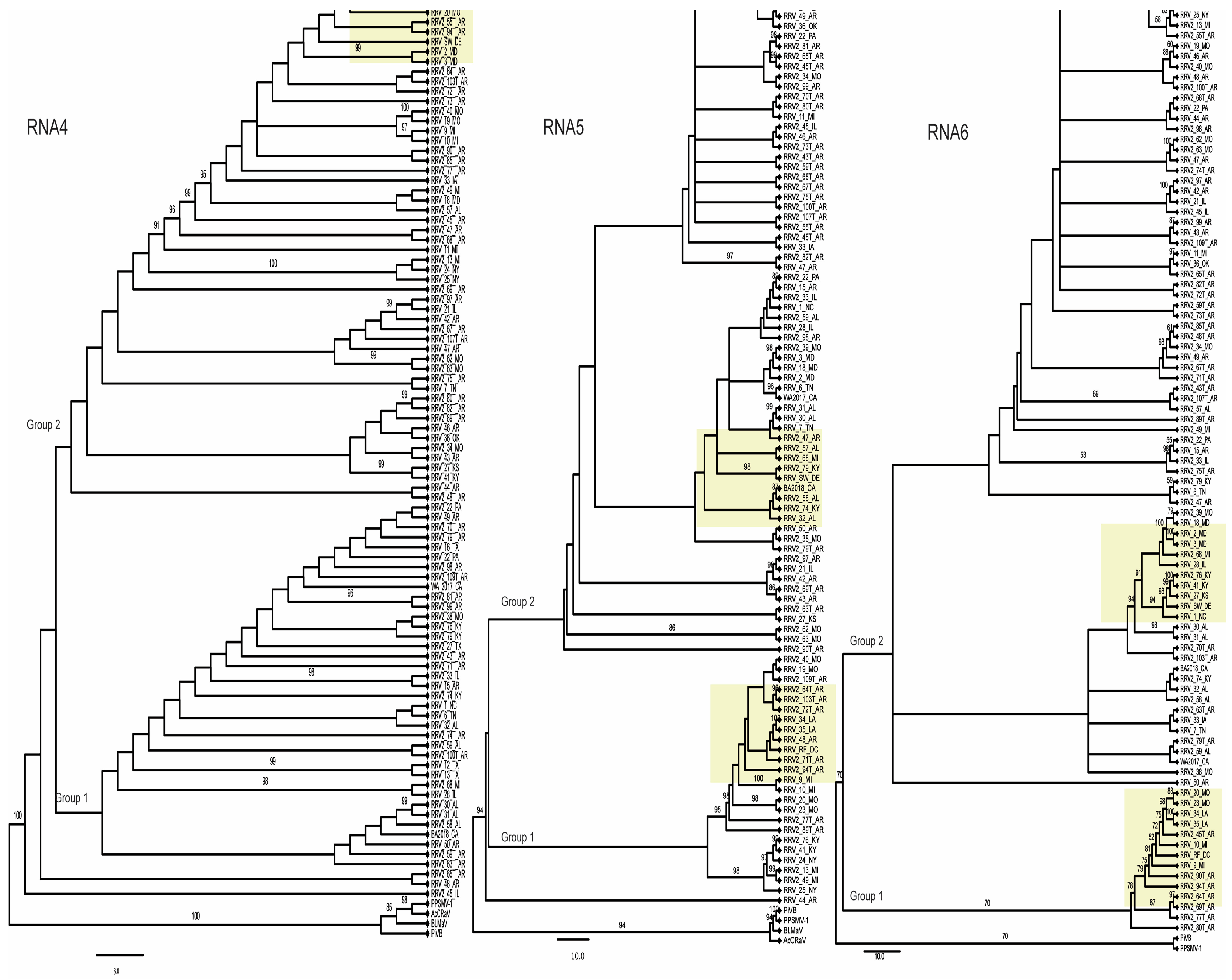
3.1. Virus Isolates and Genetic Variants
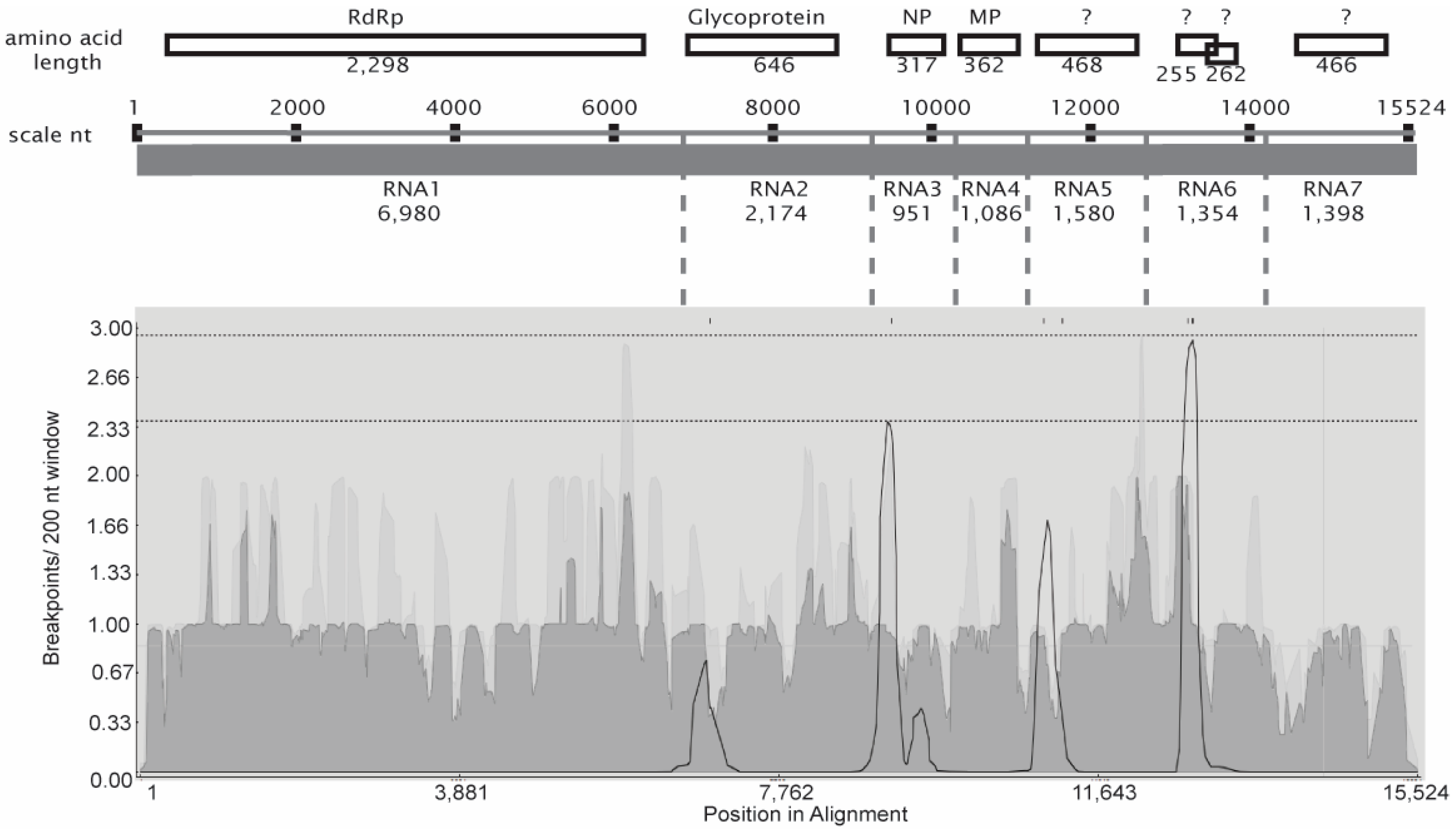
3.2. Evidence for RRV Segment Reassortment
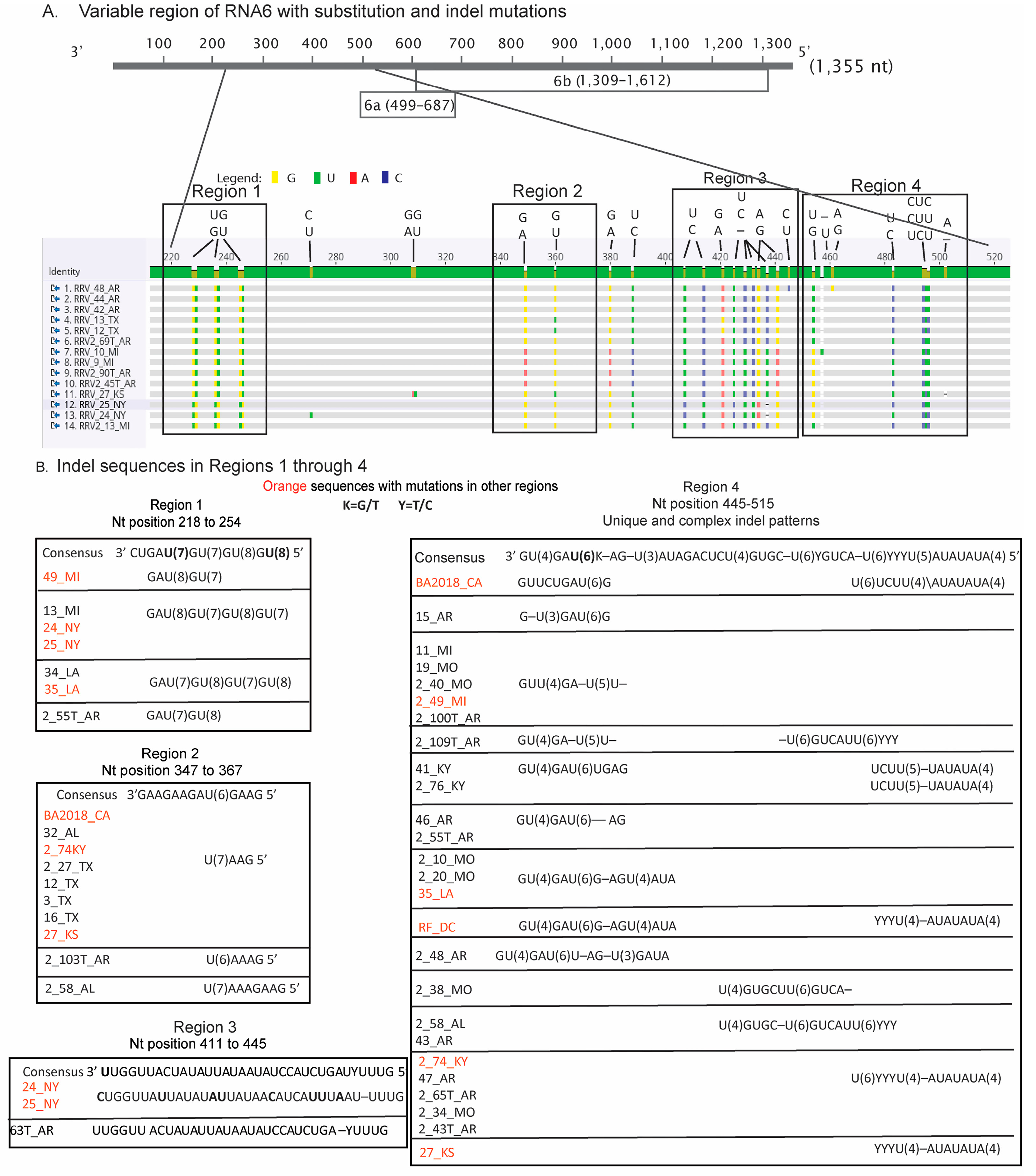
3.3. Insertion and Deletions in RNA6 Occurring along Highly Homopolymeric Tracts
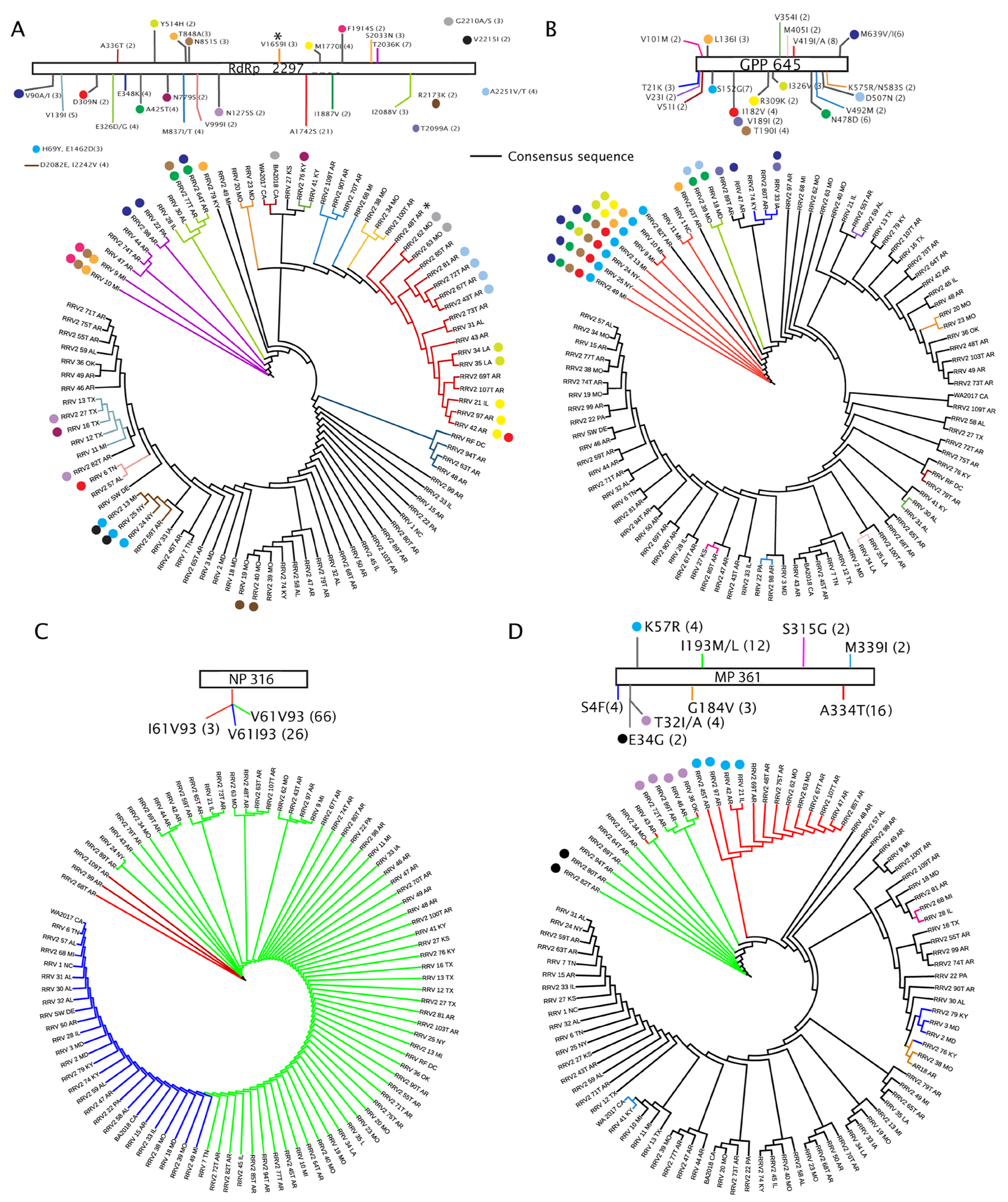
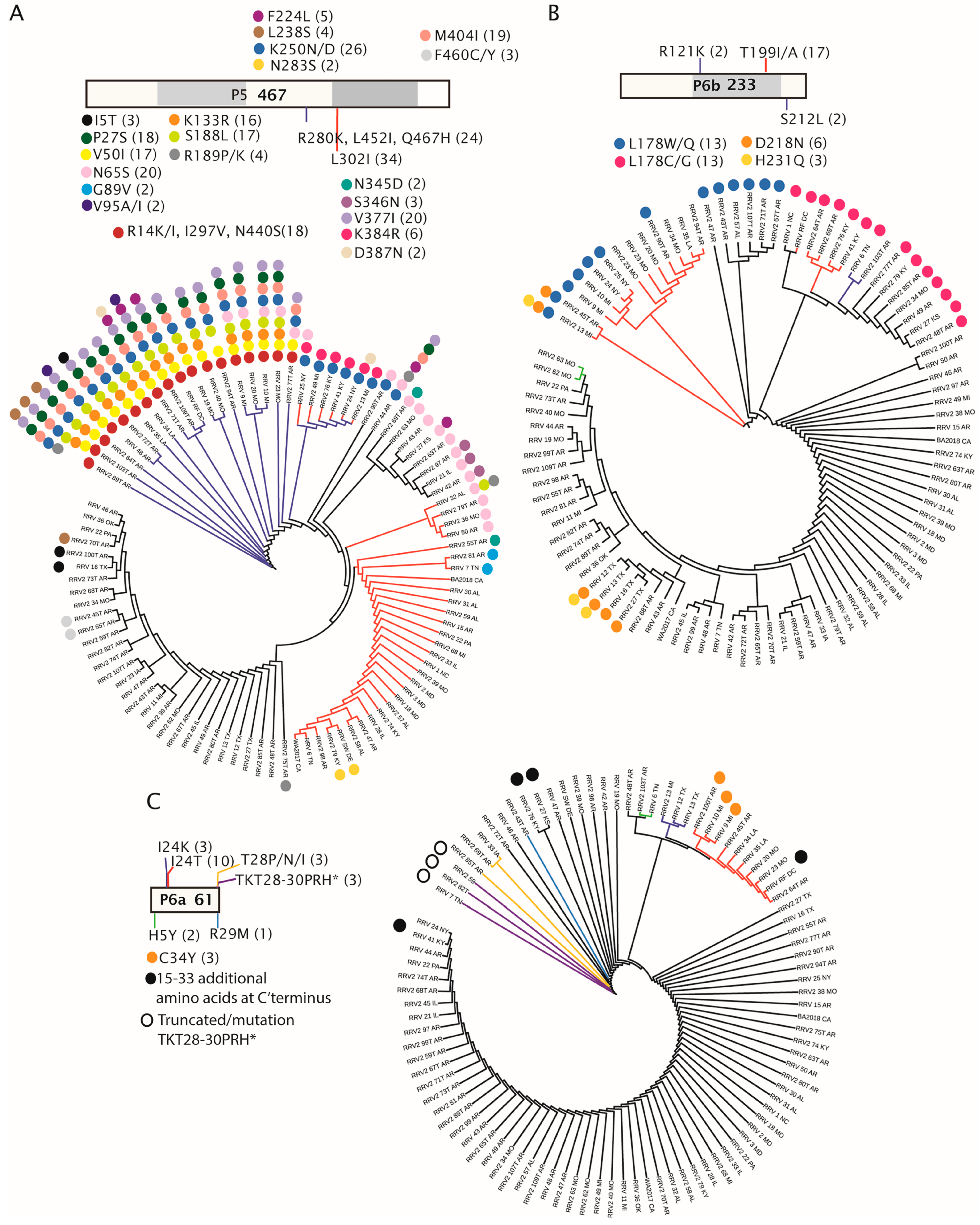
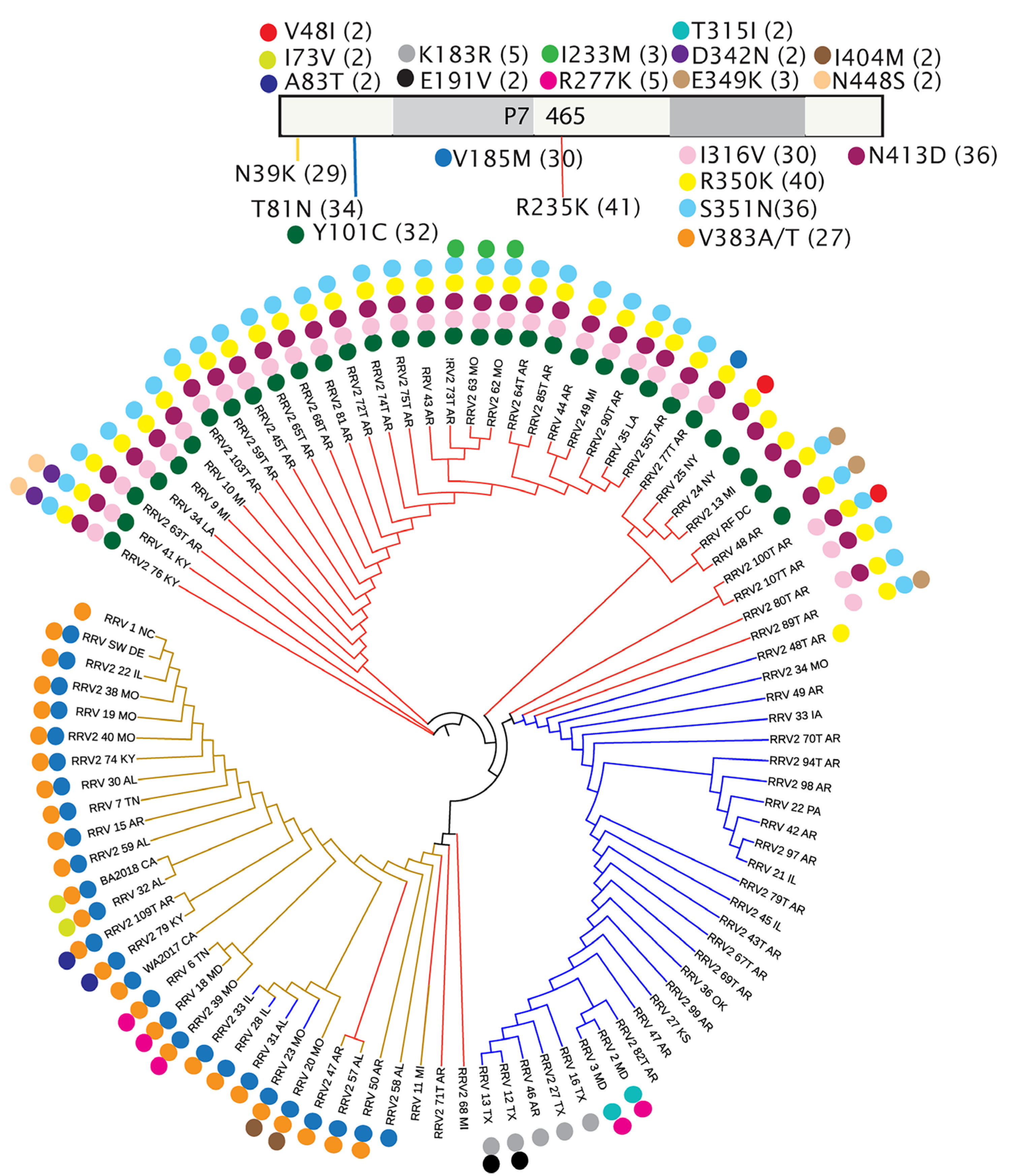
3.4. Amino Acid Changes within RRV Coding Sequences and Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kormelink, R.; Verchot, J.; Tao, X.; Desbiez, C. The Bunyavirales: The plant-infecting counterparts. Viruses 2021, 13, 842. [Google Scholar] [CrossRef] [PubMed]
- Laney, A.G.; Keller, K.E.; Martin, R.R.; Tzanetakis, I.E. A discovery 70 years in the making: Characterization of the Rose rosette virus. J. Gen. Virol. 2011, 92, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Di Bello, P.L.; Ho, T.; Tzanetakis, I.E. The evolution of emaraviruses is becoming more complex: Seven segments identified in the causal agent of Rose rosette disease. Virus Res. 2015, 210, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Maejima, K.; Komatsu, K.; Netsu, O.; Keima, T.; Shiraishi, T.; Okano, Y.; Hashimoto, M.; Yamaji, Y.; Namba, S. Fig mosaic emaravirus p4 protein is involved in cell-to-cell movement. J. Gen. Virol. 2013, 94, 682–686. [Google Scholar] [CrossRef]
- Verchot, J.; Herath, V.; Urrutia, C.D.; Gayral, M.; Lyle, K.; Shires, M.K.; Ong, K.; Byrne, D. Development of a reverse genetic system for studying rose rosette virus in whole plants. Mol. Plant-Microbe Interact. 2020, 33, 1209–1221. [Google Scholar] [CrossRef]
- Urrutia, C.D.; Romay, G.; Shaw, B.D.; Verchot, J. Advancing the rose rosette virus minireplicon and encapsidation system by incorporating GFP, mutations, and the CMV 2b silencing suppressor. Viruses 2022, 14, 836. [Google Scholar] [CrossRef]
- Babu, B.; Washburn, B.K.; Poduch, K.; Knox, G.W.; Paret, M.L. Identification and characterization of two novel genomic RNA segments RNA5 and RNA6 in rose rosette virus infecting roses. Acta Virol. 2016, 60, 156–165. [Google Scholar] [CrossRef]
