

HIV Infection Elicits Differential Transcriptomic Remodeling in CD4+ T Cells with Variable Proliferative Responses to the T Cell Receptor Stimulus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Preparation of Cells for scRNA-Seq and Sequencing
2.3. ScRNA-Seq Read Mapping and Counting
2.4. ScRNA-Seq Data Pre-Processing
2.5. Integrating Triplicate Experiments into One Dataset and Phenotypic Assessment of Cells
2.6. Identification of Genes Modulated by HIV Infection
2.7. Identification of Gene Ontology (GO) Terms and Pathways Over-Represented for Differentially Expressed Genes
3. Results
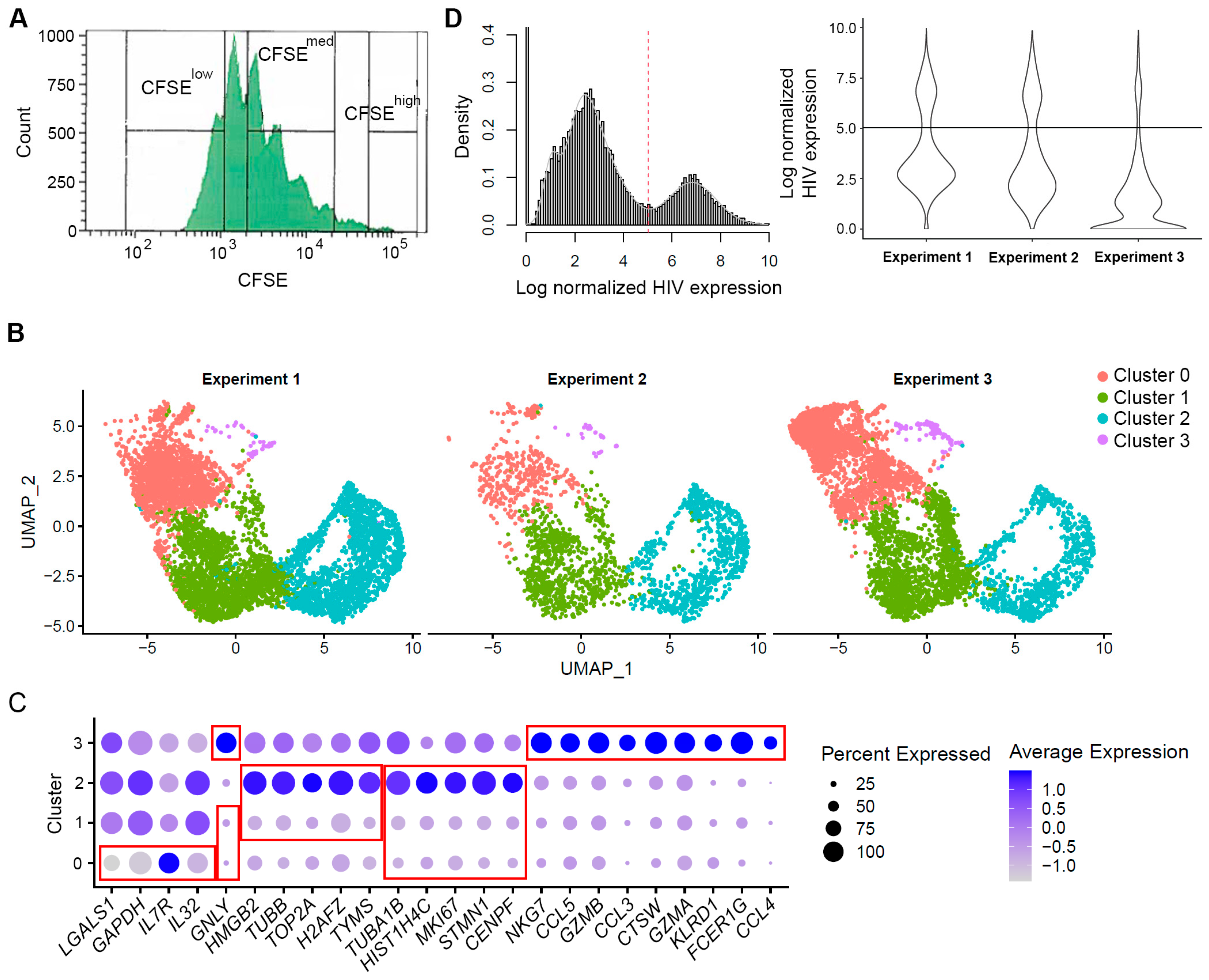
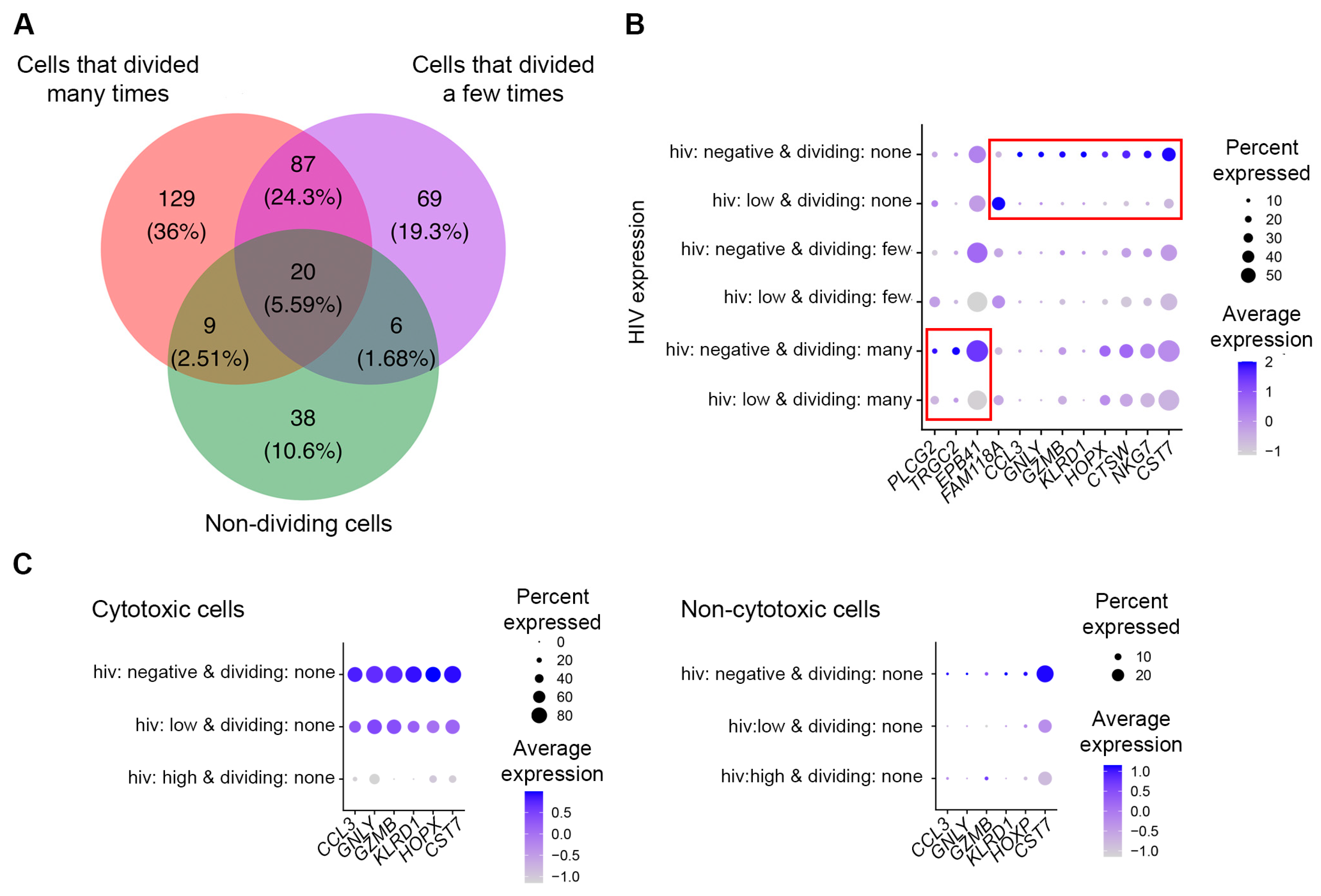
3.1. Cell Filtering and Phenotypic Assessement
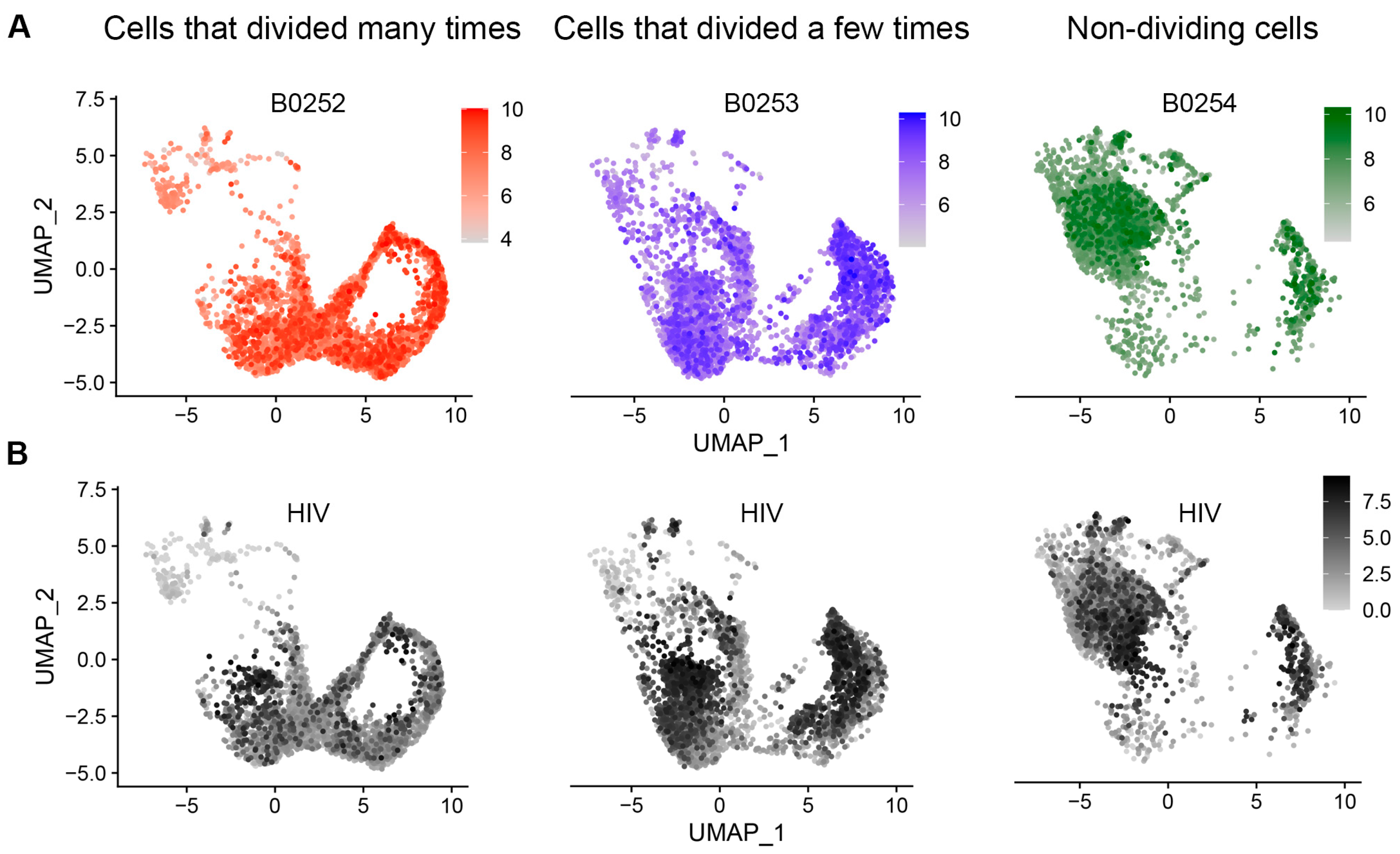
3.2. Evaluation of HIV Expression Levels in Different Cell Subsets
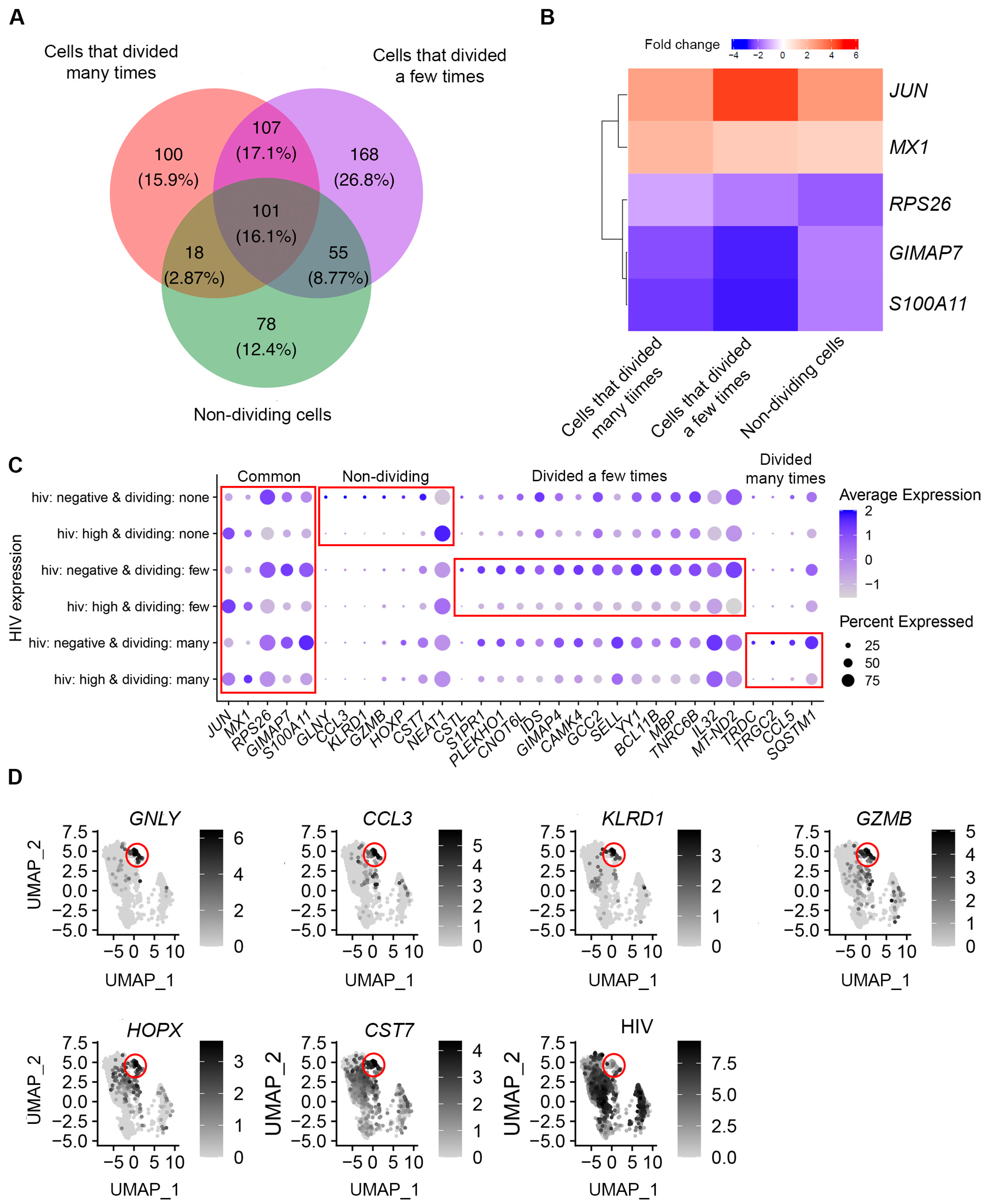
3.3. Identification of Genes Modulated by Productive HIV Infection in Different CD4+ T Cell Subsets
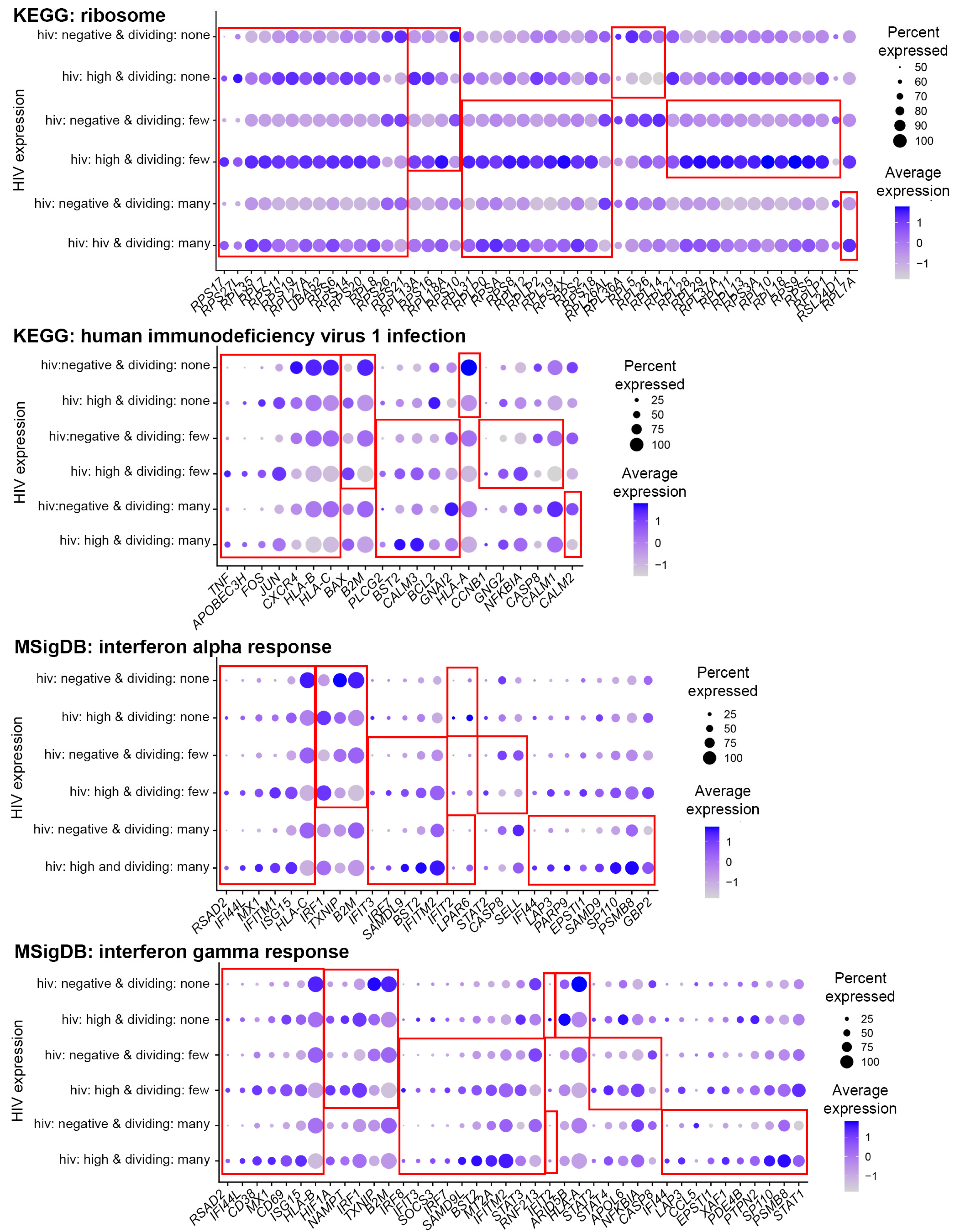
3.4. Functional Analysis of Genes Modulated by HIV during Productive Infection
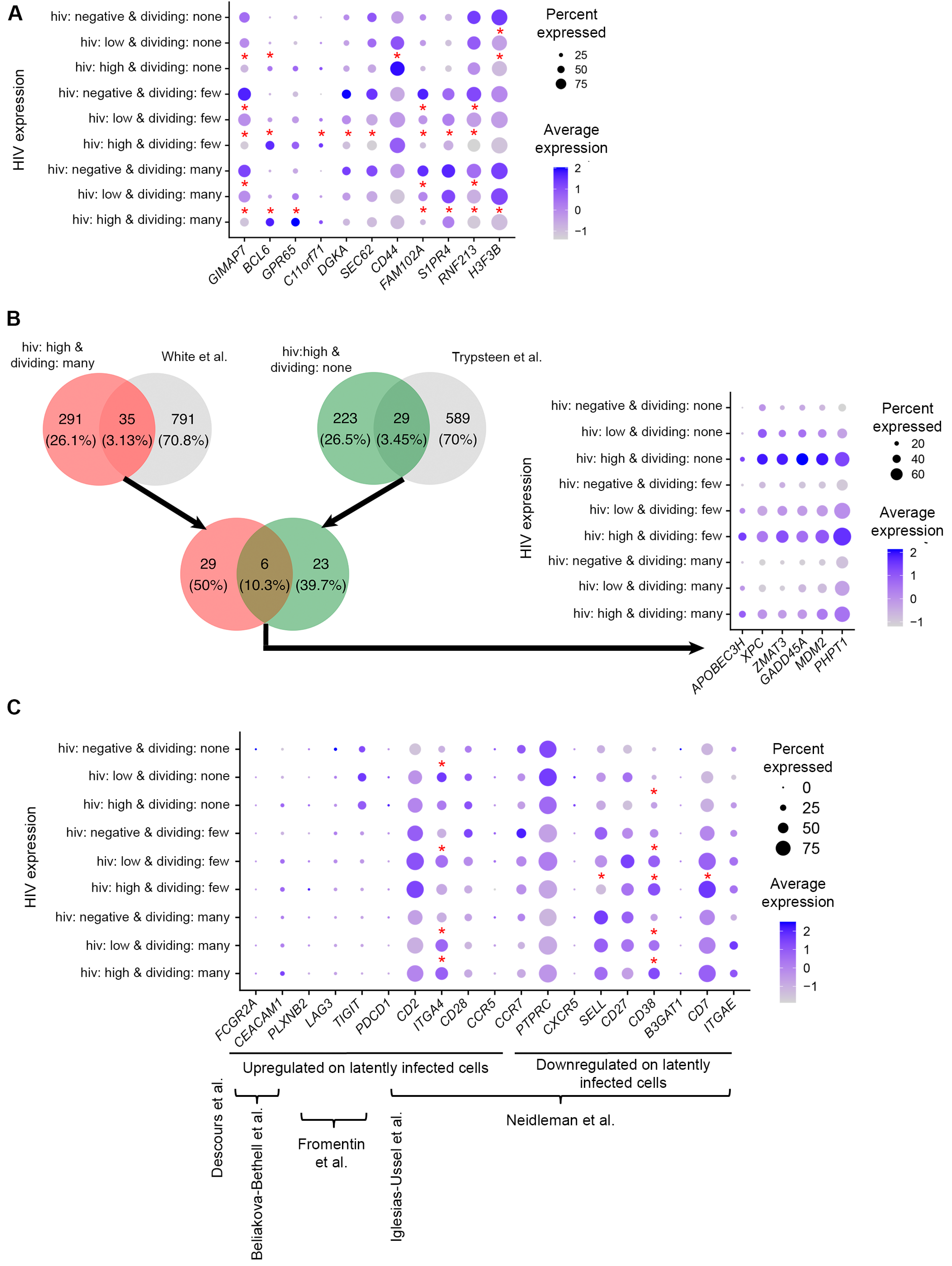
3.5. Identification of Genes Differentially Expressed in Cells with Low Levels of HIV RNA in Different CD4+ T Cell Subsets
3.6. Functional Analysis of Genes Modulated by HIV in Cells with Low Levels of HIV RNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef]
