
Pediatric Primary Hepatic Tumors: Diagnostic Considerations

Abstract
:1. Introduction
2. Benign Tumors in Infants and Early Childhood
2.1. Hepatic Hemangiomas
2.1.1. Clinical Presentation and Laboratory Findings
2.1.2. Associated Syndromes and Risk Factors
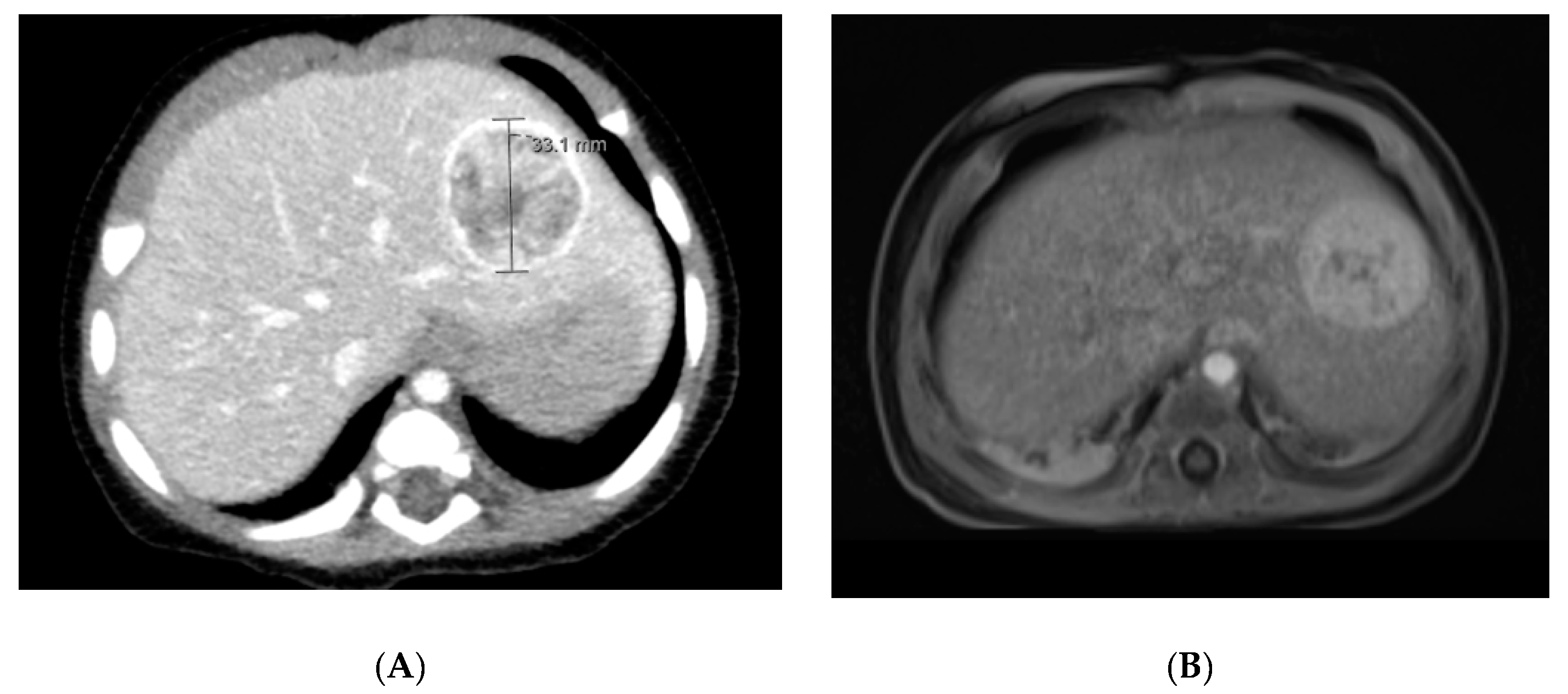
2.1.3. Diagnostic Imaging
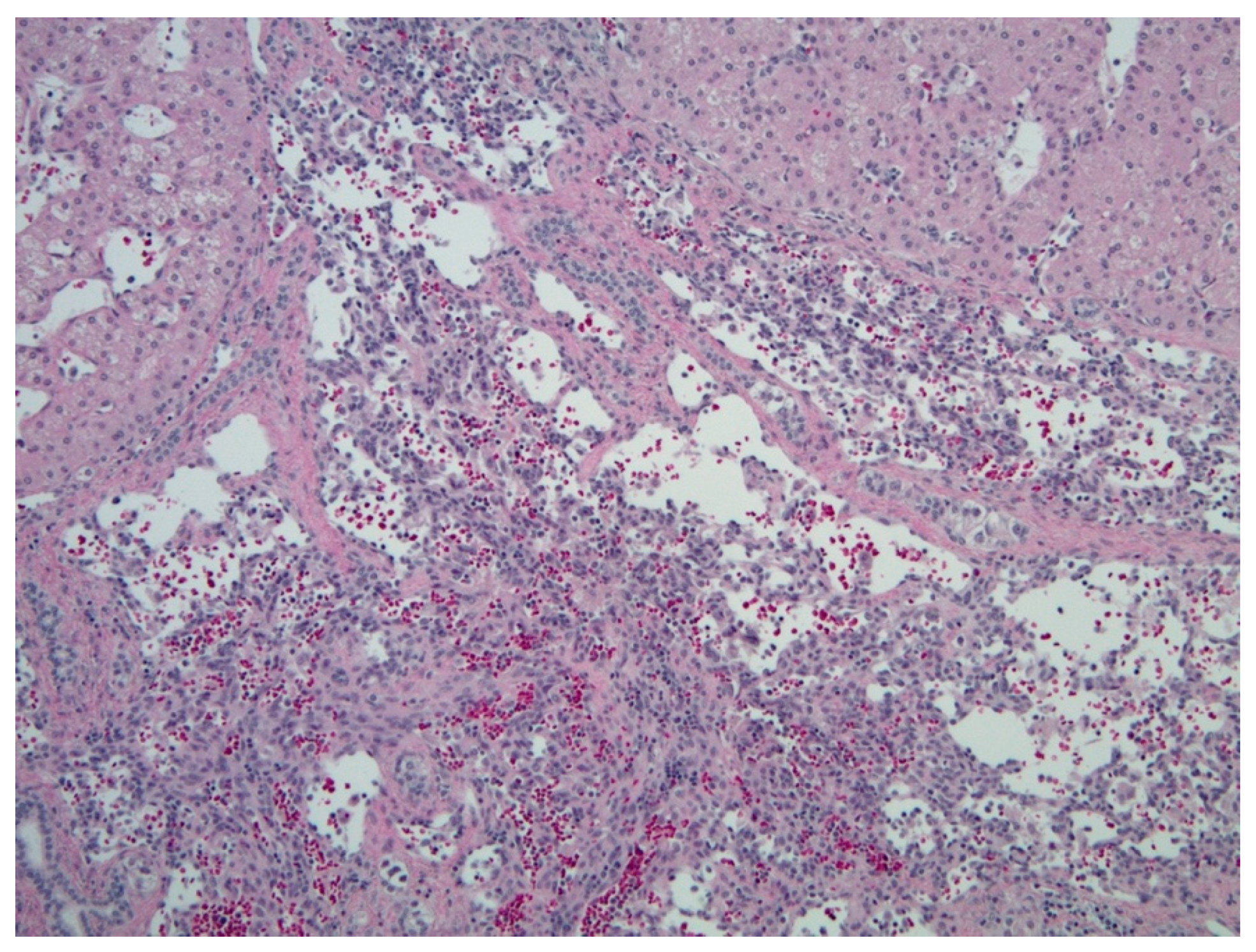
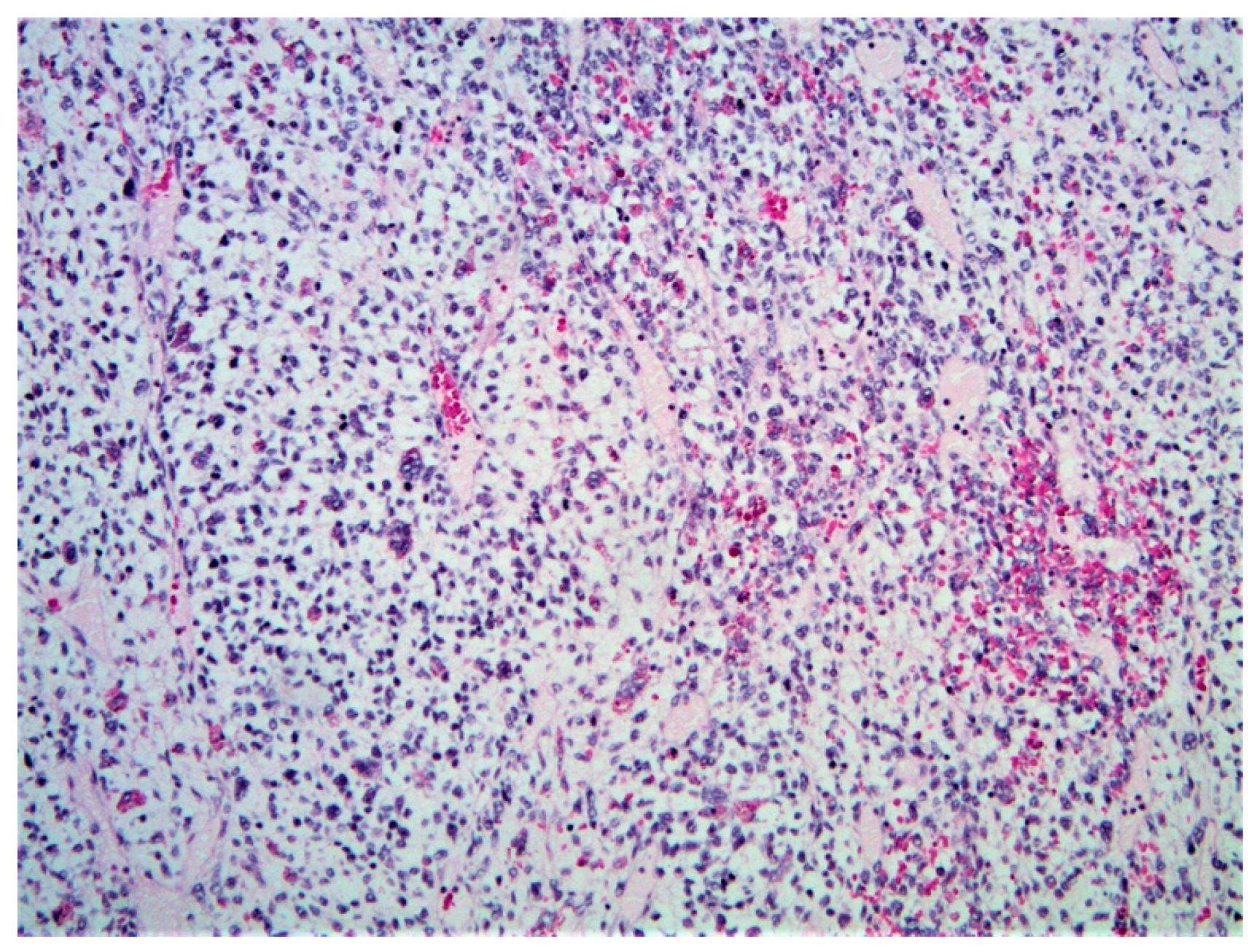
2.1.4. Histopathology and Immunohistochemistry
2.2. Mesenchymal Hamartoma
2.2.1. Clinical Presentation and Laboratory Findings
2.2.2. Associated Syndromes and Risk Factors
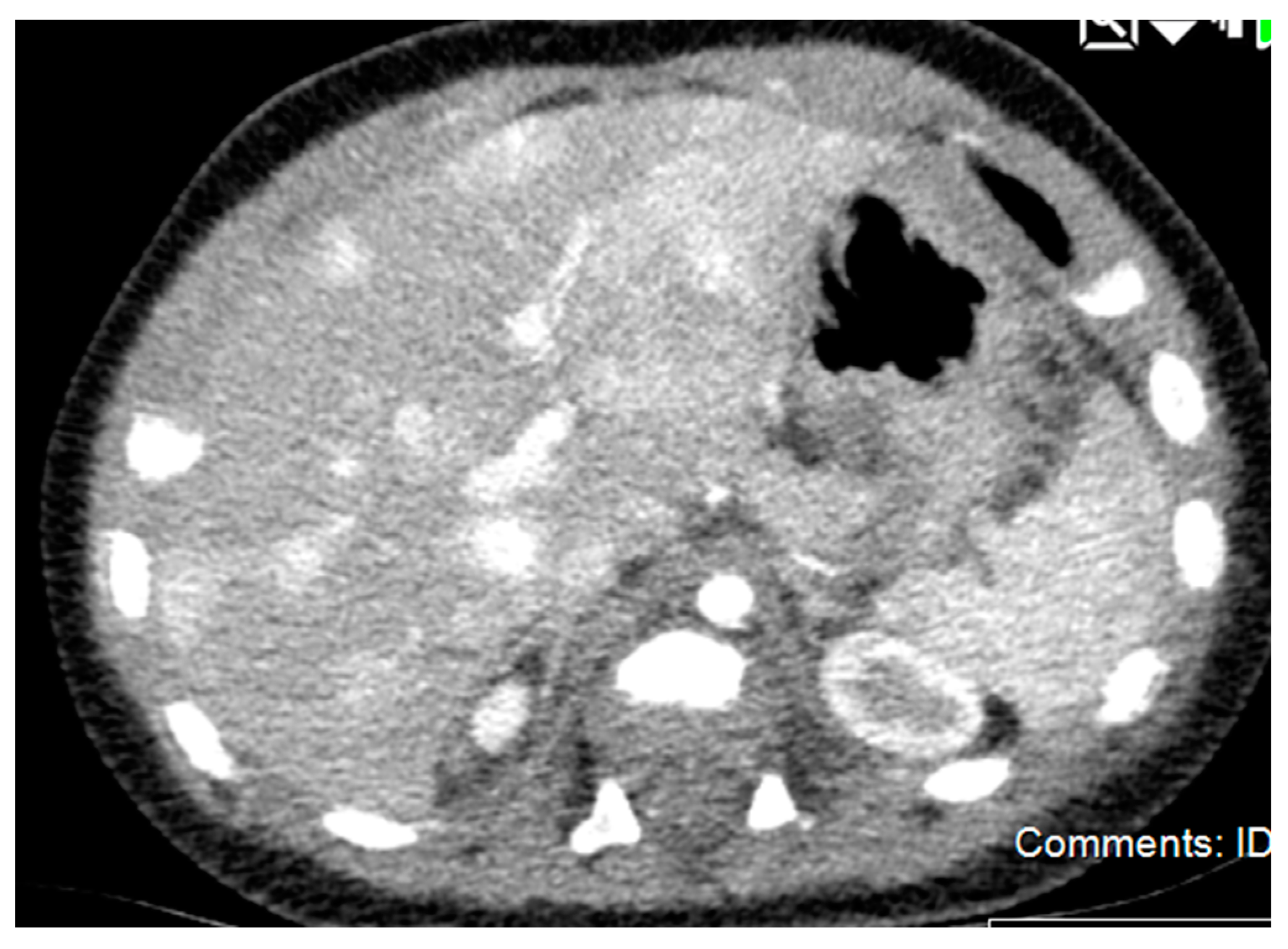
2.2.3. Diagnostic Imaging
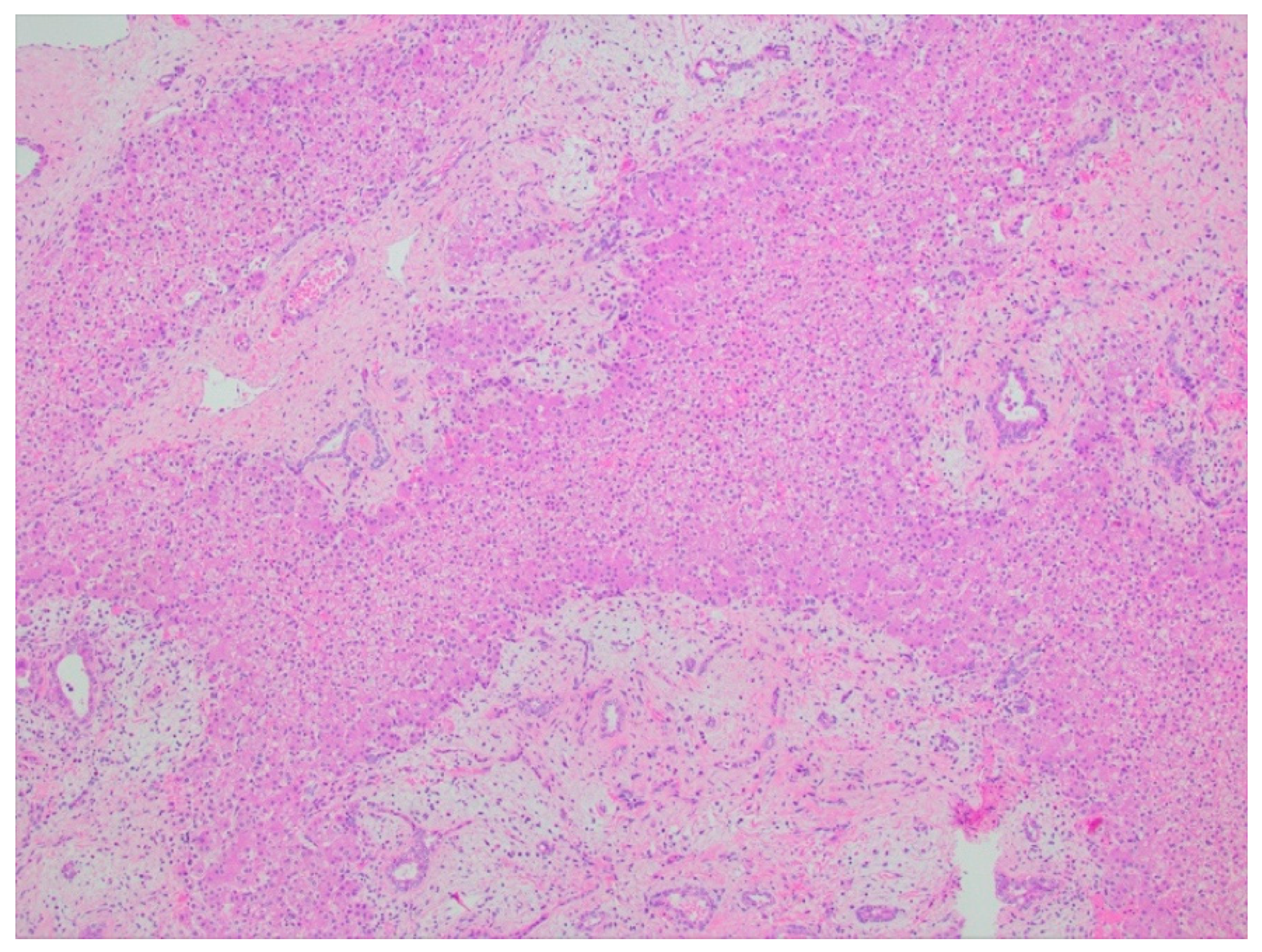
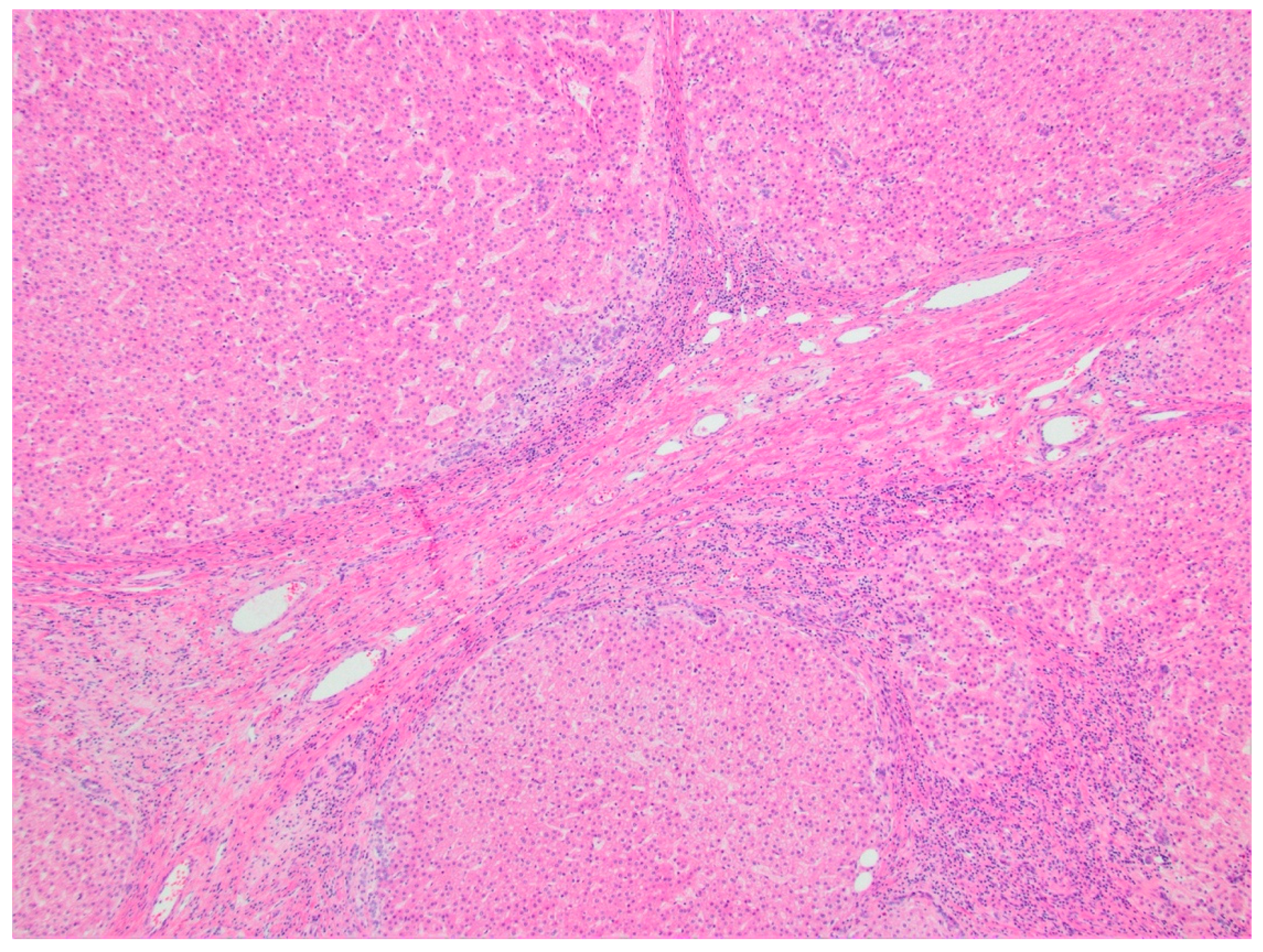
2.2.4. Histopathology
3. Malignant Tumors in Infants and Early Childhood
3.1. Hepatoblastoma
3.1.1. Clinical Presentation and Laboratory Findings
3.1.2. Associated Syndromes and Risk Factors
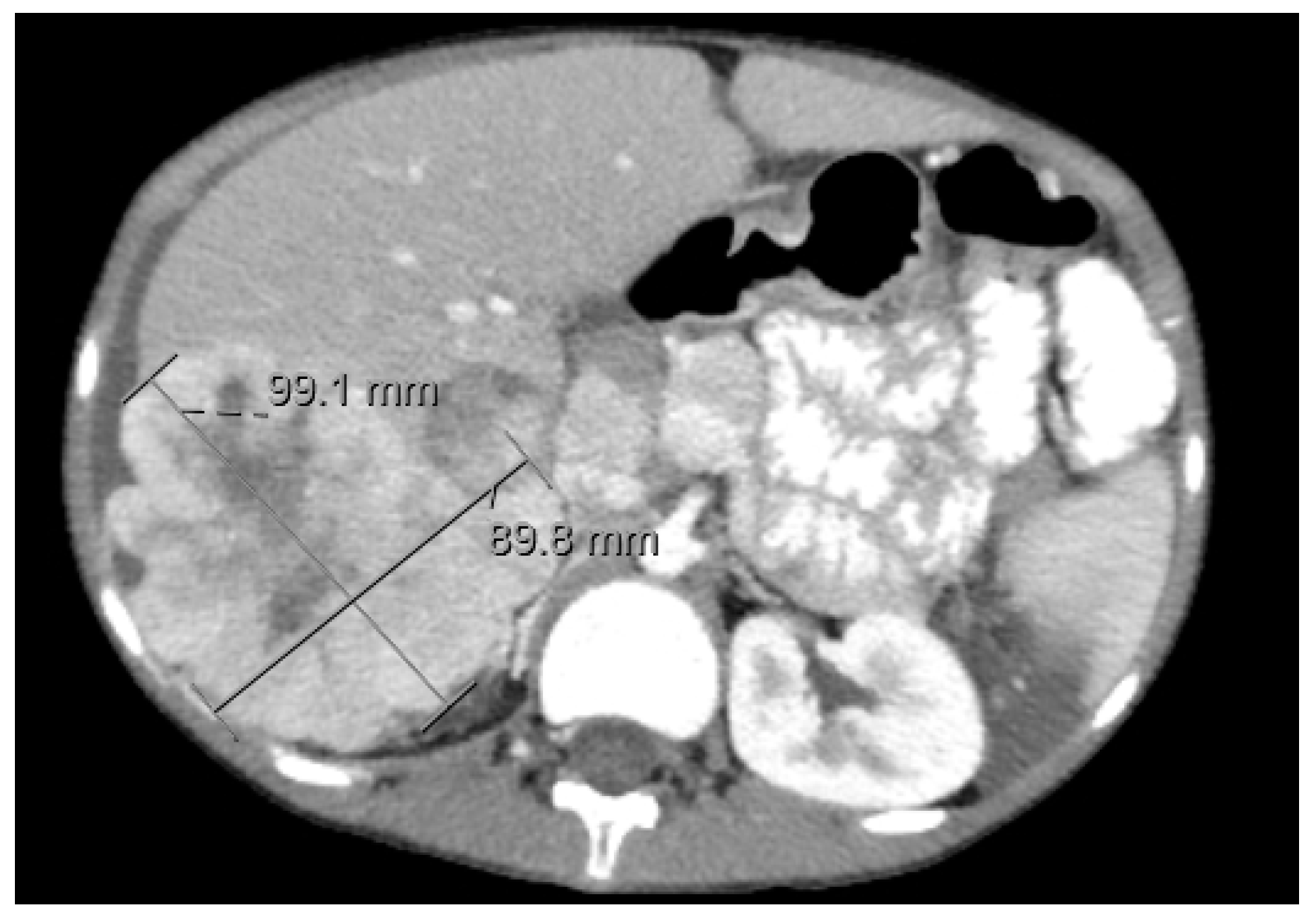
3.1.3. Diagnostic Imaging
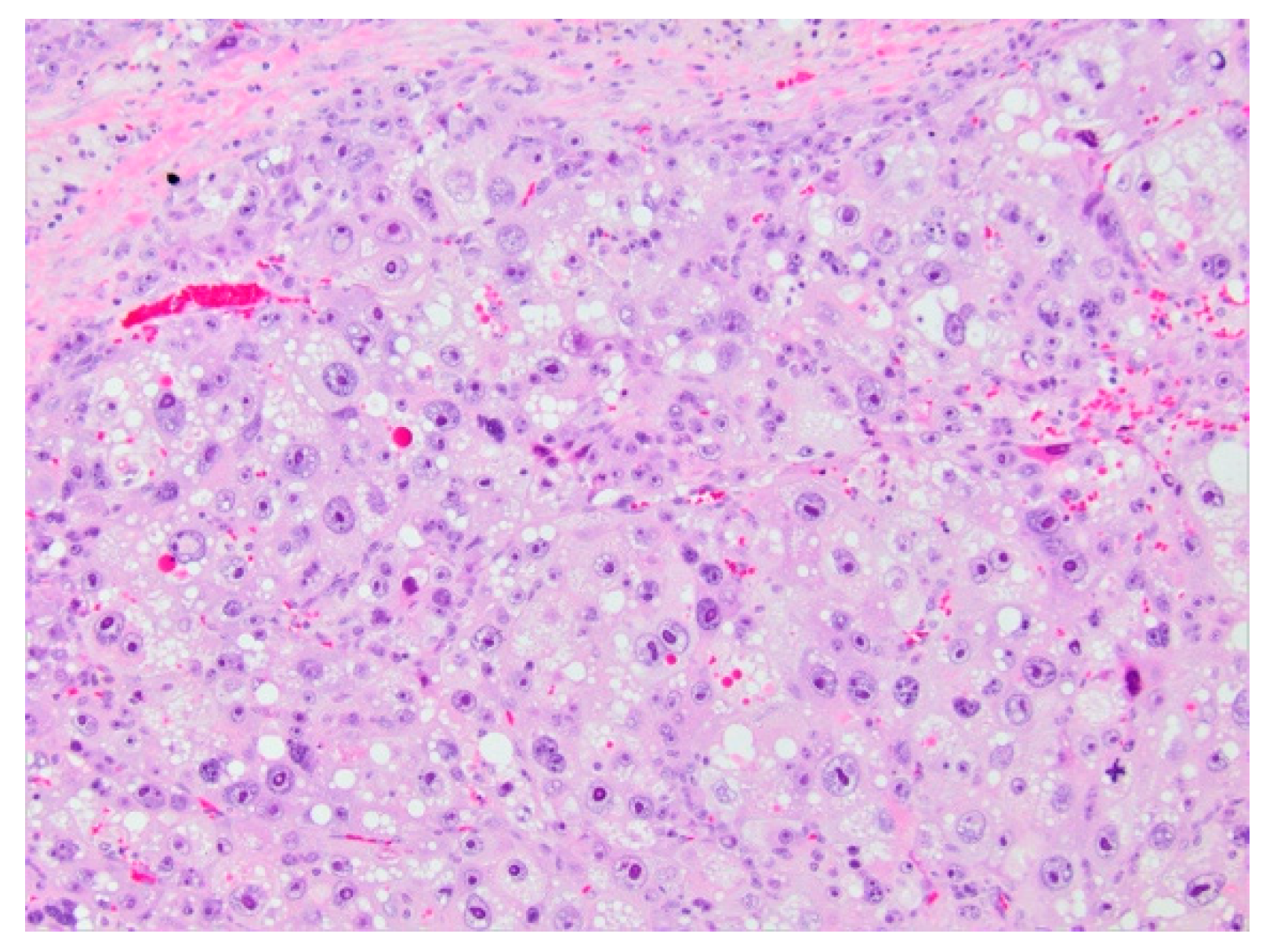
3.1.4. Histopathology and Immunohistochemistry
3.2. Malignant Rhabdoid Tumors
3.2.1. Clinical Presentation and Laboratory Studies
3.2.2. Diagnostic Imaging
3.2.3. Histopathology
3.3. Angiosarcoma
3.3.1. Clinical Presentation and Laboratory Findings
3.3.2. Associated Syndromes and Risk Factors
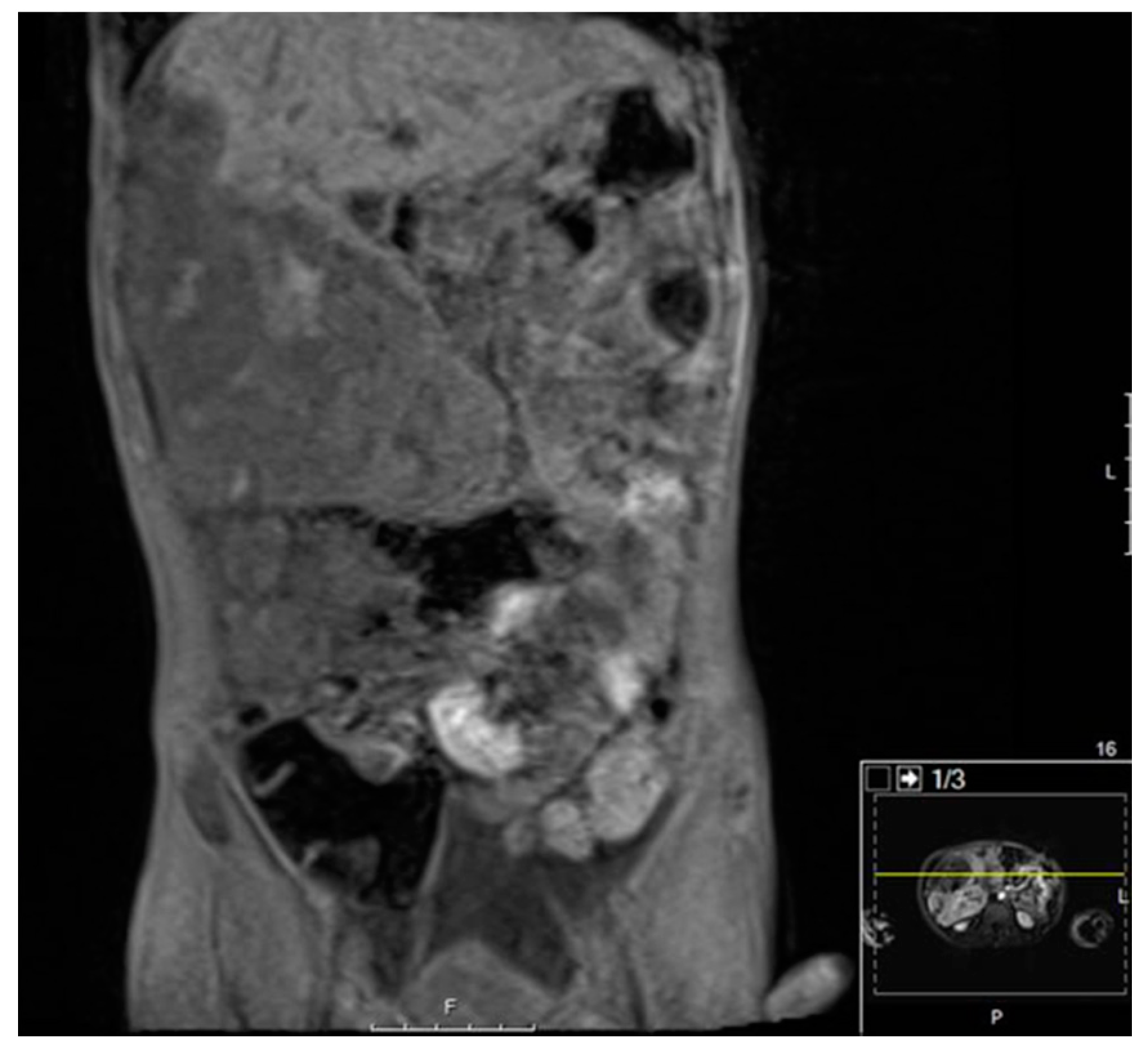
3.3.3. Diagnostic Imaging
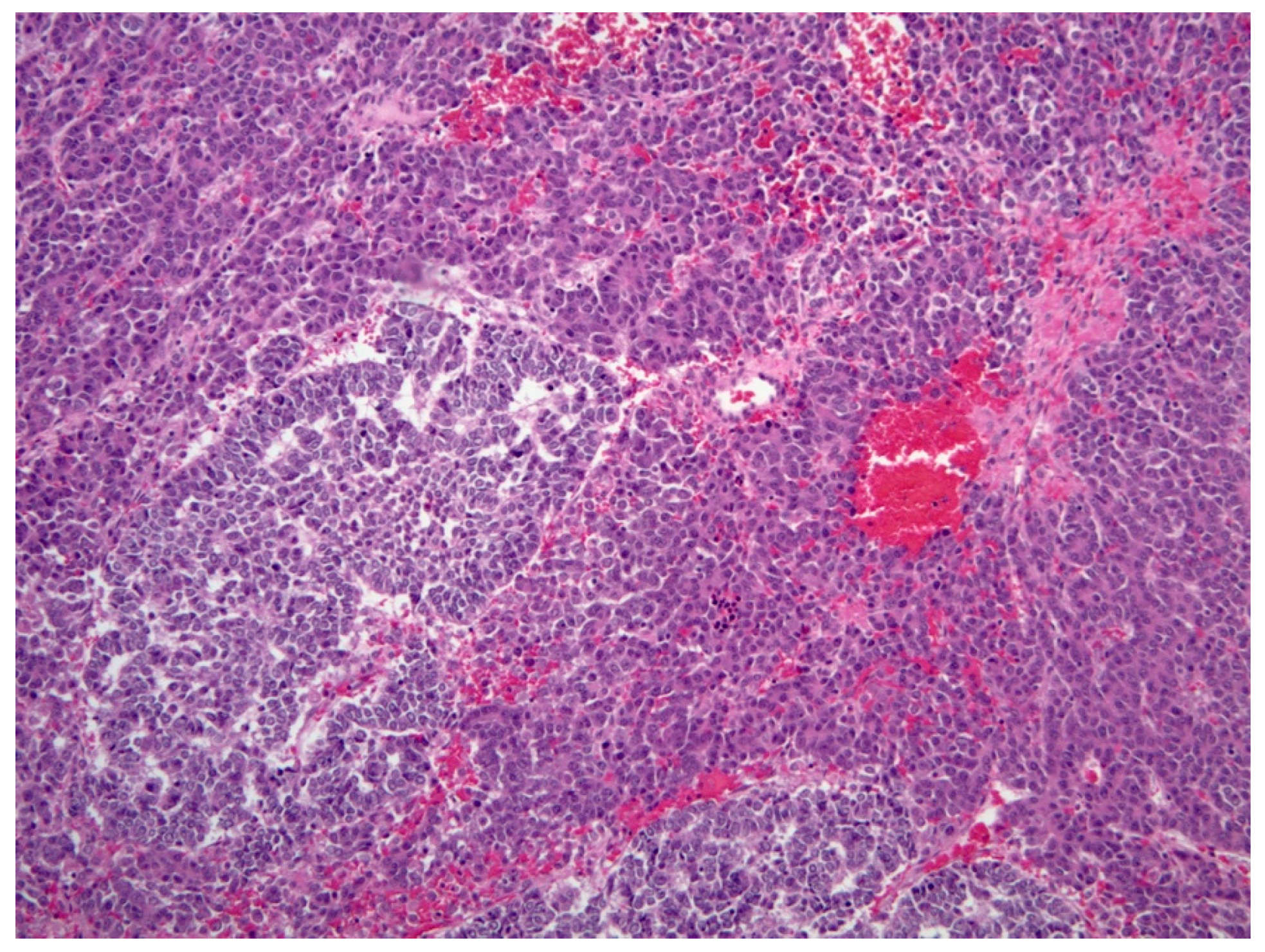
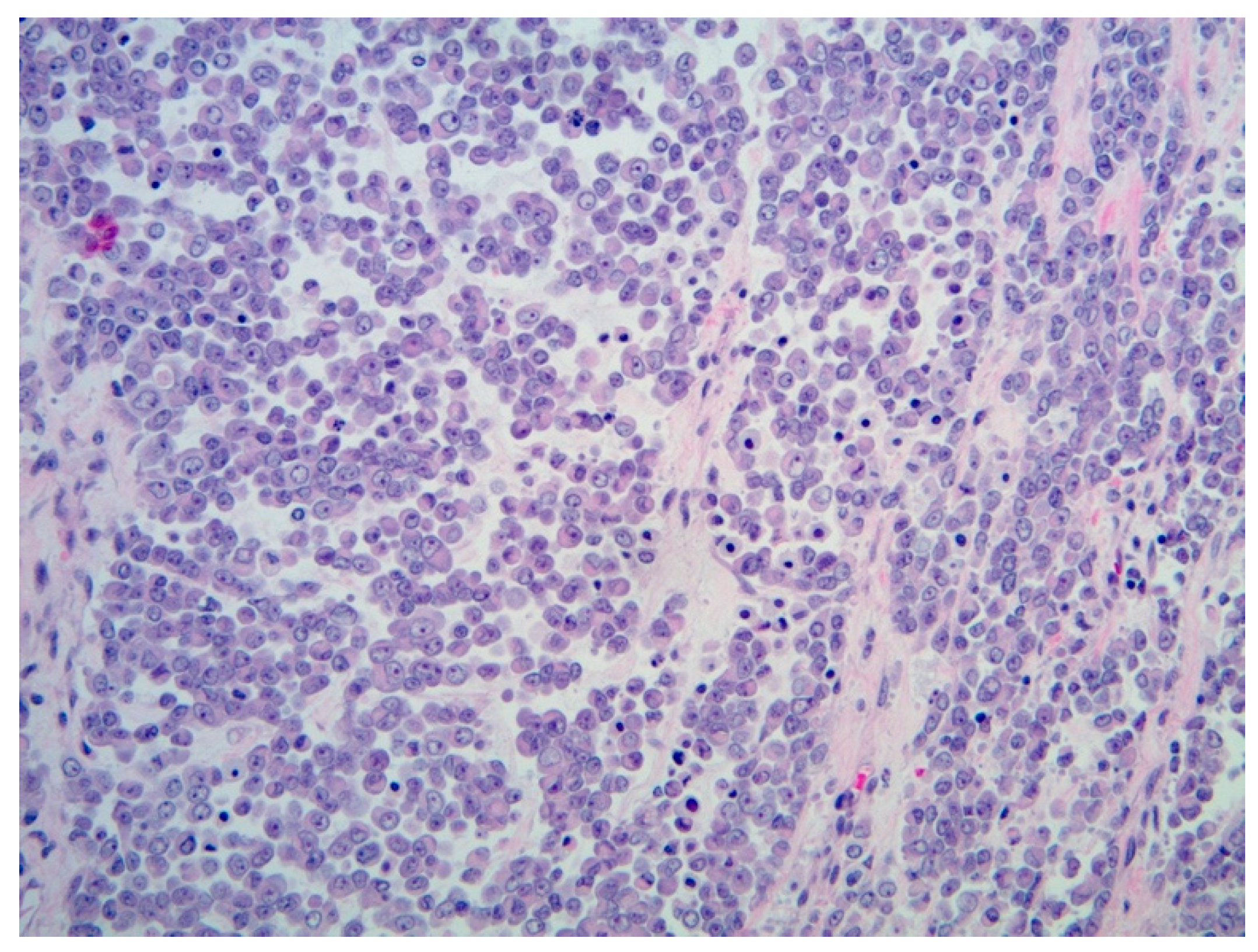
3.3.4. Histopathology
4. Benign Tumors in School-Aged Children and Adolescents
4.1. Hepatocellular Adenomas
4.1.1. Clinical Presentation