- Rehanek, M.; Karlin, D.G.; Bandte, M.; Al Kubrusli, R.; Nourinejhad Zarghani, S.; Candresse, T.; Büttner, C.; von Bargen, S. The complex world of emaraviruses—Challenges, insights, and prospects. Forests 2022, 13, 1868. [Google Scholar] [CrossRef]
- Gupta, A.K.; Hein, G.L.; Graybosch, R.A.; Tatineni, S. Octapartite negative-sense RNA genome of High Plains wheat mosaic virus encodes two suppressors of RNA silencing. Virology 2018, 518, 152–162. [Google Scholar] [CrossRef]
- Gupta, A.K.; Hein, G.L.; Tatineni, S. P7 and P8 proteins of High plains wheat mosaic virus, a negative-strand RNA virus, employ distinct mechanisms of RNA silencing suppression. Virology 2019, 535, 20–31. [Google Scholar] [CrossRef]
- Domingo, E. Virus as Populations; Elsevier: Waltham, MA, USA, 2016; ISBN 978-0-12-800837-9. [Google Scholar]
- Katsiani, A.; Stainton, D.; Lamour, K.; Tzanetakis, I.E. The population structure of Rose rosette virus in the USA. J. Gen. Virol. 2020, 101, 676–684. [Google Scholar] [CrossRef]
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef]
- Domingo, E. Viruses at the edge of adaptation. Virology 2000, 270, 251–253. [Google Scholar] [CrossRef]
- Lyons, D.M.; Lauring, A.S. Evidence for the selective basis of transition-to-transversion substitution bias in two RNA viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef]
- Ivancic-Jelecki, J.; Slovic, A.; Šantak, M.; Tešović, G.; Forcic, D. Common position of indels that cause deviations from canonical genome organization in different measles virus strains. Virol. J. 2016, 13, 134. [Google Scholar] [CrossRef]
- Walia, J.J.; Willemsen, A.; Elci, E.; Caglayan, K.; Falk, B.W.; Rubio, L. Genetic variation and possible mechanisms driving the evolution of worldwide fig mosaic virus isolates. Phytopathology 2014, 104, 108–114. [Google Scholar] [CrossRef]
- Patil, B.L.; Dangwal, M.; Mishra, R. Variability of emaravirus species associated with sterility mosaic disease of pigeonpea in India provides evidence of segment reassortment. Viruses 2017, 9, 183. [Google Scholar] [CrossRef]