- Iglesias-Ussel, M.; Vandergeeten, C.; Marchionni, L.; Chomont, N.; Romerio, F. High levels of CD2 expression identify HIV-1 latently infected resting memory CD4+ T cells in virally suppressed subjects. J. Virol. 2013, 87, 9148–9158. [Google Scholar] [CrossRef] [Green Version]
- Descours, B.; Petitjean, G.; López-Zaragoza, J.-L.; Bruel, T.; Raffel, R.; Psomas, C.; Reynes, J.; Lacabaratz, C.; Levy, Y.; Schwartz, O.; et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature 2017, 543, 564–567. [Google Scholar] [CrossRef]
- Darcis, G.; Kootstra, N.A.; Hooibrink, B.; van Montfort, T.; Maurer, I.; Groen, K.; Jurriaans, S.; Bakker, M.; van Lint, C.; Berkhout, B.; et al. CD32(+)CD4+ T cells are highly enriched for HIV DNA and can support transcriptional latency. Cell Rep. 2020, 30, 2284–2296. [Google Scholar] [CrossRef] [Green Version]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T cells expressing PD-1, TIGIT and LAG-3 contribute to HIV persistence during ART. PLoS Pathog. 2016, 12, e1005761. [Google Scholar] [CrossRef] [Green Version]
- Beliakova-Bethell, N.; Manousopoulou, A.; Deshmukh, S.; Mukim, A.; Richman, D.D.; Garbis, S.D.; Spina, C.A. Integrated proteomics and transcriptomics analyses identify novel cell surface markers of HIV latency. Virology 2022, 573, 50–58. [Google Scholar] [CrossRef]
