1. Introduction
The economic importance of roses (
Rosa L. sp.), as one of the most popular cut flowers and ornamental plants, is estimated at USD 28 billion globally [
1]. Unfortunately, roses are vulnerable to diseases such as black spot, cercospora leaf spot, downy and powdery mildew, and rust, which threaten roses’ value as a landscape crop. Rose rosette disease (RRD) is the most important viral disease of roses in the USA [
2]. RRD is caused by the rose rosette emaravirus (RRV), which is spread by an eriophyid mite,
Phyllocoptes fructiphilus Keifer. The most common control is the removal and destruction of infected plants. Management practices include exclusion of the virus or controlling the mite to limit movement [
3]. Chemical control of the mite is generally ineffective at controlling the virus due to the short inoculation access period [
4]. The disease can be fatal and shows symptoms of elongated shoots, known as witches’ broom, rosetting, leaf distortion, red or yellow leaves, excessive prickles, and increased susceptibility to other stresses or diseases. Plant death is expected 3–5 years after the initial infection [
4,
5]. Due to the high disease pressure in some areas and the lack of control methods, finding naturally resistant materials is essential.
This disease has become more prevalent and causes significant losses commercially in production and publicly in rose plantings. Therefore, research efforts to discover rose cultivars and species that display RRD resistance have become more extensive in the past couple of decades. This requires multiyear trials to determine the level of resistance [
6]. Although the level of susceptibility to RRD varies, no complete resistance to RRD has been found among the major commercial rose cultivars. However, high resistance has been found among a few cultivars with parentage originating from various resistant North American (
R. acicularis,
R. arkansana,
R. blanda,
R. californica,
R. carolina,
R. palustris,
R. pisocarpa, and
R. setigera) and Asian species (
R. spinossisima,
R. wichurana, and
R. bracteata). Most of these species and species hybrids have not been significantly explored for commercial breeding [
7].
Plants have evolved to protect themselves when invaded by pathogens. For viruses specifically, mechanisms include RNA silencing, innate immunity, ubiquitination–mediation, and translational repression as a few of the major defenses [
8]. Plant hormones are also key in defense against pathogens. Salicylic acid, jasmonic acid, and ethylene alter a plant’s response against biotic stresses. Hormones related to plant growth and development (auxin, brassinosteroids, cytokinins, and abscisic acid) are also involved in plant–pathogen interactions [