- Jones, R.A.C.; Vazquez-Iglesias, I.; McGreig, S.; Fox, A.; Gibbs, A.J. Genomic High Plains wheat mosaic virus sequences from Australia: Their phylogenetics and evidence for emaravirus recombination and reassortment. Viruses 2023, 15, 401. [Google Scholar] [CrossRef]
- Tatineni, S.; McMechan, A.J.; Wosula, E.N.; Wegulo, S.N.; Graybosch, R.A.; French, R.; Hein, G.L. An Eriophyid Mite-Transmitted Plant Virus Contains Eight Genomic RNA Segments with Unusual Heterogeneity in the Nucleocapsid Protein. J. Virol. 2014, 88, 11834–11845. [Google Scholar] [CrossRef]
- Stewart, L.R. Sequence diversity of wheat mosaic virus isolates. Virus Res. 2016, 213, 299–303. [Google Scholar] [CrossRef]
- Valkonen, J.P.T.; Jones, R.A.C.; Slack, S.A.; Watanabe, K.N. Resistance specificities to viruses in potato: Standardization of nomenclature. Plant Breed. 1996, 115, 433–438. [Google Scholar] [CrossRef]
- García-Arenal, F.; McDonald, B.A. An analysis of the durability of resistance to plant viruses. Phytopathology 2003, 93, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Malcuit, I.; De Jong, W.; Baulcombe, D.C.; Shields, D.C.; Kavanagh, T.A. Acquisition of multiple virulence/avirulence determinants by potato virus X (PVX) has occurred through convergent evolution rather than through recombination. Virus Genes 2000, 20, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gallois, J.L.; Moury, B.; German-Retana, S. Role of the genetic background in resistance to plant viruses. Int. J. Mol. Sci. 2018, 19, 2856. [Google Scholar] [CrossRef] [PubMed]
- Solomon-Blackburn, R.M.; Barker, H. Breeding virus resistant potatoes (Solanum tuberosum): A review of traditional and molecular approaches. Heredity 2001, 86, 17–35. [Google Scholar] [CrossRef]
- Windham, M.T.; Evans, T.; Collins, S.; Lake, J.A.; Lau, J.; Riera-Lizarazu, O.; Byrne, D.H. Field resistance to rose rosette disease as determined by multi-year evaluations in Tennessee and Delaware. Pathogens 2023, 12, 439. [Google Scholar] [CrossRef]
- Byrne, D.H.; Klein, P.; Yan, M.; Young, E.; Lau, J.; Ong, K.; Shires, M.; Olson, J.; Windham, M.; Evans, T.; et al. Challenges of breeding rose rosette–resistant roses. HortScience 2018, 53, 604–608. [Google Scholar] [CrossRef]
