
Maternal Copy Number Imbalances in Non-Invasive Prenatal Testing: Do They Matter?
 , , ,
, , ,
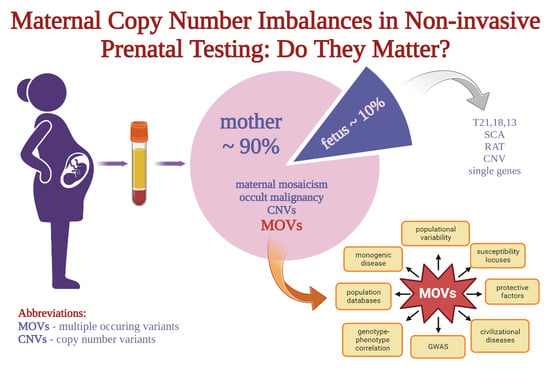
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
2.2. Bioinformatic Analysis for CNVs
2.3. Statistical Analysis
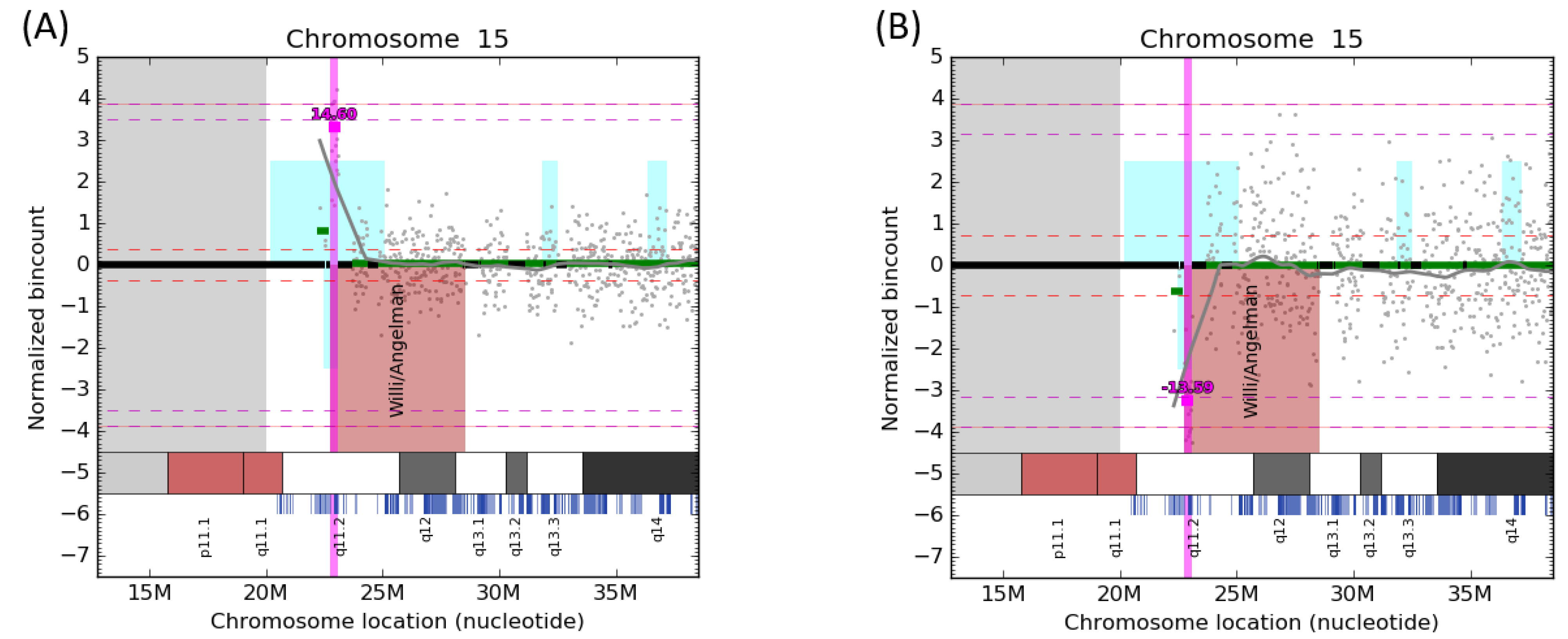
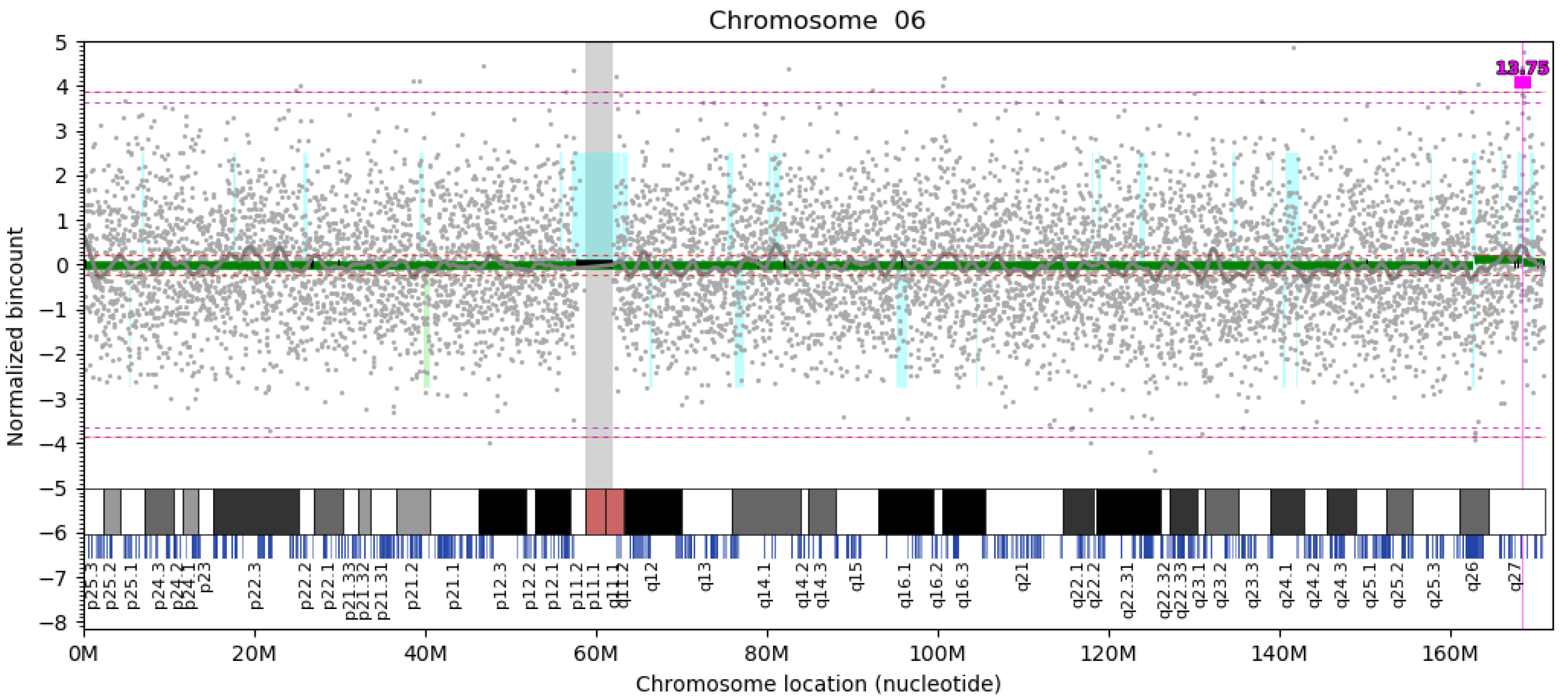
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luo, T.; Chen, H.-Y.; Zou, Q.-X.; Wang, T.; Cheng, Y.-M.; Wang, H.-F.; Wang, F.; Jin, Z.-L.; Chen, Y.; Weng, S.-Q.; et al. A novel copy number variation in CATSPER2 causes idiopathic male infertility with normal semen parameters. Hum. Reprod. 2019, 34, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, T.; Shomron, N. Genome-wide noninvasive prenatal diagnosis of monogenic disorders: Current and future trends. Comput. Struct. Biotechnol. J. 2020, 18, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Q.; Mao, X.; Lei, W.; He, M.; Lu, W. Noninvasive prenatal testing for chromosome aneuploidies and subchromosomal microdeletions/microduplications in a cohort of 42,910 single pregnancies with different clinical features. Hum. Genom. 2019, 13, 1–8. [Google Scholar] [CrossRef]
- Yang, J.; Wu, J.; Peng, H.; Hou, Y.; Guo, F.; Wang, D.; Ouyang, H.; Wang, Y.; Yin, A. Performances of NIPT for copy number variations at different sequencing depths using the semiconductor sequencing platform. Hum. Genom. 2021, 15, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Wang, W.; Dong, Y.; Zhang, M.; Wang, X.; Yin, C. Cell-free DNA screening for sex chromosome aneuploidies by non-invasive prenatal testing in maternal plasma. Mol. Cytogenet. 2020, 13, 1–8. [Google Scholar] [CrossRef]
- Mihaylova, M.; Staneva, R.; Toncheva, D.; Pancheva, M.; Hadjidekova, S. Benign, pathogenic and copy number variations of unknown clinical significance in patients with congenital malformations and developmental delay. Balk. J. Med. Genet. 2017, 20, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Zarrei, M.; MacDonald, J.; Merico, D.; Scherer, S. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Lukic, A.; Uphill, J.; Brown, C.A.; Beck, J.; Poulter, M.; Campbell, T.; Adamson, G.; Hummerich, H.; Whitfield, J.; Ponto, C.; et al. Rare structural genetic variation in human prion diseases. Neurobiol. Aging 2015, 36, 2004.e1–2004.e8. [Google Scholar] [CrossRef] [PubMed]
- Thean, L.F.; Low, Y.S.; Lo, M.; Teo, Y.-Y.; Koh, W.-P.; Yuan, J.-M.; Chew, M.H.; Tang, C.L.; Cheah, P.Y. Genome-wide association study identified copy number variants associated with sporadic colorectal cancer risk. J. Med. Genet. 2017, 55, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.S.; Benna, C.; Goldin, E.; Di Nisio, A.; De Toni, L.; Cosci, I.; Marchet, A.; Nitti, D.; Foresta, C. E2F1 copy number variations in germline and breast cancer: A retrospective study of 222 Italian women. Mol. Med. 2021, 27, 1–7. [Google Scholar] [CrossRef]