9]. Chaperonins have also been found to be involved in the regulation of virus reproduction and movement [
10]. They can be components of R-mediated resistance [
9], which produces a hypersensitive response to prevent the spread of the infection.
Although QTL for resistance to some emaraviruses in species other than roses have been described [
11,
12,
13,
14], R genes, per se, have not been identified. Two recent studies, one on inter-related diploid populations by Young et al. [
6] and another on two interconnected tetraploid populations by Lau et al. [
15], identified multiple quantitative trait loci (QTL) related to RRD partial resistance. Both studies performed QTL analysis on disease severity and virus RT-qPCR Ct values [
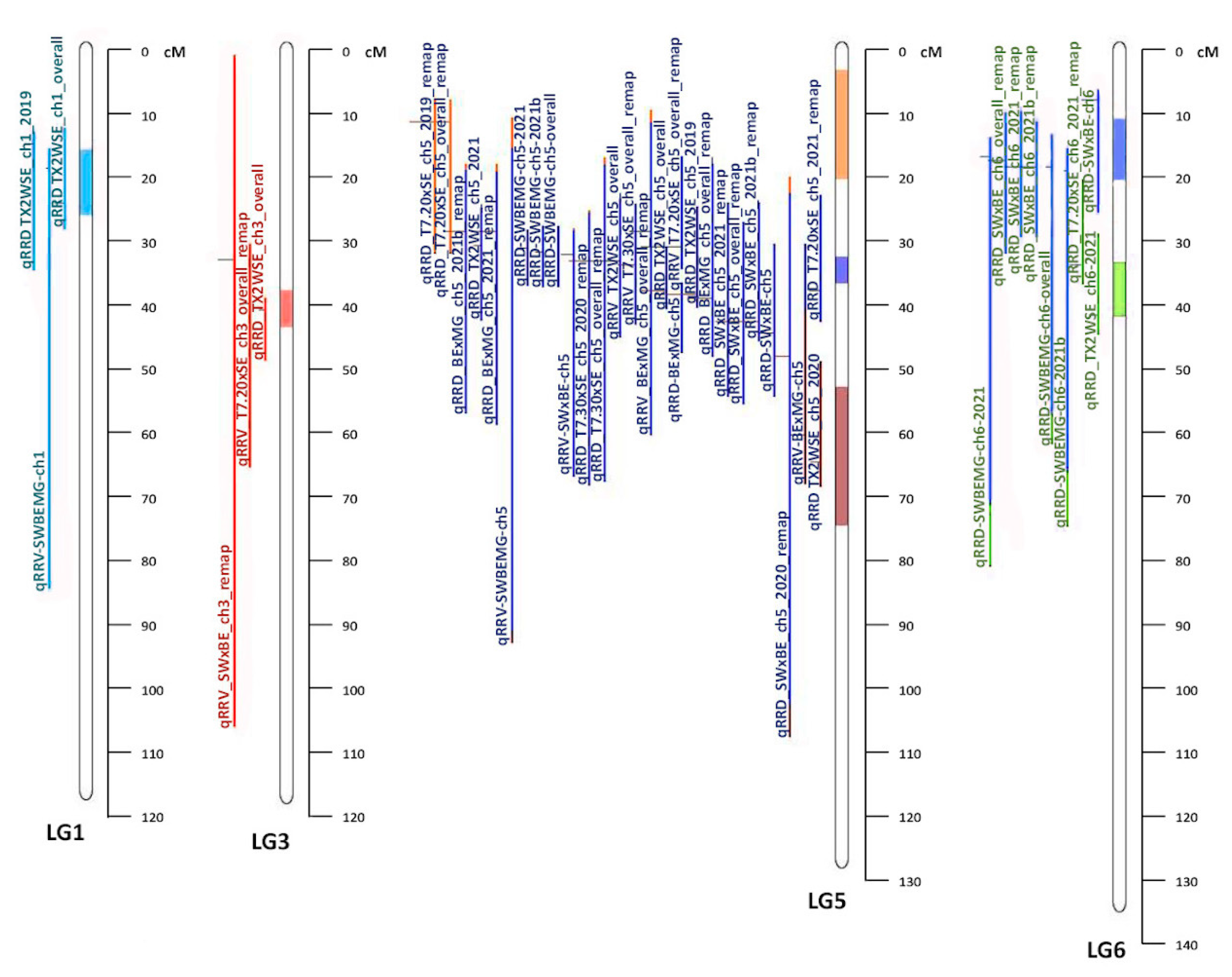
16]. Lau et al. identified QTL on linkage groups (LGs) 5, 6, and 7, accounting for 18–40% of the phenotypic variance, with the greatest effect for resistance attributed to LG 5 [
15]. By performing a joint QTL analysis of two interconnected tetraploid populations using diaQTL [
17], an additional QTL on LG 1 for Ct value was found. Young et al. found QTL on LGs 1, 3, 5, and 6, with the QTL on LG 5 being the most consistent, accounting for approximately 20–40% of the phenotypic variance [
6].
QTL mapping can be influenced by many factors, such as population size, the germplasm base, environmental conditions, and the number and density of genetic markers [
18,
19]. Using a meta-analysis, one identifies consistent QTL with major effects called meta-QTL. The objective of a meta-analysis is multifaceted. It increases QTL detection power, increases sample size, and uses data and information between studies, thereby gaining insight into the genetic architecture of the trait of interest [
20]. In addition, meta-QTL with smaller and better-defined confidence intervals and consistent effects across populations are useful for marker-assisted selection [
19]. This method has been widely used in a variety of disciplines, including medical, social, and behavioral sciences [
20], and within horticulture and agriculture on a multitude of crops [
19,
21,
22].
In this study, we conducted a meta-analysis to combine the diploid [
6] and tetraploid [
15] datasets to understand the relationship between RRD-resistant QTL in these two germplasm groups. In addition to using the data directly from these studies, we remapped all populations to improve the marker order and genetic distance estimation in both the diploid and tetraploid linkage maps to better define RRD-resistant QTL intervals and focus our candidate gene search within these RRD meta-QTL regions.