- Zlesak, D. Pollen diameter and guard cell length as predictors of ploidy in diverse rose cultivars, species, and breeding lines. Floric. Ornam. Biotechnol. 2009, 3, 53–70. [Google Scholar]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C.; et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772–777. [Google Scholar] [CrossRef]
- Young, E.L.; Lau, J.; Bentley, N.B.; Rawandoozi, Z.; Collins, S.; Windham, M.T.; Klein, P.E.; Byrne, D.H.; Riera-Lizarazu, O. Identification of QTLs for reduced susceptibility to rose rosette disease in diploid roses. Pathogens 2022, 11, 660. [Google Scholar] [CrossRef]
- Lau, J.; Young, E.L.; Collins, S.; Windham, M.T.; Klein, P.E.; Byrne, D.H.; Riera-Lizarazu, O. Rose rosette disease resistance loci detected in two interconnected tetraploid garden rose populations. Front. Plant Sci. 2022, 13, 2036. [Google Scholar] [CrossRef]
- Henderson, D.C.; Hammond, J. CKC: Isolation of Nucleic Acids from a Diversity of Plants Using CTAB and Silica Columns. Mol. Biotechnol. 2013, 53, 109–117. [Google Scholar] [CrossRef]
- Jordan, R.; Korolev, E.; Grinstead, S.; Mollov, D. First complete genome sequence of carnation latent virus, the type member of the genus Carlavirus. Arch. Virol. 2021, 166, 1501–1505. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef]
- Forslund, K.; Huson, D.H.; Moulton, V. VisRD—Visual recombination detection. Bioinformatics 2004, 20, 3654–3655. [Google Scholar] [CrossRef]
- Samson, S.; Lord, É.; Makarenkov, V. Sequence analysis SimPlot++: A Python application for representing sequence similarity and detecting recombination. arXiv 2021, arXiv:2112.09755. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Di Bello, P.L.; Thekke-Veetil, T.; Druciarek, T.; Tzanetakis, I.E. Transmission attributes and resistance to rose rosette virus. Plant Pathol. 2018, 67, 499–504. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Karlik, J.; Diaz-Lara, A.; Ong, K.; Mollov, D.; Haviland, D.; Golino, D. First report of rose rosette virus associated with rose rosette disease affecting roses in California. Plant Dis. 2019, 103, 380. [Google Scholar] [CrossRef]