- Landry, L.G.; Ali, N.; Williams, D.R.; Rehm, H.L.; Bonham, V.L. Lack Of Diversity In Genomic Databases Is A Barrier To Translating Precision Medicine Research Into Practice. Health Aff. 2018, 37, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Hyblova, M.; Harsanyova, M.; Nikulenkov-Grochova, D.; Kadlecova, J.; Kucharik, M.; Budis, J.; Minarik, G. Validation of Copy Number Variants Detection from Pregnant Plasma Using Low-Pass Whole-Genome Sequencing in Noninvasive Prenatal Testing-Like Settings. Diagnostics 2020, 10, 569. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Speed, T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Tynan, J.; Ehrich, M.; Hannum, G.; McCullough, R.; Saldivar, J.-S.; Oeth, P.; Boom, D.V.D.; Deciu, C. Detection of Fetal Subchromosomal Abnormalities by Sequencing Circulating Cell-Free DNA from Maternal Plasma. Clin. Chem. 2015, 61, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Seshan, V.E.; Olshen, A.B. DNAcopy: A Package for Analyzing DNA Copy Data. Bioconduct. Vignette 2014. [Google Scholar] [CrossRef]
- Kucharik, M.; Gnip, A.; Hyblova, M.; Budis, J.; Strieskova, L.; Harsanyova, M.; Pös, O.; Kubiritova, Z.; Radvanszky, J.; Minarik, G.; et al. Non-invasive prenatal testing (NIPT) by low coverage genomic sequencing: Detection limits of screened chromosomal microdeletions. PLoS ONE 2020, 15, e0238245. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shi, J.; Ouyang, J.; Zhang, R.; Tao, Y.; Yuan, D.; Lv, C.; Wang, R.; Ning, B.; Roberts, R.; et al. X-CNV: Genome-wide prediction of the pathogenicity of copy number variations. Genome Med. 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Hinrichs, A.S. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budisteanu, M.; Papuc, S.; Streata, I.; Cucu, M.; Pirvu, A.; Serban-Sosoi, S.; Erbescu, A.; Andrei, E.; Iliescu, C.; Ioana, D.; et al. The Phenotypic Spectrum of 15q13.3 Region Duplications: Report of 5 Patients. Genes 2021, 12, 1025. [Google Scholar] [CrossRef] [PubMed]
- Maya, I.; Perlman, S.; Shohat, M.; Kahana, S.; Yacobson, S.; Tenne, T.; Agmon-Fishman, I.; Matar, R.T.; Basel-Salmon, L.; Sukenik-Halevy, R. Should We Report 15q11.2 BP1-BP2 Deletions and Duplications in the Prenatal Setting? J. Clin. Med. 2020, 9, 2602. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chai, H.; DiAdamo, A.; Grommisch, B.; Wen, J.; Zhang, H.; Li, P. Genotype–Phenotype Correlations for Putative Haploinsufficient Genes in Deletions of 6q26-q27: Report of Eight Patients and Review of Literature. Glob. Med Genet. 2022, 9, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Gimelli, S.; Capra, V.; Di Rocco, M.; Leoni, M.; Mirabelli-Badenier, M.; Schiaffino, M.C.; Fiorio, P.; Cuoco, C.; Gimelli, G.; Tassano, E. Interstitial 7q31.1 copy number variations disrupting IMMP2L gene are associated with a wide spectrum of neurodevelopmental disorders. Mol. Cytogenet. 2014, 7, 54. [Google Scholar] [CrossRef] [Green Version]