4.1.2. Associated Syndromes and Risk Factors
4.1.3. Diagnostic Imaging
4.1.4. Histopathology
4.2. Focal Nodular Hyperplasia
4.2.1. Clinical Presentation and Laboratory Findings
4.2.2. Associated Syndromes and Risk Factors
4.2.3. Diagnostic Imaging
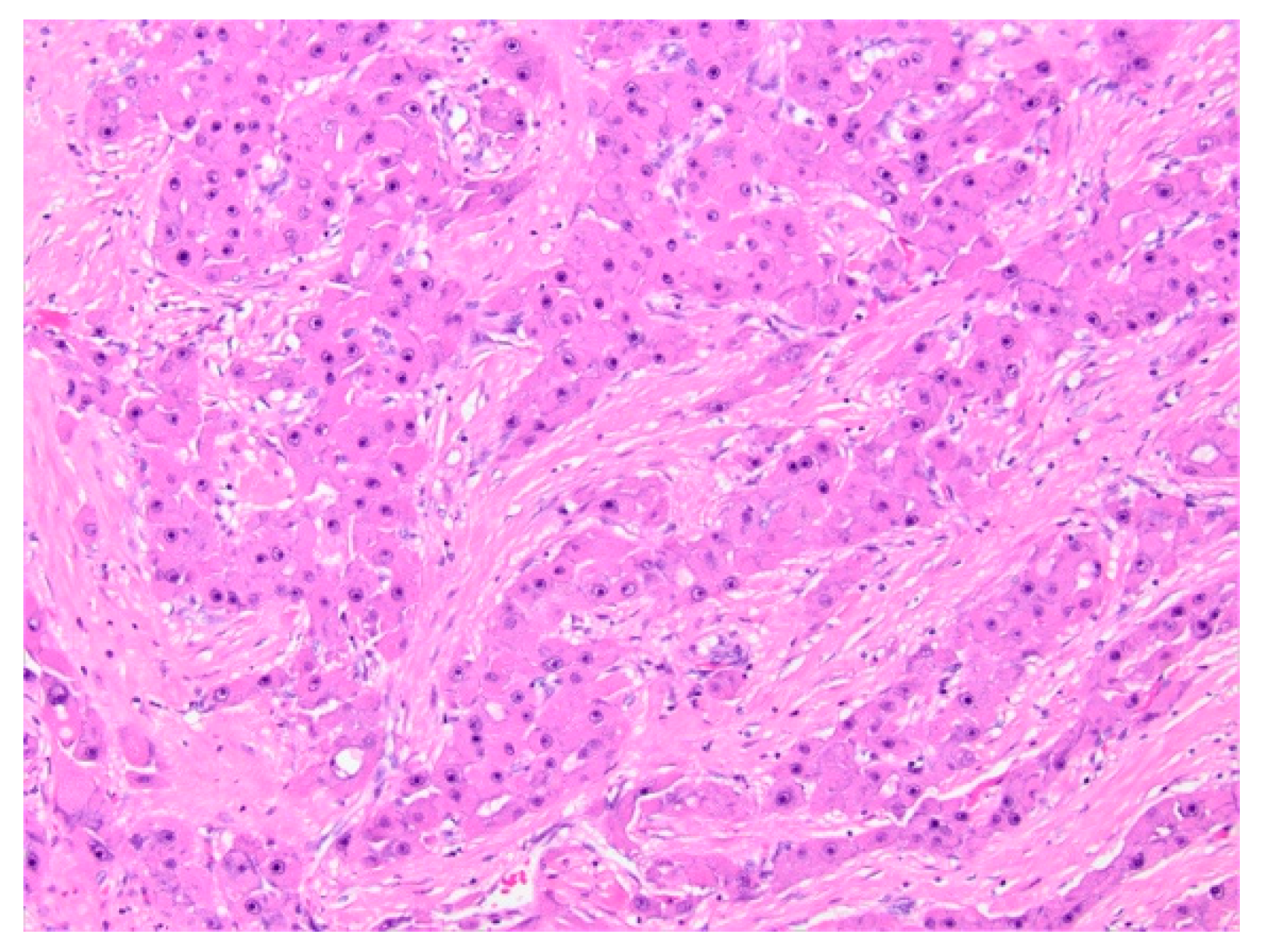
Histopathology and Immunohistochemistry
5. Malignant Tumors in School-Aged Children and Adolescents
5.1. Hepatocellular Carcinoma
5.1.1. Clinical Presentation
5.1.2. Laboratory Findings
5.1.3. Associated Syndromes and Risk Factors
5.1.4. Diagnostic Imaging
5.1.5. Histopathology and Immunohistochemistry
5.2. Undifferentiated Embryonal Sarcoma
5.2.1. Clinical Presentation
5.2.2. Laboratory Findings
5.2.3. Associated Syndromes and Risk Factors
5.2.4. Diagnostic Imaging
5.2.5. Histopathology and Immunohistochemistry
6. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Potisek, N.M.; Antoon, J.W. Abdominal Masses. Pediatr. Rev. 2017, 38, 101–103. [Google Scholar] [CrossRef]
- Ng, K.; Mogul, D.B. Pediatric Liver Tumors. Clin. Liver Dis. 2018, 22, 753–772. [Google Scholar] [CrossRef]
- Lewis, D.; Hachey, K.; Fitzgerald, S.; Vaidya, R. Rapidly Involuting Congenital Haemangioma of the Liver. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Lewis, D.; Vaidya, R. Hepatic Hemangioma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- McGuire, A.; Fernandez-Pineda, I.; Fishman, S.J.; Dickie, B.H. Pediatric Hepatic Vascular Tumors. Semin. Pediatric Surg. 2020, 29, 150970. [Google Scholar] [CrossRef]