- Ali, A.; Roossinck, M.J. Genetic Bottlenecks. In Plant Virus Evolution; Roosinck, M.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 123–131. [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Schirmer, M.; Sloan, W.T.; Quince, C. Benchmarking of viral haplotype reconstruction programmes: An overview of the capacities and limitations of currently available programmes. Brief. Bioinform. 2014, 15, 431–442. [Google Scholar] [CrossRef]
- Agol, V.I.; Gmyl, A.P. Emergency services of viral RNAs: Repair and remodeling. Microbiol. Mol. Biol. Rev. 2018, 82, e00067-17. [Google Scholar] [CrossRef]
- Alisoltani, A.; Jaroszewski, L.; Iyer, M.; Iranzadeh, A.; Godzik, A. Increased frequency of indels in hypervariable regions of SARS-CoV-2 proteins—A possible signature of adaptive selection. Front. Genet. 2022, 13, 875406. [Google Scholar] [CrossRef]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 2014, 505, 686–690. [Google Scholar] [CrossRef]
- Hily, J.M.; Poulicard, N.; Mora, M.Á.; Pagán, I.; García-Arenal, F. Environment and host genotype determine the outcome of a plant-virus interaction: From antagonism to mutualism. New Phytol. 2016, 209, 812–822. [Google Scholar] [CrossRef]
- Simmonds, P.; Aiewsakun, P.; Katzourakis, A. Prisoners of war—Host adaptation and its constraints on virus evolution. Nat. Rev. Microbiol. 2019, 17, 321–328. [Google Scholar] [CrossRef]
- Elena, S.F.; Fraile, A.; García-Arenal, F. Evolution and Emergence of Plant Viruses. Adv. Virus Res. 2014, 88, 161–191. [Google Scholar] [CrossRef]
- Moury, B.; Fabre, F.; Hébrard, E.; Froissart, R. Determinants of host species range in plant viruses. J. Gen. Virol. 2017, 98, 862–873. [Google Scholar] [CrossRef]
- García-Arenal, F.; Fraile, A. Trade-offs in host range evolution of plant viruses. Plant Pathol. 2013, 62, 2–9. [Google Scholar] [CrossRef]
- Duffy, S.; Burch, C.L.; Turner, P.E. Evolution of host specificity drives reproductive isolation among RNA viruses. Evolution 2007, 61, 2614–2622. [Google Scholar] [CrossRef]
- Schirmer, M.; D’Amore, R.; Ijaz, U.Z.; Hall, N.; Quince, C. Illumina error profiles: Resolving fine-scale variation in metagenomic sequencing data. BMC Bioinform. 2016, 17, 125. [Google Scholar] [CrossRef]