- Haraksingh, R.R.; Abyzov, A.; Gerstein, M.; Urban, A.E.; Snyder, M. Genome-Wide Mapping of Copy Number Variation in Humans: Comparative Analysis of High Resolution Array Platforms. PLoS ONE 2011, 6, e27859. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, D.; Chuchana, P.; Brockly, F.; Blancher, A.; Lefranc, G.; Lefranc, M.-P. First genomic sequence of a human Ig variable λ gene belonging to subgroup I. Functional genes, pseudogenes and vestigial sequences are interspersed in the IGLV locus. Nucleic Acids Res. 1989, 17, 3975. [Google Scholar] [CrossRef]
- Makoff, A.J.; Flomen, R.H. Detailed analysis of 15q11-q14 sequence corrects errors and gaps in the public access sequence to fully reveal large segmental duplications at breakpoints for Prader-Willi, Angelman, and inv dup(15) syndromes. Genome Biol. 2007, 8, R114. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Schaaf, C.P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 2015, 97, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguro-Ando, A.; Bamford, R.A.; Sital, W.; Sprengers, J.J.; Zuko, A.; Matser, J.M.; Oppelaar, H.; Sarabdjitsingh, A.; Joëls, M.; Burbach, J.P.H.; et al. Cntn4, a risk gene for neuropsychiatric disorders, modulates hippocampal synaptic plasticity and behavior. Transl. Psychiatry 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Fleischer, J.; Al-Kateb, H.; Mito, Y.; Amarillo, I.; Shinawi, M. Intragenic CNTN4 copy number variants associated with a spectrum of neurobehavioral phenotypes. Eur. J. Med Genet. 2019, 63, 103736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lu, Y.; Liu, X.; Yu, D.; Lv, Z.; Yang, M. Functional Evaluation ofZNF350Missense Genetic Variants Associated with Breast Cancer Susceptibility. DNA Cell Biol. 2018, 37, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.-F.; Li, C.-F.; Wang, W.-J.; Yang, W.-M.; Wang, D.D.-H.; Chang, W.-C.; Lee, W.-H.; Wang, J.-M. Loss of ZBRK1 Contributes to the Increase of KAP1 and Promotes KAP1-Mediated Metastasis and Invasion in Cervical Cancer. PLoS ONE 2013, 8, e73033. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Kuwano, Y.; Nishikawa, T.; Rokutan, K.; Nishida, K. Research Paper ZNF350 promoter methylation accel-erates colon cancer cell migration. Oncotarget 2018, 9, 36750. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Pei, D.; Liu, Y.; Yu, Y.; Guo, J.; Liu, N.; Kang, Z. Identification of a Novel Tumor Microenvironment Prognostic Signature for Bladder Urothelial Carcinoma. Front. Oncol. 2022, 12, 818860. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, R.; Huang, Y.; Liao, Y.; Su, P.; Wang, H.; Chang, C.; Lin, Y.; Yu, M.; Chu, T.; et al. Methylomics analysis identifies epigenetically silenced genes and implies an activation of β-catenin signaling in cervical cancer. Int. J. Cancer 2013, 135, 117–127. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, W.; Zhao, J.; Zhang, H. Integrated analysis of genome-wide gene expression and DNA methylation microarray of diffuse large B-cell lymphoma with TET mutations. Mol. Med. Rep. 2017, 16, 3777–3782. [Google Scholar] [CrossRef] [Green Version]
- Rafi, S.K.; Butler, M.G. The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders. Int. J. Mol. Sci. 2020, 21, 3296. [Google Scholar] [CrossRef]
- Brcic, L.; Underwood, J.F.; Kendall, K.M.; Caseras, X.; Kirov, G.; Davies, W. Medical and neurobehavioural phenotypes in carriers of X-linked ichthyosis-associated genetic deletions in the UK Biobank. J. Med. Genet. 2020, 57, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, N.F.; Janniger, C.K.; Schwartz, R.A. X-linked ichthyosis: An oculocutaneous genodermatosis. J. Am. Acad. Dermatol. 2010, 62, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Simard-Tremblay, E.; Saint-Martin, C. X-Linked Familial Focal Epilepsy Associated With Xp22.31 Deletion. Pediatr. Neurol. 2020, 108, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Gubb, S.J.A.; Brcic, L.; Underwood, J.F.G.; Kendall, K.M.; Caseras, X.; Kirov, G.; Davies, W. Medical and neurobehavioural phenotypes in male and female carriers of Xp22.31 duplications in the UK Biobank. Hum. Mol. Genet. 2020, 29, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Denison, S.R.; Callahan, G.; Becker, N.A.; Phillips, L.A.; Smith, D.I. Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosom. Cancer 2003, 38, 40–52. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2022. [Google Scholar] [CrossRef]
- Robertson, M.M. The prevalence and epidemiology of Gilles de la Tourette syndrome. J. Psychosom. Res. 2008, 65, 473–486. [Google Scholar] [CrossRef]
- Rooney, K.; Sadikovic, B. DNA Methylation Episignatures in Neurodevelopmental Disorders Associated with Large Structural Copy Number Variants: Clinical Implications. Int. J. Mol. Sci. 2022, 23, 7862. [Google Scholar] [CrossRef]
- Vasilyev, S.A.; Skryabin, N.A.; Kashevarova, A.A.; Tolmacheva, E.N.; Savchenko, R.R.; Vasilyeva, O.Y.; Lopatkina, M.E.; Zarubin, A.A.; Fishman, V.S.; Belyaeva, E.O.; et al. Differential DNA Methylation of the IMMP2L Gene in Families with Maternally Inherited 7q31.1 Microdeletions is Associated with Intellectual Disability and Developmental Delay. Cytogenet. Genome Res. 2021, 161, 105–119. [Google Scholar] [CrossRef]
- Davis, K.W.; Serrano, M.; Loddo, S.; Robinson, C.; Alesi, V.; Dallapiccola, B.; Novelli, A.; Butler, M.G. Parent-of-Origin Effects in 15q11.2 BP1-BP2 Microdeletion (Burnside-Butler) Syndrome. Int. J. Mol. Sci. 2019, 20, 1459. [Google Scholar] [CrossRef]