- Neidleman, J.; Luo, X.; Frouard, J.; Xie, G.; Hsiao, F.; Ma, T.; Morcilla, V.; Lee, A.; Telwatte, S.; Thomas, R.; et al. Phenotypic analysis of the unstimulated in vivo HIV CD4 T cell reservoir. eLife 2020, 9, e60933. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Wind-Rotolo, M.; Yang, H.C.; Siliciano, J.D.; Siliciano, R.F. Experimental approaches to the study of HIV-1 latency. Nat. Rev. Microbiol. 2007, 5, 95–106. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, D.A.; Penn, M.L.; Korin, Y.D.; Scripture-Adams, D.D.; Zack, J.A.; Kreisberg, J.F.; Roederer, M.; Sherman, M.P.; Chin, P.S.; Goldsmith, M.A. HIV-1 actively replicates in naive CD4+ T cells residing within human lymphoid tissues. Immunity 2001, 15, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Cameron, P.U.; Saleh, S.; Sallmann, G.; Solomon, A.; Wightman, F.; Evans, V.A.; Boucher, G.; Haddad, E.K.; Sekaly, R.-P.; Harman, A.N.; et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2010, 107, 16934–16939. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.; Solomon, A.; Wightman, F.; Xhilaga, M.; Cameron, P.U.; Lewin, S.R. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: A novel model of HIV-1 latency. Blood 2007, 110, 4161–4164. [Google Scholar] [CrossRef]