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genomics Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- Tomimura, K.; Gibbs, A.J.; Jenner, C.E.; Walsh, J.A.; Ohshima, K. The phylogeny of Turnip mosaic virus; comparisons of 38 genomic sequences reveal a Eurasian origin and a recent “emergence” in east Asia. Mol. Ecol. 2003, 12, 2099–2111. [Google Scholar] [CrossRef]
- Fraile, A.; McLeish, M.J.; Pagán, I.; González-Jara, P.; Piñero, D.; García-Arenal, F. Environmental heterogeneity and the evolution of plant-virus interactions: Viruses in wild pepper populations. Virus Res. 2017, 241, 68–76. [Google Scholar] [CrossRef]
- Ali, A.; Li, H.; Schneider, W.L.; Sherman, D.J.; Gray, S.; Smith, D.; Roossinck, M.J. Analysis of genetic bottlenecks during horizontal transmission of cucumber mosaic virus. J. Virol. 2006, 80, 8345–8350. [Google Scholar] [CrossRef]
- McLeish, M.J.; Fraile, A.; García-Arenal, F. Ecological complexity in plant virus host range evolution. Adv. Virus Res. 2018, 101, 293–339. [Google Scholar] [CrossRef] [PubMed]
- Conners, I.L. Twentieth Annual Report of the Canadian Plant Disease Survey; Department of Agriculture: Ottawa, ON, Canada, 1941; p. 98. [Google Scholar]
- Thomas, E.A.; Scott, C.E. Rosette of rose. Phytopathology 1953, 43, 218–219. [Google Scholar]
- Keifer, H.H. Eroiphyid studies VIII. Bull. Cali. Dept. Agric. 1940, 29, 21–46. [Google Scholar]
- Amrine, J.W., Jr.; Hindal, D.F.; Williams, R.; Appel, J.; Stasny, T.; Kassar, A. Rose rosette as a biocontrol of multiflora rose, 1987–1989. In Proceedings of the 43rd Annual Meeting of the Southern Weed Science Society, Atlanta, GA, USA, 15–17 January 1990; Volume 43, pp. 316–319. [Google Scholar]
- Hindal, D.F.; Amrine, J.W.; Williams, R.L.; Stasny, T.A. Rose rosette disease on multiflora rose (Rosa multiflora) in Indiana and Kentucky. Weed Technol. 1988, 2, 442–444. [Google Scholar] [CrossRef]
- Allington, W.B.; Staples, R.; Viehmeyer, G. Transmission of rose rosette virus by the eriophyid mite Phyllocoptes fructiphilus. J. Econ. Entomol. 1968, 61, 1137–1140. [Google Scholar] [CrossRef]