- Harris, R.A.; Raveendran, M.; Worley, K.C.; Rogers, J. Unusual sequence characteristics of human chromosome 19 are conserved across 11 nonhuman primates. BMC Evol. Biol. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimwood, J.; Gordon, L.A.; Olsen, A.; Terry, A.; Schmutz, J.; Lamerdin, J.; Hellsten, U.; Goodstein, D.; Couronne, O.; Tran-Gyamfi, M.; et al. The DNA sequence and biology of human chromosome 19. Nature 2004, 428, 529–535. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| CNV | Size | Genomic Coordinates (GRCh37/hg19) | Protein Coding Genes | ACMG Prediction | X CNV Prediction | n | Frequency (%) | dbVAR/ DGV Europe Frequency (%) | gnomAD Europe SV 2v.1 Frequency (%) |
|---|---|---|---|---|---|---|---|---|---|
| dup 6q27 | 240 kb | chr6:168,340,000–168,580,000 | AFDN, FRMD1, KIF25 | VUS | VUS | 126 | 1.962 | 3.1 (NF) | 1.4 |
| dup 22q11.22 | 260 kb | chr22:22,300,000–22,560,000 | IGLV11-55, IGLV4-60, IGLV4-69, IGLV6-57, IGLV8-61, PPM1F, TOP3D | VUS | LB | 98 | 1.526 | 0.7 (NF) | 0 |
| dup 8p23.2 | 200 kb | chr8:2,360,000–2,560,000 | - | VUS | B | 58 | 0.903 | 2.9 (F); 1 (NF) | 0.48 |
| dup 11q25 | 360 kb | chr11:134,360,000–134,720,000 | - | B | B | 55 | 0.856 | 1.9 (F); 0.9 (NF) | 0.39 |
| dup 15q13.3 | 480 kb | chr15:32,020,000–32,500,000 | OTUD7A, CHRNA7 | VUS | LB | 42 | 0.654 | 0.8 (NF) | 0.23 |
| dup 15q11.2 | 320 kb | chr15:22,760,000–23,080,000 | NIPA1, NIPA2, CYFIP1, TUBGCP5 | VUS | VUS | 38 | 0.592 | 0.3 (NF) | 0 |
| dup Xp22.31 | 1.66 Mb | chrX:6,220,000–8,960,000 | PUDP, STS, VCX, PNPLA4 | VUS | P | 31 | 0.483 | 0.1 (NF) | 0 |
| dup 1q25.1 | 320 kb | chr1:175,420,000–175,740,000 | TNR | VUS | B | 30 | 0.467 | 0.1 (NF) | 0.06 |
| del 17q22 | 340 kb | chr17:50,960,000–51,300,000 | - | VUS | B | 28 | 0.436 | 0 (NF) | 0.026 |
| del 9p23 | 260 kb | chr9:11,920,000–12,180,000 | - | VUS | B | 27 | 0.420 | 5.8 (F); 1.2 (NF) | 0.01 |
| dup 12p11.1 | 500 kb | chr12:34,300,000–34,800,000 | - | VUS | B | 24 | 0.374 | 0.1 (NF) | 0.026 |
| dup 12q24.13-q24.21 | 280 kb | chr12:114,260,000–114,540,000 | RBM19 | VUS | LB | 20 | 0.311 | 0 (NF) | 0 |
| dup 19q13.41 | 320 kb | chr19:52,280,000–52,600,000 | FPR1, FPR3, ZNF577, ZNF649, ZNF613, ZNF350, ZNF615, ZNF614, ZNF432, ZNF841 | VUS | LB | 20 | 0.311 | 0.4 (NF) | 0.026 |
| dup 6p11.2 | 600 kb | chr6:57,400,000–58,000,000 | - | VUS | B | 20 | 0.311 | 0.1 (NF) | 0.026 |
| dup 7q11.21 | 200 kb | chr7:64,680,000–64,880,000 | ZNF92 | VUS | B | 19 | 0.296 | 2.9 (F); 0.4 (NF) | 0.26 |
| dup2p22.3 | 680 kb | chr2:32,640,000–33,320,000 | BIRC6, TTC27, LTBP1 | VUS | VUS | 18 | 0.28 | 1 (F); 0.1 (NF) | 0.18 |
| dup 14q21.2 | 440 kb | chr14:43,820,000–44,260,000 | - | VUS | B | 17 | 0.265 | 0.4 (NF) | 0.15 |
| del 7q11.21 | 220 kb | chr7:64,680,000–64,880,000 | ZNF92 | B | B | 17 | 0.265 | 0.4 (NF) | 0.26 |