- Gnarra, M.; Behr, G.; Kitajewski, A.; Wu, J.K.; Anupindi, S.A.; Shawber, C.J.; Zavras, N.; Schizas, D.; Salakos, C.; Economopoulos, K.P. History of the Infantile Hepatic Hemangioma: From Imaging to Generating a Differential Diagnosis. World J. Clin. Pediatr. 2016, 5, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, S.; Xiang, B.; Xu, Z.; Jiang, X.; Liu, X.; Wang, Q.; Lu, G.; Yang, L. Clinical Features and Management of Multifocal Hepatic Hemangiomas in Children: A Retrospective Study. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, S.; Yang, K.; Xiang, B.; Xu, X.; Li, L.; Qiu, T.; Zhou, J.; Dai, S.; Zhang, X.; et al. Screening for Infantile Hepatic Hemangioma in Patients with Cutaneous Infantile Hemangioma: A Multicenter Prospective Study. J. Am. Acad. Dermatol. 2020. [Google Scholar] [CrossRef]
- Rialon, K.L.; Murillo, R.; Fevurly, R.D.; Kulungowski, A.M.; Christison-Lagay, E.R.; Zurakowski, D.; Kozakewich, H.P.W.; Alomari, A.I.; Fishman, S.J. Risk Factors for Mortality in Patients with Multifocal and Diffuse Hepatic Hemangiomas. J. Pediatric Surg. 2015, 50, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Aouni, S.; Vlieghe, V.; Segers, V.; Christophe, C. Congenital Hepatic Haemangioma Leading to Multiple Organ Failure in a Neonate. BJR Case Rep. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, I. Guidance Document for Hepatic Hemangioma (Infantile and Congenital) Evaluation and Monitoring. J. Pediatr. 2018, 203, 9. [Google Scholar] [CrossRef]
- Christison-Lagay, E.R.; Burrows, P.E.; Alomari, A.; Dubois, J.; Kozakewich, H.P.; Lane, T.S.; Paltiel, H.J.; Klement, G.; Mulliken, J.B.; Fishman, S.J. Hepatic Hemangiomas: Subtype Classification and Development of a Clinical Practice Algorithm and Registry. J. Pediatric Surg. 2007, 42, 62–68. [Google Scholar] [CrossRef]
