
Genome-Wide Gene-Set Analysis Approaches in Amyotrophic Lateral Sclerosis

Abstract
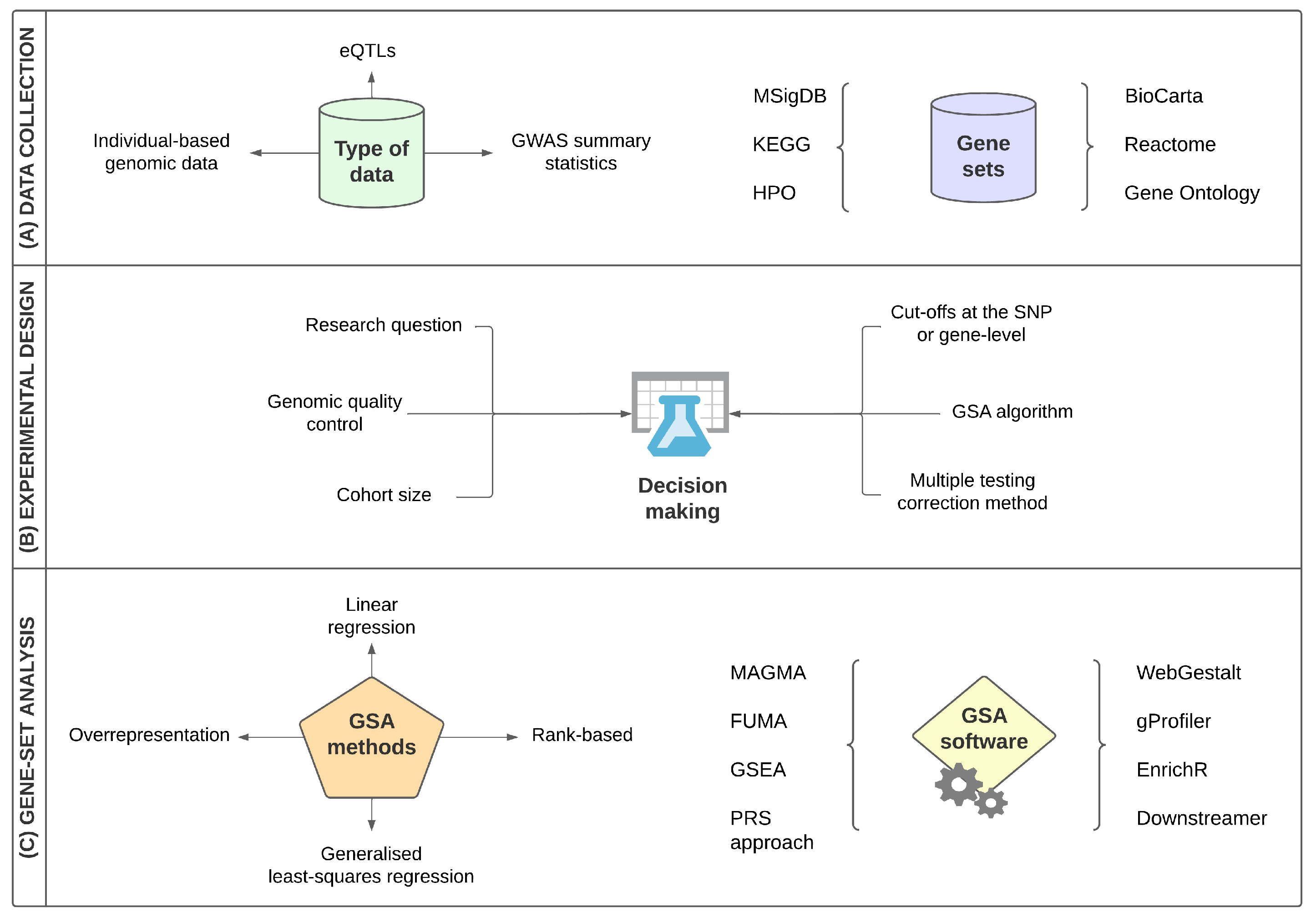
:1. Introduction
2. Limitations on Single-Gene Analysis
3. A Brief Overview of Gene-Set Analysis
3.1. The Structure of Gene-Set Analysis
- Each SNP is assigned to a gene—using specific annotation files in order to map each SNP into a gene region based on a kilobase window around the gene so that the researcher can additionally include regulatory elements—and each gene is then tested for its association with the phenotype.