| dup 3p26.3 | 360 kb | chr3:2,660,000–3,020,000 | CNTN4 | VUS | VUS | 16 | 0.249 | 0 (NF) | 0 |
| dup 7q11.21 | 480 kb | chr7:62,040,000–62,640,000 | - | B | B | 16 | 0.249 | 0 (NF) | 0.31 |
| dup Xq27.2 | 380 kb | chrX: 140,360,000–140,740,000 | SPANXA1, SPANXA2 | B | B | 14 | 0.218 | 0.3 (NF) | 0.44 |
| del 15q11.2 | 240 kb | chr15:22,840,000–23,080,000 | NIPA1, NIPA2, CYFIP1, TUBGCP5 | VUS | P | 13 | 0.202 | 0.1 (NF) | 0.18 |
| del 2p22.3 | 260 kb | chr2:35,760,000–36,080,000 | - | B | B | 12 | 0.187 | 0.2 (NF) | 0.066 |
| del 4q35.2 | 1.6 Mb | chr4:188,280,000–189,920,000 | ZFP42, TRIML2, TRIML1 | VUS | VUS | 12 | 0.187 | 0 (NF) | 0.026 |
| dup 4q35.2 | 480 kb | chr4:188,700,000–189,180,000 | ZFP42, TRIML2, TRIML1 | VUS | VUS | 12 | 0.187 | 0 (NF) | 0 |
| del 6q26 | 800 kb | chr6:162,340,000–163,140,000 | PRKN | VUS | LP | 12 | 0.187 | 0 (NF) | 0 |
| del 7q31.1 | 460 kb | chr7:110,840,000–111,300,000 | IMMP2L | B | LP | 12 | 0.187 | 0.2 (NF) | 0 |
| dup Xp21.1 | 240 kb | chrX:33,000,000–33,920,000 | - | B | B | 12 | 0.187 | 0 (NF) | 0.017 |
| CNV | Size | Genomic Coordinates (GRCh37/hg19) | Protein Coding Genes | ACMG Prediction | X CNV Prediction | n | Frequency (%) | dbVAR/ DGV Europe Frequency (%) | gnomAD Europe SV 2v.1 Frequency (%) |
|---|---|---|---|---|---|---|---|---|---|
| del 15q11.2 | 240 kb | chr15:22,840,000–23,080,000 | NIPA1, NIPA2, CYFIP1, TUBGCP5 | VUS | P | 13 | 0.202 | 0.1 (NF) | 0.18 |
| del 6q26 | 800 kb | chr6:162,340,000–163,140,000 | PRKN | VUS | LP | 12 | 0.187 | 0 (NF) | 0 |
| del 7q31.1 | 460 kb | chr7:110,840,000–111,300,000 | IMMP2L | B | LP | 12 | 0.187 | 0.2 (NF) | 0 |
| del 20p12.1 | 540 kb | chr20: 14,640,000–15,180,000 | MACROD2 | B | LP | 11 | 0.171 | 0.1 | 0 |
| del 22q11.21-q11.22 | 280 kb | chr22: 22,300,000–22,580,000 | PPM1F, TOP3B | VUS | LP | 8 | 0.125 | 1.9 (F); 0.7 (NF) | 0 |
| del Xq12 | 280 kb | chrX:65,640,000–66,020,000 | EDA2R | B | LP | 8 | 0.125 | 0.1 | 0.017 |
| del 5q23.1 | 260 kb | chr5:118,860,000–119,120,000 | HSD17B4, FAM170A | VUS | LP | 6 | 0.093 | 0 | 0.026(partial overlay) |
| del 5q23.1-q23.2 | 1.38 Mb | chr5:119,960,000–121,340,000 | SRFBP1, PRR16,FTMT | VUS | LB | 5 | 0.078 | 0 | 0 |
| dup 5p15.33 | 1.48 Mb | chr5:40,000–1,520,000 | SLC6A3, LRRC14B, ZDHHC11B, ZDHHC11, CCDC127, SLC9A3, TPPP, PLEKHG4B, SLC12A7, SLC6A19, SLC6A18, LPCAT1, PDCD6, TERT, AC026740.1, NKD2, AHRR, CLPTM1L, BRD9, TRIP13, EXOC3, CEP72, SDHA | VUS | P | 5 | 0.078 | 0 | 0 |
| del 10q21.3 | 380 kb | chr10:68,260,000–68,640,000 | CTNNA3 | VUS | LP | 5 | 0.078 | 0.1 | 0 |
| del 4q22.3 | 320 kb | chr4:98,520,000–98,840,000 | STPG2 | B | LP | 4 | 0.062 | 0 | 0.013 (overlay) |
| dup 16p13.11 | 1.16 Mb | chr16:15,120,000–16,280,000 | MYH11, NDE1, MARF1, PDXDC1, MPV17L, NTAN1, RRN3, FOPNL, AC140504.1, C16orf45, NPIPA5, ABCC6, ABCC1 | VUS | LP | 4 | 0.062 | 0.2 | 0 |