- Rodrigues, A.; Forno, A.; Costa, E.; Berenguer, A.; Pilar, C.; Loureiro, R.; Rufino, D.; Barros, A.; Teixeira, F. Diffuse Infantile Hepatic Haemangioma—How to Manage an Incidental but Potentially Lethal Finding. Oxf. Med. Case Rep. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Sekej, M.; Vadnjal Đonlagić, S.; Ključevšek, D. Contrast-Enhanced Ultrasound for the Characterization of Infantile Hepatic Hemangioma in Premature Neonate. Cureus 2020, 12. [Google Scholar] [CrossRef]
- Chung, E.M.; Cube, R.; Lewis, R.B.; Conran, R.M. Pediatric Liver Masses: Radiologic-Pathologic Correlation Part 1. Benign Tumors. RadioGraphics 2010, 30, 801–826. [Google Scholar] [CrossRef]
- Edmondson, H.A. Differential Diagnosis of Tumors and Tumor-like Lesions of Liver in Infancy and Childhood. AMA J. Dis. Child. 1956, 91, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.C.; Pienaar, J.A.; Sofianos, Z.; Varghese, J.; Warnich, I. Hepatic Mesenchymal Hamartoma: An Uncommon but Important Paediatric Diagnosis. SA J. Radiol. 2020, 24. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.D.; Alizai, N.K. Mesenchymal Hamartoma of the Liver: A Systematic Review. J. Pediatric Surg. 2005, 40, 1681–1690. [Google Scholar] [CrossRef]
- Khan, M.R.; Binkovitz, L.A.; Smyrk, T.C.; Potter, D.D.; Furuya, K.N. Mesenchymal Hamartoma in Children: A Diagnostic Challenge. Available online: https://www.hindawi.com/journals/cripe/2019/4132842/ (accessed on 7 December 2020).