- Brady, T.; Agosto, L.M.; Malani, N.; Berry, C.C.; O’Doherty, U.; Bushman, F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. Aids 2009, 23, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Soto, P.C.; Terry, V.H.; Lewinski, M.K.; Deshmukh, S.; Beliakova-Bethell, N.; Spina, C.A. HIV-1 latency is established preferentially in minimally activated and non-dividing cells during productive infection of primary CD4 T cells. PLoS ONE 2022, 17, e0271674. [Google Scholar] [CrossRef]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S.Y. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Card, C.M.; Abrenica, B.; McKinnon, L.R.; Ball, T.B.; Su, R.C. Endothelial cells promote productive HIV infection of resting CD4+ T cells by an integrin-mediated cell adhesion-dependent mechanism. AIDS Res. Hum. Retrovir. 2022, 38, 111–126. [Google Scholar] [CrossRef]
- Kinter, A.; Moorthy, A.; Jackson, R.; Fauci, A.S. Productive HIV infection of resting CD4+ T cells: Role of lymphoid tissue microenvironment and effect of immunomodulating agents. AIDS Res. Hum. Retrovir. 2003, 19, 847–856. [Google Scholar] [CrossRef]
- Gagne, M.; Michaels, D.; Schiralli Lester, G.M.; Gummuluru, S.; Wong, W.W.; Henderson, A.J. Strength of T cell signaling regulates HIV-1 replication and establishment of latency. PLoS Pathog. 2019, 15, e1007802. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Dolgalev, I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. Available online: https://cran.r-project.org/web/packages/msigdbr/ (accessed on 26 January 2023).