| del 3p26.2-p26.1 | 600 kb | chr3: 3,840,000–4,440,000 | SUMF1, SETMAR, LRRN1 | VUS | P | 4 | 0.062 | 0 | 0 |
| del 3q26.31-q26.32 | 660 kb | chr3:175,080,000–175,740,000 | NAALADL2 | B | LP | 4 | 0.062 | 0.1 | 0.039 |
| del 5q12.1 | 860 kb | chr5:59,360,000–60,220,000 | ERCC8, FKSG52, DEPDC1B, PDE4D, ELOVL7 | VUS | P | 3 | 0.047 | 0 | 0 |
| del 8p22 | 1.4 Mb | chr8:13,860,000–15,260,000 | SGCZ | VUS | LP | 3 | 0.047 | 0 | 0 |
| del 10p12.31 | 400 kb | chr10:19,420,000–19,820,000 | MALRD1 | B | LP | 3 | 0.047 | 0.1 | 0 |
| del 16p12.2 | 480 kb | chr16:21,940,000–22,420,000 | EEF2K, CDR2, MOSMO, SDR42E2, POLR3E, UQCRC2, PDZD9, VWA3A | VUS | LP | 3 | 0.047 | 0 | 0.039 |
| dup 22q11.21 | 2.17 Mb | chr22:18,900,000–21,440,000 | RIMBP3, PI4KA, KLHL22, AC007326.4, GNB1L, TBX1, TRMT2A, TANGO2, FAM230A, RTL10, GP1BB, AC002472.1, ZNF74, P2RX6, DGCR8, ESS2, CRKL, SLC7A4, TMEM191B, DGCR2, USP41, DGCR6, C22orf39, RTN4R, DGCR6L, MED15, UFD1, TXNRD2, CLDN5, GGTLC3, TSSK2, GSC2, ARVCF, SLC25A1, COMT, CLTCL1, SERPIND1, LRRC74B, AC007731.5, SCARF2, HIRA, CCDC188, RANBP1, THAP7, SNAP29, PRODH, MRPL40, ZDHHC8, CDC45, AIFM3, SEPT5, LZTR1, SEPT5-GP1BB | P | P | 3 | 0.047 | 0.1 | 0 |
| dup 5p13.2-p13.1 | 1.48 Mb | chr5:37,100,000–38,580,000 | EGFLAM, CPLANE1, LIFR, NUP155, WDR70, GDNF | LP | P | 2 | 0.031 | 0 | 0 |
| del 7p21.2 | 360 kb | chr7:16,120,000–16,480,000 | ISPD | VUS | LP | 2 | 0.031 | 0 | 0 |
| del 7q21.11 | 740 kb | chr7:84,340,000–85,080,000 | SEMA3D | VUS | LP | 2 | 0.031 | 0 | 0 |
| del 8q22.2 | 400 kb | chr8:100,340,000–100,740,000 | VPS13B | VUS | LP | 2 | 0.031 | 0.1 | 0 |
| del 8q24.23-q24.3 | 1.74 Mb | chr8: 139,540,000–141,280,000 | TRAPPC9, COL22A1, KCNK9 | VUS | P | 2 | 0.031 | 0 | 0 |
| del 9q32 | 520 kb | chr9:119,220,000–119,740,000 | ASTN2, TRIM32 | VUS | P | 2 | 0.031 | 0 | 0 |
| dup 10q11.22-q11.23 | 3.9 Mb | chr10:47,640,000–51,580,000 | PARG, AL591684.1, TMEM273, ERCC6, AL603965.1, CHAT, VSTM4, NCOA4, GDF2, TIMM23B, ARHGAP22, C10orf71, FAM21B, FAM25C, MAPK8, OGDHL, FAM25G, GDF10, ANXA8, DRGX, ANXA8L2, RBP3, FRMPD2, AGAP8, AGAP9, ZNF488, MSMB, FAM170B, LRRC18, FAM21D, ASAH2C, WDFY4, PTPN20B, SLC18A3, ANTXRL, C10orf53 | VUS | P | 2 | 0.031 | 0 | 0 |
| del 11p15.3 | 260 kb | chr11: 11,360,000–11,620,000 | GALNT18, CSNK2A3 | B | LP | 2 | 0.031 | 0 | 0 |
| del 12p13.31 | 320 kb | chr12:5,860,000–6,180,000 | VWF, ANO2 | B | LP | 2 | 0.031 | 0 | 0 |
| del 12p11.23 | 480 kb | chr12:27,280,000–27,760,000 | SMCO2, ARNTL2, STK38L, PPFIBP1 | B | LP | 2 | 0.031 | 0 | 0.026 |
| del 18p11.32 | 800 kb | chr18:1,820,000–2,620,000 | NDC80, METTL4 | B | LP | 2 | 0.031 | 0 | 0 |
| del 20p12.3 | 480 kb | chr20:8,100,000–8,560,000 | PLCB1 | VUS | LP | 2 | 0.031 | 0.1 | 0 |