- Wu, H.; Ferguson, W.; Castro, E.; Finegold, M.; Patel, K. Pediatric Mesenchymal Hamartomas of the Liver Can Show Both Foregut and Hindgut Phenotype. Pediatric Dev. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cignini, P.; Coco, C.; Giorlandino, M.; Bagolan, P.; Morini, F.; Giorlandino, C. Fetal Hepatic Mesenchymal Hamartoma: A Case Report. J. Prenat. Med. 2007, 1, 45–46. [Google Scholar] [PubMed]
- Armanian, A.-M.; Nazem, M. Mesenchymal Hamartoma of Liver in a Neonate. J. Neonatol. Biol. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Kamata, S.; Nose, K.; Sawai, T.; Hasegawa, T.; Kuroda, S.; Sasaki, T.; Okada, A.; Tawara, M. Fetal Mesenchymal Hamartoma of the Liver: Report of a Case. J. Pediatric Surg. 2003, 38, 639–641. [Google Scholar] [CrossRef]
- Hassan, M.A. Giant Solid Mesenchymal Hamartoma of the Liver in a Neonate: Case Report. Ann. Pediatric Surg. 2020, 16, 29. [Google Scholar] [CrossRef]
- Cajaiba, M.M.; Sarita-Reyes, C.; Zambrano, E.; Reyes-Múgica, M. Mesenchymal Hamartoma of the Liver Associated with Features of Beckwith-Wiedemann Syndrome and High Serum Alpha-Fetoprotein Levels. Pediatric Dev. Pathol. 2007, 10, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Abrahao-Machado, L.F.; de Macedo, F.C.; Dalence, C.; Stambo, G.; Abrahao-Machado, E.F.; Abrahao-Machado, E.C.F.; Bahrami, A.; Nascimento, A.G. Mesenchymal Hamartoma of the Liver in an Infant with Beckwith-Wiedemann Syndrome: A Rare Condition Mimicking Hepatoblastoma. ACG Case Rep. J. 2015, 2, 258–260. [Google Scholar] [CrossRef]
- Pan, E.T.; Yoeli, D.; Kueht, M.L.; Galvan, N.T.N.; Cotton, R.T.; O’Mahony, C.A.; Rana, A.; Goss, J.A. Liver Transplantation as Definitive Treatment of an Unresectable Mesenchymal Hamartoma in a Child with Beckwith–Wiedemann Syndrome. J. Surg. Case Rep. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apellaniz-Ruiz, M.; Segni, M.; Kettwig, M.; Glüer, S.; Pelletier, D.; Nguyen, V.-H.; Wagener, R.; López, C.; Muchantef, K.; Bouron-Dal Soglio, D.; et al. Mesenchymal Hamartoma of the Liver and DICER1 Syndrome. N. Engl. J. Med. 2019, 380, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Anil, G.; Fortier, M.; Low, Y. Cystic Hepatic Mesenchymal Hamartoma: The Role of Radiology in Diagnosis and Perioperative Management. Br. J. Radiol. 2011, 84, e091–e094. [Google Scholar] [CrossRef] [Green Version]
- Martins-Filho, S.N.; Putra, J. Hepatic Mesenchymal Hamartoma and Undifferentiated Embryonal Sarcoma of the Liver: A Pathologic Review. Hepatic Oncol. 2020, 7, HEP19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, S.; Lopez-Terrada, D.; Alaggio, R. Hepatoblastoma and Pediatric Hepatocellular Carcinoma: An Update. Pediatric Dev. Pathol. 2020, 23, 79–95. [Google Scholar] [CrossRef]
- Pruente, J.R.; Deike, D.E.; Lockart, B.; Gaebler-Spira, D. The Association of Hepatoblastoma, Prematurity and Cerebral Palsy: Case Reports. PRM 2020, 13, 185–188. [Google Scholar] [CrossRef]
- Li, L.; Liu, W.; Wen, R.; Jin, K. Computed Tomography Imaging and Clinical Features of Congenital Hepatoblastoma. Medicine 2020, 99. [Google Scholar] [CrossRef] [PubMed]
- Yoo, G.H.Y.; Mugarab-Samedi, V.; Hansen, G.; Miller, G.; Givelichian, L.; Kalaniti, K.; Daspal, S. Rare Cause of Emergency in the First Week of Life: Congenital Hepatoblastoma (Case Report). Oxf. Med. Case Rep. 2020, 2020, omaa002. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; George, C.; Thomas, P. A Case Report on Neonatal Hepatoblastoma. J. Pediatric Disord. Neonatal Care 2018, 1. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Burjonrappa, S. Perinatal Hemorrhage Complicating Neonatal Hepatoblastoma: Case Report. J. Pediatric Surg. 2012, 47, e29–e32. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Mukherjee, S.; Boler, A.; Sharma, A.; Biswas, S.K. Hepatoblastoma in the Neonatal Period: An Unusual Presentation. J. Cytol. 2012, 29, 252–254. [Google Scholar] [CrossRef]
- Sharma, D.; Subbarao, G.; Saxena, R. Hepatoblastoma. Semin. Diagn. Pathol. 2017, 34, 192–200. [Google Scholar] [CrossRef]
- Towbin, A.J.; Braga, F.D.C.B.; Zhang, B.; Geller, J.I.; Tiao, G.M.; Podberesky, D.J. Fractures in Children with Newly Diagnosed Hepatoblastoma. Pediatric Radiol. 2018, 48, 581–585. [Google Scholar] [CrossRef]
- Al-Jumaily, U.; Sammour, I.; Al-Muhaisen, F.; Ajlouni, F.; Sultan, I. Precocious Puberty in an Infant with Hepatoblastoma: A Case Report. J. Med. Case Rep. 2011, 5, 422. [Google Scholar] [CrossRef] [Green Version]
- Komura, E.; Matsumura, T.; Kato, T.; Tahara, T.; Tsunoda, Y.; Sawada, T. Thrombopoietin in Patients with Hepatoblastoma. Stem Cells 1998, 16, 329–333. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Ishii, E.; Hayashida, Y.; Hirata, Y.; Sakai, R.; Miyazaki, S. Mechanism of Thrombocytosis in Hepatoblastoma: A Case Report. Pediatric Hematol. Oncol. 1996, 13, 539–544. [Google Scholar] [CrossRef]
- Turcotte, L.M.; Georgieff, M.K.; Ross, J.A.; Feusner, J.H.; Tomlinson, G.E.; Malogolowkin, M.H.; Krailo, M.D.; Miller, N.; Fonstad, R.; Spector, L.G. Neonatal Medical Exposures and Characteristics of Low Birth Weight Hepatoblastoma Cases: A Report from the Children’s Oncology Group. Pediatric Blood Cancer 2014, 61, 2018–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, P.; Urayama, K.Y.; Behren, J.V.; Feusner, J. Birth Characteristics and Hepatoblastoma Risk in Young Children. Cancer 2004, 100, 1070–1076. [Google Scholar] [CrossRef]
- Sorahan, T.; Lancashire, R.J. Parental Cigarette Smoking and Childhood Risks of Hepatoblastoma: OSCC Data. Br. J. Cancer 2004, 90, 1016–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musselman, J.R.; Georgieff, M.K.; Ross, J.A.; Tomlinson, G.E.; Feusner, J.; Krailo, M.; Spector, L.G. Maternal Pregnancy Events and Exposures and Risk of Hepatoblastoma: A Children’s Oncology Group (COG) Study. Cancer Epidemiol. 2013, 37, 318–320. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Zheng, Q.-Z.; Cao, D.-Y.; Xie, Y.-N.; Song, T.; Jiang, Q.-P.; Chen, M. Prenatal Diagnosis of Congenital Hepatoblastoma. Matern.-Fetal Med. 2020, 2, 115–118. [Google Scholar] [CrossRef]
- Beckwith, J.B.; Palmer, N.F. Histopathology and Prognosis of Wilms Tumor Results from the First National Wilms’ Tumor Study. Cancer 1978, 41, 1937–1948. [Google Scholar] [CrossRef]
- Gonzalez-Crussi, F.; Goldschmidt, R.A.; Hsueh, W.; Trujillo, Y.P. Infantile Sarcoma with Intracytoplasmic Filamentous Inclusions: Distinctive Tumor of Possible Histiocytic Origin. Cancer 1982, 49, 2365–2375. [Google Scholar] [CrossRef]
- Cornet, M.; De Lambert, G.; Pariente, D.; Planchon, J.M.; Guettier, C.; Martelli, H.; Guérin, F.; Branchereau, S. Rhabdoid Tumor of the Liver: Report of 6 Pediatric Cases Treated at a Single Institute. J. Pediatric Surg. 2018, 53, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Kapral, N.; Melmer, P.; Druzgal, C.H.; Lancaster, L. Pediatric Hepatic Rhabdoid Tumor: A Rare Cause of Abdominal Mass in Children. Radiol. Case Rep. 2018, 13, 724–727. [Google Scholar] [CrossRef]
- Kupeli, S.; Yöntem, A.; Narli, N.; Doran, F.; Soyupak, S. Infantile Rhabdoid Tumor Mimicking Hepatic Hemangioendothelioma. J. Cancer Res. Ther. 2016, 12. [Google Scholar] [CrossRef]
- Oita, S.; Terui, K.; Komatsu, S.; Hishiki, T.; Saito, T.; Mitsunaga, T.; Nakata, M.; Yoshida, H. Malignant Rhabdoid Tumor of the Liver: A Case Report and Literature Review. Pediatric Rep. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Stelling, A.; Kuramoto, A.; Patel, C.; Keller, J. Malignant Rhabdoid Tumor of the Liver: Findings at US, CT, and MRI, with Histopathologic Correlation. Radiol. Case Rep. 2014, 9, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwala, S.; Jindal, B.; Jana, M.; Bhatnagar, V.; Gupta, A.K.; Iyer, V.K. Malignant Rhabdoid Tumor of Liver. J. Indian Assoc. Pediatric Surg 2014, 19, 38–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, M.; Liu, C. Malignant Rhabdoid Tumour of the Liver in a Seven-Month-Old Female Infant: A Case Report and Literature Review. Afr. J. Paediatr. Surg. AJPS 2013, 10, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Patel, Y.; Lewis, T.J.; Elsamaloty, H.; Strobel, S. Extrarenal Malignant Rhabdoid Tumors: Radiologic Findings with Histopathologic Correlation. Cancer Imaging 2010, 10, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Jayaram, A.; Finegold, M.J.; Parham, D.M.; Jasty, R. Successful Management of Rhabdoid Tumor of the Liver. J. Pediatric Hematol. Oncol. 2007, 29, 406–408. [Google Scholar] [CrossRef]
- Trobaugh-Lotrario, A.D.; Finegold, M.J.; Feusner, J.H. Rhabdoid Tumors of the Liver: Rare, Aggressive, and Poorly Responsive to Standard Cytotoxic Chemotherapy. Pediatric Blood Cancer 2011, 57, 423–428. [Google Scholar] [CrossRef]