- Preglej, T.; Ellmeier, W. CD4+ cytotoxic T cells—Phenotype, function and transcriptional networks controlling their differentiation pathways. Immunol. Lett. 2022, 247, 27–42. [Google Scholar] [CrossRef]
- Furler, R.L.; Ali, A.; Yang, O.O.; Nixon, D.F. Nef-induced differential gene expression in primary CD4+ T cells following infection with HIV-1 isolates. Virus Genes 2019, 55, 541–544. [Google Scholar] [CrossRef]
- Hyrcza, M.D.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Heisler, L.; Yang, S.; Wilkins, O.; Ostrowski, M.; Der, S.D. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J. Virol. 2007, 81, 3477–3486. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat, A.R.; German, J.; Teslovich, T.M.; Cofrancesco, J., Jr.; Jie, C.C.; Talbot, C.C., Jr.; Siliciano, R.F. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: Type I interferon-mediated disruption of T-cell dynamics. J. Virol. 2008, 82, 1870–1883. [Google Scholar] [CrossRef] [Green Version]
- Olvera-García, G.; Aguilar-García, T.; Gutiérrez-Jasso, F.; Imaz-Rosshandler, I.; Rangel-Escareño, C.; Orozco, L.; Aguilar-Delfín, I.; Vázquez-Pérez, J.A.; Zúñiga, J.; Pérez-Patrigeon, S.; et al. A transcriptome-based model of central memory CD4 T cell death in HIV infection. BMC Genom. 