| del 1q21.1 | 880 kb | chr1:144,880,000–145,720,000 | ANKRD35, ITGA10, POLR3C, AC239799.1, TXNIP, RBM8A, AC243547.3, PIAS3, RNF115, NOTCH2NLA, ANKRD34A, AL590452.1, POLR3GL, LIX1L, PEX11B, NUDT17, NBPF10, NUDT4B, SEC22B, HJV, CD160, PDE4DIP | VUS | LP | 2 | 0.031 | 0 | 0 |
| del 2p16.3 | 320 kb | chr2: 50,700,000–5,1020,000 | NRXN1 | LP | P | 2 | 0.031 | 0 | 0 |
| del 2p16.3 | 400 kb | chr2: 51,080,000–51,480,000 | NRXN1 | LP | P | 2 | 0.031 | 0 | 0 |
| del 2p13.2 | 440 kb | chr2: 72,500,000–72,940,000 | EXOC6B | VUS | LP | 2 | 0.031 | 0 | 0 |
| del 2q12.3 | 420 kb | chr2: 108,600,000–109,020,000 | SLC5A7, SULT1C3, SULT1C2, SULT1C4 | B | LP | 2 | 0.031 | 0 | 0 |
| del 3q26.1 | 800 kb | chr3:164,920,000–165,720,000 | BCHE | VUS | LP | 2 | 0.031 | 0 | 0 |
| Chromosome | Effective Length (GRCh38/hg20) * | Number of Variants | Number of Variants per Megabase | p-Value (<0.05) * |
|---|---|---|---|---|
| 1 | 231,223,641 | 64 | 0.276788306 | 0.309486126 |
| 2 | 240,863,511 | 71 | 0.294772752 | 0.39856174 |
| 3 | 198,255,541 | 67 | 0.337947679 | 0.625386241 |
| 4 | 189,962,376 | 58 | 0.305323618 | 0.453773416 |
| 5 | 181,358,067 | 55 | 0.303267458 | 0.442908357 |
| 6 | 170,078,524 | 60 | 0.352778226 | 0.697688611 |
| 7 | 158,970,135 | 60 | 0.377429383 | 0.801517446 |
| 8 | 144,768,136 | 54 | 0.37301026 | 0.78465846 |
| 9 | 122,084,564 | 41 | 0.335832792 | 0.614630158 |
| 10 | 133,263,006 | 45 | 0.33767811 | 0.624020457 |
| 11 | 134,634,058 | 37 | 0.274819021 | 0.300273823 |
| 12 | 133,137,821 | 34 | 0.255374466 | 0.216709904 |
| 13 | 97,983,128 | 29 | 0.295969322 | 0.404743508 |
| 14 | 91,660,769 | 31 | 0.338203578 | 0.626681306 |
| 15 | 85,089,576 | 26 | 0.305560343 | 0.455026684 |
| 16 | 83,378,703 | 32 | 0.38379105 | 0.824340228 |
| 17 | 83,481,871 | 24 | 0.287487567 | 0.36152445 |
| 18 | 80,089,650 | 24 | 0.299664189 | 0.4239761 |
| 19 | 58,440,758 | 8 | 0.136890764 | 0.008989368 |
| 20 | 63,944,268 | 15 | 0.234579275 | 0.144314325 |
| 21 | 40,088,623 | 9 | 0.224502598 | 0.115899249 |
| 22 | 40,181,019 | 14 | 0.348423219 | 0.677094783 |
| X | 154,893,034 | 84 | 0.542309734 | 0.998853971 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyblova, M.; Gnip, A.; Kucharik, M.; Budis, J.; Sekelska, M.; Minarik, G. Maternal Copy Number Imbalances in Non-Invasive Prenatal Testing: Do They Matter? Diagnostics 2022, 12, 3056. https://doi.org/10.3390/diagnostics12123056
Hyblova M, Gnip A, Kucharik M, Budis J, Sekelska M, Minarik G. Maternal Copy Number Imbalances in Non-Invasive Prenatal Testing: Do They Matter? Diagnostics. 2022; 12(12):3056. https://doi.org/10.3390/diagnostics12123056
Chicago/Turabian StyleHyblova, Michaela, Andrej Gnip, Marcel Kucharik, Jaroslav Budis, Martina Sekelska, and Gabriel Minarik. 2022. "Maternal Copy Number Imbalances in Non-Invasive Prenatal Testing: Do They Matter?" Diagnostics 12, no. 12: 3056. https://doi.org/10.3390/diagnostics12123056