- Genes are mapped to gene sets, and an association measure is computed for each gene set.
3.2. Main Categories of Gene-Set Analysis Methods
- Mean-based, where the gene-set association measures are summarized using the mean or sum of the gene associations.
- Count-based, where the genes are labelled as “significant” or “not significant”, and only “significant” genes determined by a specific cut-off are considered in the gene-set test statistic.
- Rank-based, where the genes in the gene-level matrix are ranked by their association with the phenotype and then an overrepresentation of the genes that belong in the gene set and also are at the top of that ranking is computed.
3.3. Gene-Set Analysis Confounding
4. Recent Approaches in ALS Genome-Wide Gene-Set Analysis Studies
5. Discussion
5.1. Cohort Size Affects the Power of Genome-Wide GSA
5.2. Limitations on Dimensionality Reduction Approaches
5.3. Comparing the Collected Gene-Set Analysis Methods
5.4. Gene-Set Analysis Deepens Our Understanding of the Implicated ALS Functional Pathways
6. Conclusions
- The use of large cohort sizes can increase the power of genome-wide gene-set analyses;
- Comprehensive, transparent and reproducible genomic quality control strategies are likely to support more consistent biological findings;
- Data-driven and holistic approaches in the selection of genes and gene-set annotation databases are preferable;
- Selection of competitive GSA methods and mean-based statistics provide a better performance, and the biological assumptions are more consistent with a real-life complex functional network;
- Detailed and transparent GSA methodology can contribute to reproducible research results and informed decision-making;
- Enhanced visualisation approaches may aid interpretation, e.g., Enrichment Networks.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Van Den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Xu, X.; Mukherjee, S.; Willis, J.; Hayes, J. Successes of Genome-wide association studies. Cell 2010, 142, 350–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, L.; Yilmaz, Z.; Gaspar, H.; Walters, R.; Goldstein, J.; Anttila, V.; Bulik-Sullivan, B.; Ripke, S.; Thornton, L.; Hinney, A.; et al. Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. Am. J. Psychiatry 2017, 174, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, U.G.; Milla, V.; Stafford, M.Y.C.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.R. A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.R.; Al-Chalabi, A.; Chio, A.; Hardiman, O.; Kiernan, M.C.; Rohrer, J.D.; Rowe, J.; Seeley, W.; Talbot, K. Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1042–1044. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Kenna, K.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257. [Google Scholar] [CrossRef] [Green Version]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; Van Der Spek, R.A.; Võsa, U.; De Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.H. Huntington’s Disease Genetics. NeuroRx 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Bhatia, G.; Gusev, A.; Finucane, H.K.; Bulik-Sullivan, B.K.; Pollack, S.J.; Lee, H.; Wray, N.R.; Kendler, K.S.; O’donovan, M.C.; et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015, 47, 1385–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilopoulou, C.; Morris, A.P.; Giannakopoulos, G.; Duguez, S.; Duddy, W. What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis? J. Pers. Med. 2020, 10, 247. [Google Scholar] [CrossRef]