2016, 17, 956. [Google Scholar] [CrossRef] [Green Version]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; De Jesus, P.D.; Herbert, K.M.; Pache, L.; Smith, N.; van der Merwe, J.A.; et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-κB-elicited antiviral immune responses. eLife 2019, 8, e41930. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209. [Google Scholar] [CrossRef]
- Kazer, S.W.; Aicher, T.P.; Muema, D.M.; Carroll, S.L.; Ordovas-Montanes, J.; Miao, V.N.; Tu, A.A.; Ziegler, C.G.K.; Nyquist, S.K.; Wong, E.B.; et al. Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat. Med. 2020, 26, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.M.; Zhang, J.Y.; Xing, X.; Huang, H.H.; Xia, P.; Dai, X.P.; Hu, W.; Zhang, C.; Song, J.W.; Fan, X.; et al. Global transcriptomic characterization of T cells in individuals with chronic HIV-1 infection. Cell Discov. 2022, 8, 29. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Q.; Hui, H.; Agrawal, K.; Karris, M.A.Y.; Rana, T.M. An atlas of immune cell exhaustion in HIV-infected individuals revealed by single-cell transcriptomics. Emerg. Microbes Infect. 2020, 9, 2333–2347. [Google Scholar] [CrossRef]
- Bauby, H.; Ward, C.C.; Hugh-White, R.; Swanson, C.M.; Schulz, R.; Goujon, C.; Malim, M.H. HIV-1 Vpr induces widespread transcriptomic changes in CD4+ T cells early postinfection. mBio 2021, 12, e0136921. [Google Scholar] [CrossRef]