- Pemberton, H.B.; Ong, K.; Windham, M.; Olson, J.; Byrne, D.H. What is rose rosette disease? HortScience 2018, 53, 592–595. [Google Scholar] [CrossRef]
- Pemberton, H.B.; Karlik, J.F. A recent history of changing trends in USA garden rose plant sales, types, and production methods. Acta Hortic. 2015, 1064, 223–234. [Google Scholar] [CrossRef]
- Cho, E.-K. Strains of soybean mosaic virus: Classification based on virulence in resistant soybean cultivars. Phytopathology 1979, 69, 467. [Google Scholar] [CrossRef]
- Drijfhout, E.; Silbernagel, M.H.; Burke, D.W. Differentiation of strains of bean common mosaic virus. Netherlands J. Plant Pathol. 1978, 84, 13–26. [Google Scholar] [CrossRef]
- Fraile, A.; García-Arenal, F. The coevolution of plants and viruses: Resistance and pathogenicity. Adv. Virus Res. 2010, 76, 1–32. [Google Scholar] [CrossRef]
- Cox, B.A.; Jones, R.A.C. Genetic variability in the coat protein gene of Potato virus X and the current relationship between phylogenetic placement and resistance groupings. Arch. Virol. 2010, 155, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Grech-Baran, M.; Witek, K.; Szajko, K.; Witek, A.I.; Morgiewicz, K.; Wasilewicz-Flis, I.; Jakuczun, H.; Marczewski, W.; Jones, J.D.G.; Hennig, J. Extreme resistance to potato virus Y in potato carrying the Rysto gene is mediated by a TIR-NLR immune receptor. Plant Biotechnol. J. 2020, 18, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Pérez, M.G.; García-Luque, I.; Fraile, A.; García-Arenal, F. Mutations that determine resistance breaking in a plant RNA virus have pleiotropic effects on its fitness that depend on the host environment and on the type, single or mixed, of infection. J. Virol. 2016, 90, 9128–9137. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; McGavin, W.; Cock, P.J.A.; Schnettler, E.; Yan, F.; Chen, J.; MacFarlane, S. Newly identified RNAs of raspberry leaf blotch virus encoding a related group of proteins. J. Gen. Virol. 2015, 96, 3432–3439. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | N | S | ƞ | π | K | Tajima’s D * | Fu and Li’s D **† | Fu and Li’s F ** |