- Gall, L.L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duguez, S. Molecular and cellular mechanisms affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Morgan, S.; Duguez, S.; Duddy, W. Personalized Medicine and Molecular Interaction Networks in Amyotrophic Lateral Sclerosis (ALS): Current Knowledge. J. Pers. Med. 2018, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- De Leeuw, C.A.; Neale, B.M.; Heskes, T.; Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 2016, 17, 353–364. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1. [Google Scholar] [CrossRef]
- Pritykin, Y.; Ghersi, D.; Singh, M. Genome-Wide Detection and Analysis of Multifunctional Genes. PLoS Comput. Biol. 2015, 11, e1004467. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Goeman, J.J.; Bühlmann, P. Analyzing gene expression data in terms of gene sets: Methodological issues. Bioinformatics 2007, 23, 980–987. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Mooney, M.A.; Wilmot, B. Gene Set Analysis: A Step-By-Step Guide. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 517–527. [Google Scholar] [CrossRef]
- Van Rheenen, W.; van der Spek, R.A.; Bakker, M.K.; van Vugt, J.J.; Hop, P.J.; Zwamborn, R.A.; de Klein, N.; Westra, H.J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef]
- Benyamin, B.; He, J.; Zhao, Q.; Gratten, J.; Garton, F.; Leo, P.J.; Liu, Z.; Mangelsdorf, M.; Al-Chalabi, A.; Anderson, L.; et al. Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat. Commun. 2017, 8, 611. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, R.; Misawa, K.; Tohnai, G.; Nakatochi, M.; Furuhashi, S.; Atsuta, N.; Hayashi, N.; Yokoi, D.; Watanabe, H.; Watanabe, H.; et al. A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun. Biol. 2020, 3, 526. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Fogh, I.; Selvackadunco, S.; Topp, S.D.; Shatunov, A.; van Rheenen, W.; Al-Khleifat, A.; Opie-Martin, S.; Ratti, A.; Calvo, A.; et al. SCFD1 expression quantitative trait loci in amyotrophic lateral sclerosis are differentially expressed. Brain Commun. 2021, 3, fcab236. [Google Scholar] [CrossRef] [PubMed]
- Saez-Atienzar, S.; Bandres-Ciga, S.; Langston, R.G.; Kim, J.J.; Choi, S.W.; Reynolds, R.H.; Abramzon, Y.; Dewan, R.; Ahmed, S.; Landers, J.E.; et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wen, Y.; Guo, X.; Hao, J.; Wang, W.; He, A.; Fan, Q.; Li, P.; Liu, L.; Liang, X.; et al. A Genome-wide Expression Association Analysis Identifies Genes and Pathways Associated with Amyotrophic Lateral Sclerosis. Cell. Mol. Neurobiol. 2018, 38, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Hou, L.; Zhang, J.; Tang, X.; Cheng, Z.; Li, G.; Fang, X.; Xu, J.; Zhang, X.; Xu, R. Polymorphism of rs3737597 in DISC1 Gene on Chromosome 1q42.2 in sALS Patients: A Chinese Han Population Case-Control Study. Mol. Neurobiol. 2017, 54, 3162–3179. [Google Scholar] [CrossRef]
- Schymick, J.C.; Scholz, S.W.; Fung, H.C.; Britton, A.; Arepalli, S.; Gibbs, J.R.; Lombardo, F.; Matarin, M.; Kasperaviciute, D.; Hernandez, D.G.; et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2007, 6, 322–328. [Google Scholar] [CrossRef]
- Cronin, S.; Berger, S.; Ding, J.; Schymick, J.C.; Washecka, N.; Hernandez, D.G.; Greenway, M.J.; Bradley, D.G.; Traynor, B.J.; Hardiman, O. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 2008, 17, 768–774. [Google Scholar] [CrossRef]
- Lee, Y.; Song, G. Genome-wide pathway analysis in amyotrophic lateral sclerosis. Genet. Mol. Res. 2015, 14, 6429–6438. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.; Liu, G.; Jiang, Y.; Fu, J.; Zhang, B.; Song, R.; Wang, W. Pathway Analysis of Two Amyotrophic Lateral Sclerosis GWAS Highlights Shared Genetic Signals with Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2014, 51, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Deng, L.; Mei, P.; Zhou, Y.; Wang, B.; Zhang, J.; Lin, J.; Wei, Y.; Zhang, X.; Xu, R. A genome-wide association study combining pathway analysis for typical sporadic amyotrophic lateral sclerosis in Chinese Han populations. Neurobiol. Aging 2014, 35, 9–1778. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [Green Version]