- Kachanov, D.; Teleshova, M.; Kim, E.; Dobrenkov, K.; Moiseenko, R.; Usychkina, A.; Filin, A.; Semenkov, A.; Mitrofanova, A.; Konovalov, D.; et al. Malignant Rhabdoid Tumor of the Liver Presented with Initial Tumor Rupture. Cancer Genet. 2014, 207, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Marzano, E.; Lermite, E.; Nobili, C.; Teyssedou, C.; Bachellier, P.; Arnaud, J.-P.; Pessaux, P. Malignant Rhabdoid Tumour of the Liver in the Young Adult: Report of First Two Cases. HPB Surg. 2009, 2009, 628206. [Google Scholar] [CrossRef] [Green Version]
- Nazir, Z.; Pervez, S. Malignant Vascular Tumors of Liver in Neonates. J. Pediatric Surg. 2006, 41, e49–e51. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.K.; Squires, J.H.; Reyes-Múgica, M.; McCormick, A.; Mahmood, B. Hepatic Vascular Tumors in the Neonate: Angiosarcoma. J. Pediatr. 2018, 193, 245–248.e1. [Google Scholar] [CrossRef] [PubMed]
- Grassia, K.L.; Peterman, C.M.; Iacobas, I.; Margolin, J.F.; Bien, E.; Padhye, B.; Meyers, R.L.; Adams, D.M. Clinical Case Series of Pediatric Hepatic Angiosarcoma. Pediatric Blood Cancer 2017, 64, e26627. [Google Scholar] [CrossRef]
- Geramizadeh, B.; Safari, A.; Bahador, A.; Nikeghbalian, S.; Salahi, H.; Kazemi, K.; Dehghani, S.-M.; Malek-Hosseini, S.-A. Hepatic Angiosarcoma of Childhood: A Case Report and Review of Literature. J. Pediatric Surg. 2011, 46, e9–e11. [Google Scholar] [CrossRef] [PubMed]
- Potanos, K.M.; Hodgkinson, N.; Fullington, N.M.; Narla, A.; Albritton, K.; Kozakewich, H.; Kim, H.B. Long Term Survival in Pediatric Hepatic Angiosarcoma (PHAS): A Case Report and Review of the Literature. J. Pediatric Surg. Case Rep. 2015, 3, 410–413. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, S. Malignant Hemangioendothelioma in Infants and Children. Cancer 1961, 14, 11. [Google Scholar] [CrossRef]
- Adam, Y.G.; Huvos, A.G.; Hajdu, S.I. Malignant Vascular Tumors of Liver. Ann. Surg. 1972, 175, 375–383. [Google Scholar] [CrossRef]
- Chabalko, J.J.; Fraumeni, J.F. Blood-Vessel Neoplasms in Children: Epidemiologic Aspects. Med. Pediatric Oncol. 1975, 1, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Falk, H.; Herbert, J.T.; Edmonds, L.; Heath, C.W.; Thomas, L.B.; Popper, H. Review of Four Cases of Childhood Hepatic Angiosarcoma—Elevated Environmental Arsenic Exposure in One Case. Cancer 1981, 47, 382–391. [Google Scholar] [CrossRef]
- Kirchner, S.G.; Heller, R.M.; Kasselberg, A.G.; Greene, H.L. Infantile Hepatic Hemangioendothelioma with Subsequent Malignant Degeneration. Pediatric Radiol. 1981, 11, 42–45. [Google Scholar] [CrossRef]
- Weinberg, A.G.; Finegold, M.J. Primary Hepatic Tumors of Childhood. Hum. Pathol. 1983, 14, 512–537. [Google Scholar] [CrossRef]
- Alt, B.; Hafez, G.R.; Trigg, M.; Shahidi, N.T.; Gilbert, E.F. Angiosarcoma of the Liver and Spleen in an Infant. Pediatric Pathol. 1985, 4, 331–339. [Google Scholar] [CrossRef]
- Selby, D.M.; Stocker, J.T.; Ishak, K.G. Angiosarcoma of the Liver in Childhood: A Clinocopathologic and Follow-up Study of 10 Cases. Pediatric Pathol. 1992, 12, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Awan, S.; Davenport, M.; Portmann, B.; Howard, E.R. Angiosarcoma of the Liver in Children. J. Pediatric Surg. 1996, 31, 1729–1732. [Google Scholar] [CrossRef]
- Gunawardena, S.W.; Trautwein, L.M.; Finegold, M.J.; Ogden, A.K. Hepatic Angiosarcoma in a Child: Successful Therapy with Surgery and Adjuvant Chemotherapy. Med. Pediatric Oncol. 1997. [Google Scholar] [CrossRef]
- Valle, P.L.; Gerald, W.L.; Tsai, J.; Meyers, P.; Quaglia, M.P.L. Malignant Vascular Tumors in Young Patients. Cancer 1998, 83, 1634–1639. [Google Scholar] [CrossRef]
- Dimashkieh, H.H.; Mo, J.Q.; Wyatt-Ashmead, J.; Collins, M.H. Pediatric Hepatic Angiosarcoma: Case Report and Review of the Literature. Pediatric Dev. Pathol. 2004, 7, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.M.; Kandel, J.; Lefkowitch, J.H.; Lobritto, S.J.; Morel, K.D.; North, P.E.; Garzon, M.C. Multiple Cutaneous Infantile Hemangiomas Associated with Hepatic Angiosarcoma: Case Report and Review of the Literature. Pediatrics 2006, 118, e907–e913. [Google Scholar] [CrossRef]
- Oanez, A.; Monique, F.; Stephanie, F.; Daniele, P.; Dominique, D.; Emmanuel, J.; Frederic, G.; Olivier, B. The Widening Spectrum of Liver Angiosarcoma in Children. J. Pediatric Gastroenterol. Nutr. 2011, 1. [Google Scholar] [CrossRef]
- Faria, F.W. Pediatric Hepatic Angiosarcoma Treated with Chemotherapy and Partial Hepatectomy: A Case Report. JCO 2013, 31, e21001. [Google Scholar] [CrossRef]
- López, R.; Castro-Villabón, D.; Álvarez, J.; Vera, A.; Andrade, R. Hepatic Angiosarcoma Presenting as Acute Liver Failure in Young Adults. Report of Two Cases and Review of Literature. Case Rep. Clin. Med. 2013, 2, 439. [Google Scholar] [CrossRef] [Green Version]
- Jeng, M.R.; Fuh, B.; Blatt, J.; Gupta, A.; Merrow, A.C.; Hammill, A.; Adams, D. Malignant Transformation of Infantile Hemangioma to Angiosarcoma: Response to Chemotherapy with Bevacizumab: Angiosarcoma: Response to Chemotherapy with Bevacizumab. Pediatric Blood Cancer 2014, 61, 2115–2117. [Google Scholar] [CrossRef]
- Olson, T.S.; Chan, E.S.; Paessler, M.E.; Sullivan, K.E.; Frantz, C.N.; Russo, P.; Bessler, M. Liver Failure Due to Hepatic Angiosarcoma in an Adolescent with Dyskeratosis Congenita. J. Pediatric Hematol. Oncol. 2014, 36, 312–315. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Masand, P.; Thompson, P.; Finegold, M.; Leung, D.H. Angiosarcoma Successfully Treated with Liver Transplantation and Sirolimus. Pediatric Transplant. 2014, 18, E114–E119. [Google Scholar] [CrossRef]
- Kamath Hepatic Angiosarcoma Developing in an Infantile Hemangioendothelioma: A Rare Case Report. Available online: https://www.cancerjournal.net/article.asp?issn=0973-1482;year=2015;volume=11;issue=4;spage=1022;epage=1022;aulast=Kamath (accessed on 7 February 2021).
- Pilbeam, K.; Eidenschink, B.; Sulciner, M.; Luquette, M.; Neglia, J.; Chinnakotla, S. Success of Chemotherapy and a Liver Transplant in a Pediatric Patient with Hepatic Angiosarcoma: A Case Report. Pediatric Transpl. 2019, 23, e13410. [Google Scholar] [CrossRef] [PubMed]
- Geramizadeh, B.; Kashkooe, A.; Bahador, A.; Dehghani, S.M.; Shamsaeefar, A.; Kazemi, K.; Malekhosseini, S.A. Pediatric Hepatocellular Carcinoma, a Single Center Study from the South of Iran: Case Series. Hepat. Mon. 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M. Vinyl Chloride and the Liver. J. Hepatol. 2009, 51, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Weihrauch, M.; Bader, M.; Lehnert, G.; Koch, B.; Wittekind, C.; Wrbitzky, R.; Tannapfel, A. Mutation Analysis of K-Ras-2 in Liver Angiosarcoma and Adjacent Nonneoplastic Liver Tissue from Patients Occupationally Exposed to Vinyl Chloride. Environ. Mol. Mutagenesis 2002, 40, 36–40. [Google Scholar] [CrossRef]
- Franchi-Abella, S.; Branchereau, S. Benign Hepatocellular Tumors in Children: Focal Nodular Hyperplasia and Hepatocellular Adenoma. Int. J. Hepatol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Hahn, E.; Putra, J. Hepatocellular Adenoma in the Paediatric Population: Molecular Classification and Clinical Associations. World J. Gastroenterol. 2020, 26, 2294–2304. [Google Scholar] [CrossRef]
- Applegate, K.E.; Ghei, M.; Perez-Atayde, A.R. Prenatal Detection of a Solitary Liver Adenoma. Pediatric Radiol. 1999, 29, 92–94. [Google Scholar] [CrossRef]
- Bioulac-Sage, P.; Balabaud, C.; Zucman-Rossi, J. Subtype Classification of Hepatocellular Adenoma. Dig. Surg. 2010, 27, 39–45. [Google Scholar] [CrossRef]
- Agrawal, S.; Agarwal, S.; Arnason, T.; Saini, S.; Belghiti, J. Management of Hepatocellular Adenoma: Recent Advances. Clin. Gastroenterol. Hepatol. 2015, 13, 1221–1230. [Google Scholar] [CrossRef]
- Sempoux, C.; Balabaud, C.; Bioulac-Sage, P. Malignant Transformation of Hepatocellular Adenoma. Hepatic Oncol. 2014, 1, 421–431. [Google Scholar] [CrossRef]
- Oliveira, S.; Samba, A.K.; Towbin, A.J.; Gupta, A.; Geller, J.I.; Nathan, J.D.; Kohli, R. Incidental Inflammatory Adenoma with β-Catenin Activation in the Setting of Paediatric NASH. Pediatric Obes. 2018, 13, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Rajalakshmi, V. Spontaneous Hepatocellular Adenoma in Paediatric Age Group—Case Report. JCDR 2013. [Google Scholar] [CrossRef]
- Brady, P.C.; Missmer, S.A.; Laufer, M.R. Hepatic Adenomas in Adolescents and Young Women with Endometriosis Treated with Norethindrone Acetate. J. Pediatric Adolesc. Gynecol. 2017, 30, 422–424. [Google Scholar] [CrossRef]
- Haring, M.P.D.; Gouw, A.S.H.; de Haas, R.J.; Cuperus, F.J.C.; de Jong, K.P.; de Meijer, V.E. The Effect of Oral Contraceptive Pill Cessation on Hepatocellular Adenoma Diameter: A Retrospective Cohort Study. Liver Int. 2019, 39, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosnier, H.; Thibaud, E.; Duflos, C.; Polak, M. Norethisterone-Induced Hepatic Adenomas Can Cause Life-Threatening Bleeding in Girls with Inherited Platelet Disorders. Fertil. Steril. 2010, 94, 2329.e1–2329.e3. [Google Scholar] [CrossRef]