|---|---|---|---|---|---|---|---|---|
| Concatenated | 95 | 1648 | 1703 | 0.0073 | 113 | −2.2586 | −4.4978 | −4.0345 |
| RNA1 | 95 | 769 | 792 | 0.0060 | 42 | −2.4870 | −4.2207 | −4.1514 |
| RNA2 | 95 | 218 | 221 | 0.0043 | 9 | −2.6536 | −4.4239 | −4.3939 |
| RNA3 | 95 | 79 | 83 | 0.0073 | 7 | −1.8977 | −4.6653 | −4.2174 |
| RNA4 | 95 | 95 | 98 | 0.0080 | 9 | −1.8117 | −4.4080 | −3.9841 |
| RNA5 | 95 | 209 | 223 | 0.0149 | 239 | −1.5531 | −3.6656 | −3.3067 |
| RNA6 | 95 | 146 | 154 | 0.0069 | 9 | −2.3190 | −4.9792 | −4.6142 |
| RNA7 | 95 | 133 | 134 | 0.0104 | 14 | −1.4900 | −3.8783 | −3.4378 |
| Minor Parent | Major Parent | Breakpoint Positions | RDP Analysis (RGCMBSPL3) ** |
|---|---|---|---|
| RRV_12_TX and RRV_13_TX | RRV_9_MI and RRV_10_MI | 1–9136 | 6.58 × 10−13 to 1.36 × 10−54 |
| RRV2_13_MI, RRV_24_NY, and RRV_25_NY, | RRV_44_AR and RRV_48_AR | 6982–12,801 | 5.68 × 10−5 to 2.09 × 10−45 |
| RRV_27_KS, RRV_42_AR, and RRV2_69T_AR | RRV_9_MI and RRV_10_MI | 6928–9134 | 3.97 × 10−9 to 5.82 × 10−20 |
| RRV_42_AR | RRV2_45T and RRV2_90T | 7184–9134 | 1.18 × 10−5 to 3.91 × 10−12 |
| Sequence | Amino Acid Length | Parsimony-Informative Amino Acids |
|---|---|---|
| RdRp | 2297 | 31 (1%) |
| GPP | 645 | 20 (3%) |
| NP | 316 | 2 (0.6%) |
| MP | 361 | 9 (2%) |
| P5 | 467 | 29 (6%) |
| P6a | 61–94 | 3 (≤0.05%) |
| P6b | 233 | 7 (3%) |
| P7 | 465 | 23 (5%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verchot, J.; Herath, V.; Jordan, R.; Hammond, J. Genetic Diversity among Rose Rosette Virus Isolates: A Roadmap towards Studies of Gene Function and Pathogenicity. Pathogens 2023, 12, 707. https://doi.org/10.3390/pathogens12050707
Verchot J, Herath V, Jordan R, Hammond J. Genetic Diversity among Rose Rosette Virus Isolates: A Roadmap towards Studies of Gene Function and Pathogenicity. Pathogens. 2023; 12(5):707. https://doi.org/10.3390/pathogens12050707
Chicago/Turabian StyleVerchot, Jeanmarie, Venura Herath, Ramon Jordan, and John Hammond. 2023. "Genetic Diversity among Rose Rosette Virus Isolates: A Roadmap towards Studies of Gene Function and Pathogenicity" Pathogens 12, no. 5: 707. https://doi.org/10.3390/pathogens12050707