- Klein, N.d.; Tsai, E.A.; Vochteloo, M.; Baird, D.; Huang, Y.; Chen, C.Y.; Dam, S.v.; Deelen, P.; Bakker, O.B.; Garwany, O.E.; et al. Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain-related diseases. bioRxiv 2021. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience 2019, 8, 1–6. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Taylor, T.; Awad, A.; Baker, D.; Giovanonni, G.; Noyce, A.J.; Dobson, R. Summary-data-based Mendelian randomization prioritizes potential druggable targets for multiple sclerosis. Brain Commun. 2020, 2, fcaa119. [Google Scholar] [CrossRef]
- Huang, H.; Ouyang, Q.; Zhu, M.; Yu, H.; Mei, K.; Liu, R. mTOR-mediated phosphorylation of VAMP8 and SCFD1 regulates autophagosome maturation. Nat. Commun. 2021, 12, 6622. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gallwitz, D.; Peng, R. Structure-based Functional Analysis Reveals a Role for the SM Protein Sly1p in Retrograde Transport to the Endoplasmic Reticulum. Mol. Biol. Cell 2005, 16, 3951. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. Am. J. Hum. Genet. 2007, 81, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.G.; Pawitan, Y.; Magnusson, P.K.; Prince, J.A. Strategies and issues in the detection of pathway enrichment in genome-wide association studies. Hum. Genet. 2009, 126, 289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Chang, S.; Cui, S.; Guo, L.; Zhang, L.; Wang, J. ICSNPathway: Identify candidate causal SNPs and pathways from genome-wide association study by one analytical framework. Nucleic Acids Res. 2011, 39, 437–443. [Google Scholar] [CrossRef]
- Xie, Y.; Luo, X.; He, H.; Tang, M. Novel Insight Into the Role of Immune Dysregulation in Amyotrophic Lateral Sclerosis Based on Bioinformatic Analysis. Front. Neurosci. 2021, 15, 657465. [Google Scholar] [CrossRef]
- Ballard, D.H.; Cho, J.; Zhao, H. Comparisons of multi-marker association methods to detect association between a candidate region and disease. Genet. Epidemiol. 2010, 34, 201–212. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, G.R.; Burke, J.E. Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr. Opin. Cell Biol. 2020, 63, 57–67. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Studies | Input Data | Ancestry | GSA Software | GS Annotation |
|---|---|---|---|---|
| [41] | 27,205 cases and 110,881 controls [41,42,43] eQTL data | European, Japanese, Chinese | FUMA, MAGMA, Downstreamer | G0, HPO, REACTOME |
| [44] | 20,806 cases and 59,804 controls [14] eQTL data dbGaP Ac. phs000424.v8.p2 | European | g:Profiler, Enrichr, GSEA | G0, KEGG |
| [45] | 12,577 cases and 23,475 controls [20] 5605 cases and 24,110 controls [14] 2411 cases and 10,322 controls [14] | European | PRS approach | MSigDB |
| [46] | 12,577 cases and 23,475 controls [20], eQTL data [47] | European | GSEA | KEGG |
| [42] | 12,577 cases and 23,475 controls [20], 1234 cases and 2850 controls, 431 cases and 567 controls, [42] | European, Chinese, Australian | MAGMA | NA |
| [48] | 276 ALS cases and 271 controls [49], 221 cases and 216 controls [50] | American, Irish | WebGestalt | GO |
| [51] | 276 cases and 271 controls [49] | American | ICSNPathway | KEGG, BioCarta, GO BP, GO MF |
| [52] | 276 cases and 271 controls [49], 221 cases and 211 controls [50] | American, Irish | WebGestalt | KEGG |
| [53] | 250 cases and 250 controls | Chinese Han | WebGestalt | KEGG |
| Software | Input Data | Null Hypothesis | GS Method | Studies |
|---|---|---|---|---|
| Downstreamer | p-values, eQTL | Competitive | Generalized least-squares regression | [41] |
| Enrichr | Gene list | Competitive | Overrepresentation/hypergeometric test | [44] |
| FUMA | eQTL & GWAS | Competitive | Overrepresentation | [41] |