- Wang, L.; Wang, C.; Li, W.; Meng, F.; Li, Y.; Fan, H.; Zhou, Y.; Bharathi, G.; Gao, S.; Yang, Y. Multiple Hepatocellular Adenomas Associated with Long-Term Administration of Androgenic Steroids for Aplastic Anemia: A Case Report and Literature Review. Medicine 2020, 99, e20829. [Google Scholar] [CrossRef]
- Triantafyllopoulou, M.; Whitington, P.F.; Melin-Aldana, H.; Benya, E.C.; Brickman, W. Hepatic Adenoma in an Adolescent with Elevated Androgen Levels. J. Pediatric Gastroenterol. Nutr. 2007, 44, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.; Fiske, L.M.; Carreras, C.T.; Weinstein, D.A. Natural History of Hepatocellular Adenoma Formation in Glycogen Storage Disease Type I. J. Pediatr. 2011, 159, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishnani, P.S.; Chuang, T.-P.; Bali, D.; Koeberl, D.; Austin, S.; Weinstein, D.A.; Murphy, E.; Chen, Y.-T.; Boyette, K.; Liu, C.-H.; et al. Chromosomal and Genetic Alterations in Human Hepatocellular Adenomas Associated with Type Ia Glycogen Storage Disease. Hum. Mol. Genet. 2009, 18, 4781–4790. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Labrune, P.; Morcrette, G.; Rebouissou, S.; Franco, D.; Prévot, S.; Quaglia, A.; Bedossa, P.; Libbrecht, L.; Terracciano, L.; et al. Molecular Characterization of Hepatocellular Adenomas Developed in Patients with Glycogen Storage Disease Type I. J. Hepatol. 2013, 58, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Glonnegger, H.; Schulze, M.; Kathemann, S.; Berg, S.; Füllgraf, H.; Tannapfel, A.; Gerner, P.; Grohmann, J.; Niemeyer, C.; Hettmer, S. Case Report: Hepatic Adenoma in a Child with a Congenital Extrahepatic Portosystemic Shunt. Front. Pediatr. 2020, 8, 501. [Google Scholar] [CrossRef]
- Franchi-Abella, S.; Branchereau, S.; Lambert, V.; Fabre, M.; Steimberg, C.; Losay, J.; Riou, J.-Y.; Pariente, D.; Gauthier, F.; Jacquemin, E.; et al. Complications of Congenital Portosystemic Shunts in Children: Therapeutic Options and Outcomes. J. Pediatric Gastroenterol. Nutr. 2010, 51, 322–330. [Google Scholar] [CrossRef]
- Tonorezos, E.S.; Barnea, D.; Abou-Alfa, G.K.; Bromberg, J.; D’Angelica, M.; Sklar, C.A.; Shia, J.; Oeffinger, K.C. Hepatocellular Adenoma among Adult Survivors of Childhood and Young Adult Cancer. Pediatric Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Cattoni, A.; Rovelli, A.; Prunotto, G.; Bonanomi, S.; Invernizzi, P.; Perego, R.; Mariani, A.M.; Balduzzi, A. Hepatic Focal Nodular Hyperplasia after Pediatric Hematopoietic Stem Cell Transplantation: The Impact of Hormonal Replacement Therapy and Iron Overload. Pediatric Blood Cancer 2020, 67, e28137. [Google Scholar] [CrossRef]
- Zhuang, L.; Ni, C.; Din, W.; Zhang, F.; Zhuang, Y.; Sun, Y.; Xi, D. Huge Focal Nodular Hyperplasia Presenting in a 6-Year-Old Child: A Case Presentation. Int. J. Surg. Case Rep. 2016, 29, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gürses, C.; Okşar, F.S.; Erol, B.; Yalçın, M.; Kahvecioğlu, N.; Alparslan, A.Ş. Natural Course of Hepatic Focal Nodular Hyperplasia from Childhood to Adulthood and Review of the Literature. Turk. J. Gastroenterol. 2017, 28, 492–497. [Google Scholar] [CrossRef]
- Ma, I.T.; Rojas, Y.; Masand, P.M.; Castro, E.C.; Himes, R.W.; Kim, E.S.; Goss, J.A.; Nuchtern, J.G.; Finegold, M.J.; Thompson, P.A.; et al. Focal Nodular Hyperplasia in Children: An Institutional Experience with Review of the Literature. J. Pediatric Surg. 2015, 50, 382–387. [Google Scholar] [CrossRef]
- Aboughalia, H.; Chisholm, K.M.; Iyer, R.S. Focal Nodular Hyperplasia Masquerading as Malignancy in an Infant with Elevated Alpha-Fetoprotein: A Case Report with Literature Review. Clin. Imaging 2021, 69, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Halder, A.; Ghosh, R.; Banerjee, S.; Mishra, P.K.; Chatterjee, U. Focal Nodular Hyperplasia of the Liver in Children: A Report of 2 Cases. Indian J. Pathol. Microbiol. 2019, 62, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, S.; Xiang, B.; Wen, T.; Yang, J.; Zhong, L.; Li, F.; Li, L. Clinical Features of Focal Nodular Hyperplasia of the Liver in Children. J. Pediatric Gastroenterol. Nutr. 2016, 62, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Bouyn, C.I.-D.; Leclere, J.; Raimondo, G.; le Pointe, H.D.; Couanet, D.; Valteau-Couanet, D.; Hartmann, O. Hepatic Focal Nodular Hyperplasia in Children Previously Treated for a Solid Tumor. Cancer 2003, 97, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Yazal Erdem, A.; Emir, S.; Demir, H.A.; Özyörük, D.; Çetin, I.; Turan, A.; Karakus, E. Focal Nodulary Hyperplasia of the Liver Due to Congenital Portosystemic Shunt: A Rare Condition Mimicking Hepatocellulary Carcinoma. J. Pediatric Hematol. Oncol. 2017, 39, e8. [Google Scholar] [CrossRef]
- Cha, D.I.; Yoo, S.-Y.; Kim, J.H.; Jeon, T.Y.; Eo, H. Clinical and Imaging Features of Focal Nodular Hyperplasia in Children. Am. J. Roentgenol. 2014, 202, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Jeon, T.Y.; Yoo, S.-Y.; Kim, J.H.; Eo, H.; Lee, S.-K.; Kim, J.S. Hepatic Tumours in Children with Biliary Atresia: Single-Centre Experience in 13 Cases and Review of the Literature. Clin. Radiol. 2014, 69, e113–e119. [Google Scholar] [CrossRef]
- Fang, C.; Bernardo, S.; Sellars, M.E.; Deganello, A.; Sidhu, P.S. Contrast-Enhanced Ultrasound in the Diagnosis of Pediatric Focal Nodular Hyperplasia and Hepatic Adenoma: Interobserver Reliability. Pediatric Radiol. 2019, 49, 82–90. [Google Scholar] [CrossRef]
- Liu, Q.-Y.; Zhang, W.-D.; Lai, D.-M.; Ou-Yang, Y.; Gao, M.; Lin, X.-F. Hepatic Focal Nodular Hyperplasia in Children: Imaging Features on Multi-Slice Computed Tomography. World J. Gastroenterol. 2012, 18, 7048–7055. [Google Scholar] [CrossRef] [PubMed]
- Khanna, G. Pediatric Hepatoblastoma and Hepatocellular Carcinoma: Lessons Learned in the Last Decade. Pediatric Radiol. 2018, 48, 531–532. [Google Scholar] [CrossRef] [Green Version]
- Ziogas, I.A.; Ye, F.; Zhao, Z.; Matsuoka, L.K.; Montenovo, M.I.; Izzy, M.; Benedetti, D.J.; Lovvorn, H.N.; Gillis, L.A.; Alexopoulos, S.P. Population-Based Analysis of Hepatocellular Carcinoma in Children: Identifying Optimal Surgical Treatment. J. Am. Coll. Surg. 2020, 230, 1035–1044.e3. [Google Scholar] [CrossRef]
- D’Souza, A.M.; Towbin, A.J.; Gupta, A.; Alonso, M.; Nathan, J.D.; Bondoc, A.; Tiao, G.; Geller, J.I. Clinical Heterogeneity of Pediatric Hepatocellular Carcinoma. Pediatric Blood Cancer 2020, 67, e28307. [Google Scholar] [CrossRef] [Green Version]
- Cowell, E.; Patel, K.; Heczey, A.; Finegold, M.; Venkatramani, R.; Wu, H.; López-Terrada, D.; Miloh, T. Predisposing Conditions to Pediatric Hepatocellular Carcinoma and Association with Outcomes: Single-Center Experience. J. Pediatric Gastroenterol. Nutr. 2019, 68, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Tiao, G. Approach to Pediatric Hepatocellular Carcinoma. Clin. Liver Dis. 2013, 2, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Sergi, C.M. Hepatocellular Carcinoma, Fibrolamellar Variant: Diagnostic Pathologic Criteria and Molecular Pathology Update. A Primer. Diagnostics 2015, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Lim, I.I.P.; Farber, B.A.; LaQuaglia, M.P. Advances in Fibrolamellar Hepatocellular Carcinoma: A Review. Eur. J. Pediatric Surg. 2014, 24, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Thakral, N.; Simonetto, D.A. Hyperammonemic Encephalopathy: An Unusual Presentation of Fibrolamellar Hepatocellular Carcinoma. Clin. Mol. Hepatol. 2020, 26, 74–77. [Google Scholar] [CrossRef]
- Tajiri, H.; Takano, T.; Tanaka, H.; Ushijima, K.; Inui, A.; Miyoshi, Y.; Ozono, K.; Abukawa, D.; Endo, T.; Brooks, S.; et al. Hepatocellular Carcinoma in Children and Young Patients with Chronic HBV Infection and the Usefulness of Alpha-Fetoprotein Assessment. Cancer Med. 2016, 5, 3102–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-L.; Ko, Y.-C. Survival and Distribution Pattern of Childhood Liver Cancer in Taiwan. Eur. J. Cancer 1998, 34, 2064–2067. [Google Scholar] [CrossRef]
- Chang, M.-H.; You, S.-L.; Chen, C.-J.; Liu, C.-J.; Lee, C.-M.; Lin, S.-M.; Chu, H.-C.; Wu, T.-C.; Yang, S.-S.; Kuo, H.-S.; et al. Decreased Incidence of Hepatocellular Carcinoma in Hepatitis B Vaccinees: A 20-Year Follow-up Study. J. Natl. Cancer Inst. 2009, 101, 1348–1355. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-F.; Chang, M.-H. Natural History of Chronic Hepatitis B Virus Infection from Infancy to Adult Life -the Mechanism of Inflammation Triggering and Long-Term Impacts. J. Biomed. Sci. 2015, 22. [Google Scholar] [CrossRef]
- Tyrosinemia Type 1. NORD (National Organization for Rare Disorders). Available online: https://rarediseases.org/rare-diseases/tyrosinemia-type-1/ (accessed on 20 December 2020).