- Pollara, J.; Khanal, S.; Edwards, R.W.; Hora, B.; Ferrari, G.; Haynes, B.F.; Bradley, T. Single-cell analysis of immune cell transcriptome during HIV-1 infection and therapy. BMC Immunol. 2022, 23, 48. [Google Scholar] [CrossRef]
- Collora, J.A.; Liu, R.; Pinto-Santini, D.; Ravindra, N.; Ganoza, C.; Lama, J.R.; Alfaro, R.; Chiarella, J.; Spudich, S.; Mounzer, K.; et al. Single-cell multiomics reveals persistence of HIV-1 in expanded cytotoxic T cell clones. Immunity 2022, 55, 1013–1031. [Google Scholar] [CrossRef]
- White, C.H.; Moesker, B.; Beliakova-Bethell, N.; Martins, L.J.; Spina, C.A.; Margolis, D.M.; Richman, D.D.; Planelles, V.; Bosque, A.; Woelk, C.H. Transcriptomic analysis implicates the p53 signaling pathway in the establishment of HIV-1 latency in central memory CD4 T cells in an In vitro model. PLoS Pathog. 2016, 12, e1006026. [Google Scholar] [CrossRef] [Green Version]
- Trypsteen, W.; White, C.H.; Mukim, A.; Spina, C.A.; De Spiegelaere, W.; Lefever, S.; Planelles, V.; Bosque, A.; Woelk, C.H.; Vandekerckhove, L.; et al. Long non-coding RNAs and latent HIV—A search for novel targets for latency reversal. PLoS ONE 2019, 14, e0224879. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment 1 | Experiment 2 | Experiment 3 | |
|---|---|---|---|
| Total number of cells | 8715 | 2913 | 6174 |
| Mean reads per cells | 23,219 | 62,097 | 32,774 |
| Median genes per cell | 1689 | 2859 | 2050 |
| Cell Subset | High HIV RNA | Low HIV RNA | HIV-Negative |