| g:Profiler | Gene list | Competitive | Overrepresentation/hypergeometric test | [44] |
| GSEA/i-GSEA | p-values | Competitive | rank-based, (KS test) | [44,46,51] |
| MAGMA | Genotypes, p-values | Competitive | Linear regression | [42] |
| WebGestalt | Gene list | Competitive | Overrepresentation/hypergeometric test | [48,52,53] |
| Studies | Multiple Testing Correction Method | Main Findings |
|---|---|---|
| [41] | Bonferroni | Cerebral cortical atrophy (p-value = 1.8 × 10−8), Abnormal nervous system electrophysiology (p-value = 4.1 × 10−7) Distal amyotrophy (p-value = 8.6 × 10−7), Membrane trafficking (p-value = 4.2 × 10−6), Intra-Golgi and retrograde Golgi-to-ER trafficking (p-value =1.4 × 10−5) Macroautophagy (p-value = 3.2 × 10−5) |
| [44] | FDR < 0.05 | Vesicle-mediated transport in synapse (adjusted p-value = 7.58 × 10−7), Glutamatergic synapse (adjusted p-value = 4.20 × 10−6) Vesicle docking involved in exocytosis (adjusted p-value = 3.30 × 10−5) |
| [45] | FDR < 0.05 | Neuron projection morphogenesis, Membrane trafficking, Signal transduction mediated by ribonucleotides |
| [46] | Empirical p-values | Peroxisome (empirical p-value = 0.006), Citrate cycle TCA cycle (empirical p-value = 0.025), Tight Junction (p-value NA) PPAR signaling pathway (empirical p-value = 0.025), SNARE interactions in vesicular transport (empirical p-value = 0.027), Arachidonic acid metabolism (empirical p-value = 0.040), Glycolysis-gluconeogenesis (empirical p-value = 0.043) |
| [42] | NA | No significant pathways were detected after multiple testing correction |
| [48] | NA | Nervous system development (adjusted p-value = 1.13 × 10−9) |
| [51] | FDR < 0.05 | Chromatin assembly (FDR = 0.001), Nucleosome assembly (FDR = 0.018) |
| [52] | FDR < 0.05 | RNA transport (adjusted p-value = 1.00 × 10−3), Vascular smooth muscle contraction (adjusted p-value = 1.80 × 10−3), Neuroactive ligand-receptor interaction (adjusted p-value = 6.30 × 10−3), Systemic lupus erythematosus (adjusted p-value = 6.30 × 10−3), Chemokine signaling pathway (adjusted p-value = 6.30 × 10−3), Hematopoietic cell lineage (adjusted p-value = 6.30 × 10−3), Cytosolic DNA-sensing pathway (adjusted p-value = 1.30 × 10−2), Protein processing in ER (adjusted p-value = 1.62 × 10−2), Alzheimer’s disease (adjusted p-value = 1.69 × 10−2), Parkinson’s disease (adjusted p-value = 3.12 × 10−2), Oxidative phosphorylation (adjusted p-value = 3.26 × 10−2), Cytokine–cytokine receptor interaction (adjusted p-value = 3.37 × 10−2) |
| [53] | FDR < 0.05 | Phosphatidylinositol signaling system (adjusted p-value = 0.0011), Pathways in cancer (adjusted p-value = 0.0011), Wnt signaling pathway (adjusted p-value = 0.0020), Axon guidance (adjusted p-value = 0.0021), MAPK signaling pathway (adjusted p-value = 0.0021), Neurotrophin signaling pathway (adjusted p-value = 0.0021), Arrhythmogenic right ventricular cardiomyopathy (adjusted p-value = 0.0044), Colorectal cancer (adjusted p-value = 0.0099), Arachidonic acid metabolism (adjusted p-value = 0.0454), T-cell receptor signaling pathway (adjusted p-value = 0.0488) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilopoulou, C.; Duguez, S.; Duddy, W. Genome-Wide Gene-Set Analysis Approaches in Amyotrophic Lateral Sclerosis. J. Pers. Med. 2022, 12, 1932. https://doi.org/10.3390/jpm12111932
Vasilopoulou C, Duguez S, Duddy W. Genome-Wide Gene-Set Analysis Approaches in Amyotrophic Lateral Sclerosis. Journal of Personalized Medicine. 2022; 12(11):1932. https://doi.org/10.3390/jpm12111932
Chicago/Turabian StyleVasilopoulou, Christina, Stephanie Duguez, and William Duddy. 2022. "Genome-Wide Gene-Set Analysis Approaches in Amyotrophic Lateral Sclerosis" Journal of Personalized Medicine 12, no. 11: 1932. https://doi.org/10.3390/jpm12111932