- McKiernan, P.J. Nitisinone in the Treatment of Hereditary Tyrosinaemia Type 1. Drugs 2006, 66, 743–750. [Google Scholar] [CrossRef]
- Komatsu, H.; Inui, A.; Kishiki, K.; Kawai, H.; Yoshio, S.; Osawa, Y.; Kanto, T.; Fujisawa, T. Liver Disease Secondary to Congenital Heart Disease in Children. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 651–666. [Google Scholar] [CrossRef]
- Oh, C.; Youn, J.K.; Han, J.-W.; Kim, G.B.; Kim, H.-Y.; Jung, S.-E. Hepatocellular Carcinoma after the Fontan Procedure in a 16-Year-Old Girl. Medicine 2016, 95. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Walker, T.T.; Bove, K.; Veldtman, G. Fontan-Associated Liver Disease: A Review. J. Cardiol. 2019, 74, 223–232. [Google Scholar] [CrossRef]
- Knisely, A.S.; Strautnieks, S.S.; Meier, Y.; Stieger, B.; Byrne, J.A.; Portmann, B.C.; Bull, L.N.; Pawlikowska, L.; Bilezikçi, B.; Özçay, F.; et al. Hepatocellular Carcinoma in Ten Children under Five Years of Age with Bile Salt Export Pump Deficiency. Hepatology 2006, 44, 478–486. [Google Scholar] [CrossRef] [PubMed]
- AlSalloom, A. Hepatocellular Carcinoma in a Boy with Progressive Familial Intrahepatic Cholestasis Type II: Challenging Identification: Case Report. Int. J. Health Sci. 2013, 7, 252–255. [Google Scholar] [CrossRef]
- Coire, C.I.; Qizilbash, A.H.; Castelli, M.F. Hepatic Adenomata in Type Ia Glycogen Storage Disease. Arch. Pathol. Lab. Med. 1987, 111, 166–169. [Google Scholar]
- Jang, H.-J.; Yang, H.R.; Ko, J.S.; Moon, J.S.; Chang, J.Y.; Seo, J.K. Development of Hepatocellular Carcinoma in Patients with Glycogen Storage Disease: A Single Center Retrospective Study. J. Korean Med. Sci. 2019, 35. [Google Scholar] [CrossRef]
- Hadžić, N.; Quaglia, A.; Portmann, B.; Paramalingam, S.; Heaton, N.D.; Rela, M.; Mieli-Vergani, G.; Davenport, M. Hepatocellular Carcinoma in Biliary Atresia: King’s College Hospital Experience. J. Pediatr. 2011, 159, 617–622.e1. [Google Scholar] [CrossRef]
- Suri, A.; Sharma, V.K.; Ranade, P.R.; Marar, S.; Nagral, A. Ruptured Hepatocellular Carcinoma in a Child with Budd-Chiari Syndrome. Indian Pediatr. 2016, 53, 2. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Suddle, A.; Quaglia, A.; Peddu, P.; Karani, J.; Satyadas, T.; Heaton, N. Congenital Extrahepatic Portosystemic Shunt Complicated by the Development of Hepatocellular Carcinoma. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 552–557. [Google Scholar] [CrossRef]
- Anupindi, S.A.; Biko, D.M.; Ntoulia, A.; Poznick, L.; Morgan, T.A.; Darge, K.; Back, S.J. Contrast-Enhanced US Assessment of Focal Liver Lesions in Children. RadioGraphics 2017, 37, 1632–1647. [Google Scholar] [CrossRef]
- Taimr, P.; Bröker, M.E.E.; Dwarkasing, R.S.; Hansen, B.E.; de Knegt, R.J.; De Man, R.A.; IJzermans, J.N.M. A Model-Based Prediction of the Probability of Hepatocellular Adenoma and Focal Nodular Hyperplasia Based on Characteristics on Contrast-Enhanced Ultrasound. Ultrasound Med. Biol. 2017, 43, 2144–2150. [Google Scholar] [CrossRef]
- Saar, B.; Kellner-Weldon, F. Radiological Diagnosis of Hepatocellular Carcinoma. Liver Int. 2008, 28, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Palm, V.; Sheng, R.; Mayer, P.; Weiss, K.-H.; Springfeld, C.; Mehrabi, A.; Longerich, T.; Berger, A.K.; Kauczor, H.-U.; Weber, T.F. Imaging Features of Fibrolamellar Hepatocellular Carcinoma in Gadoxetic Acid-Enhanced MRI. Cancer Imaging 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, D.; Szklaruk, J.; Kundra, V.; Kaseb, A.; Rashid, A.; Elsayes, K.M. Imaging Features of Fibrolamellar Hepatocellular Carcinoma. Am. J. Roentgenol. 2014, 202, 544–552. [Google Scholar] [CrossRef]
- Weeda, V.B.; Aronson, D.C.; Verheij, J.; Lamers, W.H. Is Hepatocellular Carcinoma the Same Disease in Children and Adults? Comparison of Histology, Molecular Background, and Treatment in Pediatric and Adult Patients. Pediatric Blood Cancer 2019, 66, e27475. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jang, M.; Park, Y.N. Histopathological Variants of Hepatocellular Carcinomas: An Update According to the 5th Edition of the WHO Classification of Digestive System Tumors. J. Liver Cancer 2020, 20, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Katzenstein, H.M.; Krailo, M.D.; Malogolowkin, M.H.; Ortega, J.A.; Qu, W.; Douglass, E.C.; Feusner, J.H.; Reynolds, M.; Quinn, J.J.; Newman, K.; et al. Fibrolamellar Hepatocellular Carcinoma in Children and Adolescents. Cancer 2003, 97, 2006–2012. [Google Scholar] [CrossRef]
- Allan, B.J.; Wang, B.; Davis, J.S.; Parikh, P.P.; Perez, E.A.; Neville, H.L.; Sola, J.E. A Review of 218 Pediatric Cases of Hepatocellular Carcinoma. J. Pediatric Surg. 2014, 49, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Schlageter, M.; Terracciano, L.M.; D’Angelo, S.; Sorrentino, P. Histopathology of Hepatocellular Carcinoma. World J. Gastroenterol. 2014, 20, 15955–15964. [Google Scholar] [CrossRef]
- Stocker, J.T.; Ishak, K.G. Undifferentiated (Embryonal) Sarcoma of the Liver: Report of 31 Cases. Cancer 1978, 42, 336–348. [Google Scholar] [CrossRef]
- Mathias, M.D.; Ambati, S.R.; Chou, A.J.; Slotkin, E.K.; Wexler, L.H.; Meyers, P.A.; Magnan, H. A Single-Center Experience with Undifferentiated Embryonal Sarcoma of the Liver. Pediatric Blood Cancer 2016, 63, 2246–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Rojas, Y.; Zhang, W.; Beierle, E.A.; Doski, J.J.; Goldfarb, M.; Goldin, A.B.; Gow, K.W.; Langer, M.; Meyers, R.L.; et al. Characteristics and Outcomes in Children with Undifferentiated Embryonal Sarcoma of the Liver: A Report from the National Cancer Database. Pediatric Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Xu, H.; Sangster, G.P.; Gu, X. Pulmonary Metastases from an Undifferentiated Embryonal Sarcoma of the Liver: A Case Report and Review. Case Rep. Oncol. Med. 2018, 2018, 7840865. [Google Scholar] [CrossRef]
- Hu, H.-M.; Zhang, W.-L.; Li, J.; Wen, Y.; Li, F.; Zhi, T.; Huang, D.-S. Report of Seven Children with Undifferentiated Embryonal Sarcoma of the Liver. Chin. Med. J. 2019, 132, 2244–2245. [Google Scholar] [CrossRef]
- Gao, J.; Fei, L.; LI, S.; Cui, K.; Zhang, J.; YU, F.; Zhang, B. Undifferentiated Embryonal Sarcoma of the Liver in a Child: A Case Report and Review of the Literature. Oncol. Lett. 2013, 5, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Putra, J.; Ornvold, K. Undifferentiated Embryonal Sarcoma of the Liver: A Concise Review. Arch. Pathol. Lab. Med. 2015, 139, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Thombare, P.; Verma, M.; Shah, K.; Doshi, H.; Verma, G.; Patkar, D. Undifferentiated Embryonal Sarcoma of Liver: Paradoxical Imaging Appearance. Radiol. Case Rep. 2020, 15, 1095–1098. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, C.-J.; Xu, C.; Sheng, Q.-J.; Dou, X.-G.; Ding, Y. Undifferentiated Embryonal Sarcoma of the Liver: Clinical Characteristics and Outcomes. World J. Clin. Cases 2020, 8, 4763–4772. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.; Melo, D.; Esteves, C.; Lima, B.; Carneiro, F.; Oliveira, P. Undifferentiated Embryonal Sarcoma of the Liver: A Rare Hepatic Tumor and Its Related Characteristic Radiological Features. Radiol. Case Rep. 2021, 16, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, W.; Song, G.; Xiao, Y.; Sun, F.; Wang, N. Infantile Hepatic Hemangiomas Associated with High-Output Cardiac Failure and Pulmonary Hypertension. BMC Cardiovasc. Disord. 2019, 19, 216. [Google Scholar] [CrossRef] [Green Version]
- Letherer, A.; Mastenbrook, J.; VanEnk, R.A.; Bauler, L.D. Undifferentiated Embryonal Sarcoma of the Liver Presents as a Molecular Mimic of Parasitic Infection. Cureus 2020, 12, e6800. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.M.; Heath, J.E.; Twaddell, W.S.; Castellani, R.J. Undifferentiated Sarcoma of the Liver: A Case Study of an Erythropoietin-Secreting Tumor. Int. J. Surg. Pathol. 2014, 22, 555–558. [Google Scholar] [CrossRef]
- Perl, R.M.; Häring, A.; Horger, M.S.; Pfannenberg, C.; Gatidis, S. Paraneoplastic Syndrome in Undifferentiated Embryonic Sarcoma of the Liver. EJNMMI Res. 2020, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Setty, B.A.; Jinesh, G.G.; Arnold, M.; Pettersson, F.; Cheng, C.-H.; Cen, L.; Yoder, S.J.; Teer, J.K.; Flores, E.R.; Reed, D.R.; et al. The Genomic Landscape of Undifferentiated Embryonal Sarcoma of the Liver Is Typified by C19MC Structural Rearrangement and Overexpression Combined with TP53 Mutation or Loss. PLoS Genet. 2020, 16, e1008642. [Google Scholar] [CrossRef] [PubMed]
- Lepreux, S.; Rebouissou, S.; Le Bail, B.; Saric, J.; Balabaud, C.; Bloch, B.; Martin-Négrier, M.-L.; Zucman-Rossi, J.; Bioulac-Sage, P. Mutation of TP53 Gene Is Involved in Carcinogenesis of Hepatic Undifferentiated (Embryonal) Sarcoma of the Adult, in Contrast with Wnt or Telomerase Pathways: An Immunohistochemical Study of Three Cases with Genomic Relation in Two Cases. J. Hepatol. 2005, 42, 424–429. [Google Scholar] [CrossRef]
- Kim, J.-H.; Sio, C.A.; Park, H.; Kim, H.; Shin, H.D.; Jung, K. Undifferentiated Embryonal Sarcoma of the Liver in a Child: A Whole Exome Sequencing Analysis. Dig. Liver Dis. 2017, 49, 944–946. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Focal Hepatic Hemangiomas (FHH) | Multifocal Hepatic Hemangiomas (MHH) | Diffuse Hepatic Hemangiomas (DHH) | |
|---|---|---|---|
| Age of Onset | Proliferates in utero Fully formed at birth | Post-natal period | Post-natal period |
| Natural Course of Progression | Undergoes involution over the first 12–14 months of life | 3 Phases:
| Significant liver involvement with near complete displacement of all liver parenchyma |
| Laboratory Findings | Anemia Mild thrombocytopenia Elevated alpha feto-protein (AFP) at birth (should downtrend) Normal thyroid studies | Low T4 High TSH | Low T4 High TSH |
| Clinical Presentation | Asymptomatic prenatally After birth may present with abdominal distention and palpable hepatic mass, unexplained anemia, or coagulopathy No association with cutaneous infantile hemangiomas | Most asymptomatic May present with hepatomegaly and abdominal distension or be identified on screening after development of congestive heart failure or hypothyroidism May be associated with cutaneous infantile hemangiomas (60%) | Massive hepatomegaly with hepatic failure May be associated with cutaneous infantile hemangiomas |
| Clinical Complications | Prenatally: fetal cardiomegaly, cardiac failure, hydrops fetalis Postnatally: cardiac failure | High output cardiac failure | High output cardiac failure Abdominal compartment syndrome Multisystem organ failure Profound consumptive hypothyroidism |
| Subtype | Cell Size, Shape, and Pattern | Nuclei and Nucleoli | Cytoplasm |
|---|---|---|---|
| Fetal | Polygonal cells between 10 and 20 μ in diameter Organized in sheets or as one to two cell thick trabecula | Centrally placed round nuclei with well delineated nuclear membranes, finely stippled chromatin and inconspicuous nucleoli | Appears clear, commonly contains clusters of hematopoietic precursors Crowded pattern may have ampophilic cytoplasm with a proportionately high nuclear-to-cytoplasmic ratio |
| Embryonal | Round or angulated cells, 10 to 15 μ in diameter Grow in sheets and commonly form tubular or acinar structures around a central lumen | High nuclear-to-cytoplasmic ratio | Scant cytoplasm |
| Small Cell | Round or oval, 7–8 μ in diameter Usually form clusters or nests in an “organoid” pattern | Fine nuclear chromatin, inconspicuous nucleoli, minimal mitotic activity | Scant cytoplasm |
| Macrotrabecular | Cell plates more than 20-cells thick that can be found in a pure form or in combination with other patterns | Variable (may be reflective of fetal or embryonal origins or be pleomorphic) | Variable |
| Histology | IHC | Genetics | |
|---|---|---|---|
| HHCA | Intralesion steatosis with a lack of associated inflammation or cellular atypia | Decreased or absent LFABP immunostaining | HNF1A inactivating mutation |
| IHCA | Inflammatory infiltrate, sinusoidal dilatation, dystrophic arteries, and variable ductular reaction in periphery of the lesions | Positive CRP Positive serum amyloid A | IL6ST mutation |
| bHCA | Mild cytologic atypia Pseudoacinar formation | Diffuse and strong expression of glutamine synthetase Nuclear positivity for β-catenin | CTNNB1 mutation |
| shHCA | Intratumoral hemorrhage | Positive Prostaglandin D2 synthase | Deletion of INHBE leading to INHBE–GLI1 fusions |
| uHCA | Nonspecific, has typical HCA findings | None | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, B.; Ravishankar, S.; Pateva, I. Pediatric Primary Hepatic Tumors: Diagnostic Considerations. Diagnostics 2021, 11, 333. https://doi.org/10.3390/diagnostics11020333
Lucas B, Ravishankar S, Pateva I. Pediatric Primary Hepatic Tumors: Diagnostic Considerations. Diagnostics. 2021; 11(2):333. https://doi.org/10.3390/diagnostics11020333
Chicago/Turabian StyleLucas, Bryony, Sanjita Ravishankar, and Irina Pateva. 2021. "Pediatric Primary Hepatic Tumors: Diagnostic Considerations" Diagnostics 11, no. 2: 333. https://doi.org/10.3390/diagnostics11020333