4. Discussion
RRD is the most important viral disease of roses in the USA [
2], causing substantial losses in the landscape rose industry. Due to this cost, increasing consideration is being invested into understanding the genetic factors that govern RRD resistance. QTL studies have been used to study resistance to emaraviruses in plants [
11,
12,
13]. This meta-analysis used the two studies performed on roses so far [
6,
15]. The objectives of this study were to understand the relationship between RRD-resistant QTL in diploid and tetraploid germplasms, better define RRD-resistant QTL intervals, and search for candidate genes in RRD meta-QTL intervals. For this, we looked at two studies that captured one multi-parental diploid and two interconnected bi-parental tetraploid populations across three years. We also remapped the populations from the studies to produce marker maps where the order of markers and genetic distance estimations were improved using the physical position of markers and a multi-point approach [
27].
The meta-analysis was conducted on LGs that contained QTL from both diploid [
6] and tetraploid [
15] populations (LGs 1, 3, 5, and 6). One meta-QTL was identified on LG 1 (MetaRRD1.1), which was roughly a third of the size of the average CI of the QTL from the original studies. All QTL from both diploid and tetraploid populations co-localized with MetaRRD1.1. One meta-QTL was identified on LG 3 (MetaRRD3.1). Three meta-QTL were discovered on LG 5; MetaRRD5.1, MetaRRD5.2, and MetaRRD5.3. All QTL on LG 5 from both studies co-localized with MetaRRD5.2, except for two QTL for severity from the T7.20 × SE population, contributing to MetaRRD5.1, as well as qRRD_TX2WSE_ch5_2021 from the diploid study and qRRV.BE × MG-ch5 from the tetraploid study, contributing to MetaRRD5.3. Thus, our meta-analysis suggests the existence of additional QTL on LG 5 (MetaRRD5.1 and 5.3), which was not evident from the results of separate analyses. However, MetaRRD5.2 was particularly interesting as it was supported by most of the QTL (22) in LG 5. For LG 6, MetaRRD6.1 and MetaRRD6.2 were discovered. The individual QTL from previous studies had an average CI larger than the two meta-QTL combined. As in the case of LG 5, our meta-analysis suggests the presence of two QTL on LG 6, a result that was not evident from the results of separate studies. More research to validate this additional QTL is needed.
When searching for candidate genes, researchers must choose from many possible factors that may be influencing the phenotype of interest [
48]. Little research has been performed in the Rosaceae family related to R genes for viral resistance. The discovery of resistance genes in apricots against the plum pox virus [
49] is one example within the family. Work has also been undertaken in the distantly related blackcurrant, where they found resistance genes for blackcurrant reversion virus resistance [
50]. The results of our meta-analysis provide us with more focused regions of interest for a search of the candidate genes underlying RRD-resistant QTL. The genes that we focused on in our search included genes encoding NBS-LRRs, NB-ARCs, genes associated with antiviral mechanisms, and general defense response. All were identified in the intervals of interest, however, to varying degrees. Genes related to anti-viral mechanisms co-localized to MetaRRD5.2, while homologs of genes encoding disease-resistant proteins with NBS-LRR and NB-ARC domains had numerous hits on all meta-QTL. Other interesting genes found within the meta-QTL regions were those involved in viral RNA translation or synthesis, as well as general defense response genes. Based on the original studies, LG 5 is a major factor in RRD-reduced susceptibility as it accounted for a large portion of the phenotypic variation. The average LG 5 QTL confidence interval from these original studies was ~26.5 cM. In this study, we were able to determine a narrower interval of ~4.3 cM representing ~4 Mbp, allowing a more focused candidate gene search in LG 5. It is important to point out that the candidate gene search we performed used the reference
R. chinensis genome assembly [
32]. Thus, additional work is clearly needed to determine the gene content in QTL intervals in our germplasm.
The results presented here and additional work to better define meta-QTL physical intervals will not only help with candidate gene searches but will allow the identification of marker haplotypes to guide the development of more robust marker-based selection tools to track and use a given QTL in a plant breeding context. Work to identify marker haplotypes in linkage disequilibrium (LD) with key QTL is underway.

 ,
, 

{kind=link}