|---|---|---|---|
| Divided many times | 406 | 3314 | 544 |
| Divided a few times | 1471 | 2351 | 314 |
| Non-dividing | 838 | 2779 | 430 |
| Total | 2715 | 8444 | 1288 |
| Cell Subset | High HIV RNA | Low HIV RNA | HIV-Negative |
|---|---|---|---|
| % cells that divided many times | 15.0 | 39.2 | 42.2 |
| % cells that divided a few times | 54.2 | 27.8 | 24.4 |
| % non-dividing cells | 30.9 | 32.9 | 33.4 |
| Total | 100 | 100 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Deshmukh, S.; Mukim, A.; Zhang, J.; Beliakova-Bethell, N. HIV Infection Elicits Differential Transcriptomic Remodeling in CD4+ T Cells with Variable Proliferative Responses to the T Cell Receptor Stimulus. Pathogens 2023, 12, 511. https://doi.org/10.3390/pathogens12040511
Zhang X, Deshmukh S, Mukim A, Zhang J, Beliakova-Bethell N. HIV Infection Elicits Differential Transcriptomic Remodeling in CD4+ T Cells with Variable Proliferative Responses to the T Cell Receptor Stimulus. Pathogens. 2023; 12(4):511. https://doi.org/10.3390/pathogens12040511
Chicago/Turabian StyleZhang, Xinlian, Savitha Deshmukh, Amey Mukim, Jasen Zhang, and Nadejda Beliakova-Bethell. 2023. "HIV Infection Elicits Differential Transcriptomic Remodeling in CD4+ T Cells with Variable Proliferative Responses to the T Cell Receptor Stimulus" Pathogens 12, no. 4: 511. https://doi.org/10.3390/pathogens12040511