Alagille Syndrome: Diagnostic Challenges and Advances in Management

Abstract
:1. Introduction
2. Clinical Overview
2.1. Hepatic Features
2.2. Cardiac Features
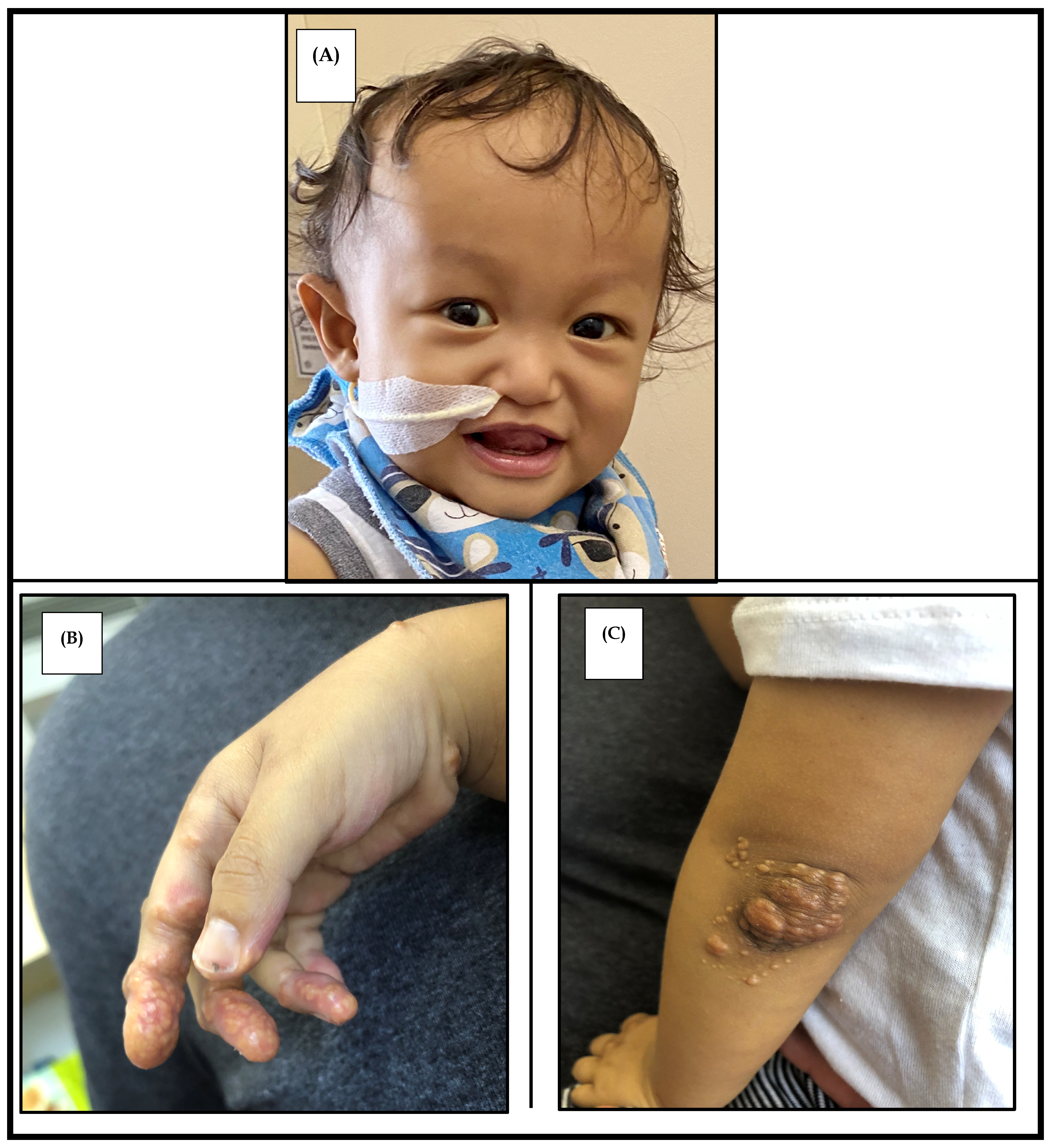
2.3. Facial Features
2.4. Ocular Features
2.5. Skeletal Features
2.6. Renal Features
2.7. Vascular Features
3. Genetics of Alagille Syndrome
3.1. Gene Identification & Mutational Analysis
3.2. The Notch Signaling Pathway & Bile Duct Development
4. Diagnostic Testing
4.1. Liver Histopathology
4.2. Genetic Testing
5. Diagnostic Challenges
5.1. Genotype-Phenotypic Variability
5.2. Bile Duct Paucity
5.3. Cholangiography
6. Management of Alagille Syndrome
6.1. Nutrition and FSVD
6.2. Medical Management of Pruritus
6.3. Surgical Management of Pruritus
6.4. Liver Transplantation
7. Advancement in Management in Alagille Syndrome
7.1. IBAT Inhibitors
7.2. Cholangiocyte Regeneration
7.3. Stem Cell Applications in ALGS
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alagille, D.; Habib, E.; Thomassin, N. L’atresie des voies biliaires intrahepatiques avec voies biliaires extrahepatiques permeables chez l’enfant. J. Par. Pediatr. 1969, 301, 301–318. [Google Scholar]
- Alagille, D.; Estrada, A.; Hadchouel, M.; Gautler, M.; Odievre, M.; Dommergues, J. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): Review of 80 cases. J. Pediatr. 1987, 110, 195–200. [Google Scholar] [CrossRef]
- Alagille, D.; Odievre, M.; Gautier, M.; Dommergues, J. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J. Pediatr. 1975, 86, 63–71. [Google Scholar] [CrossRef]
- Danks, D.; Campbell, P.; Jack, I.; Rogers, J.; Smith, A. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch. Dis. Child. 1977, 52, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Krantz, I.D.; Deng, Y.; Genin, A.; Banta, A.B.; Collins, C.C.; Qi, M.; Trask, B.J.; Kuo, W.L.; Cochran, J. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 1997, 16, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.; Bason, L.; Piccoli, D.; Krantz, I.; Spinner, N. Consequences of JAG1 mutations. J. Med. Genet. 2003, 40, 891–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.; Kamath, B.M.; Chitayat, D. Alagille syndrome: Clinical perspectives. Appl. Clin. Genet. 2016, 9, 75. [Google Scholar] [PubMed] [Green Version]
- Deprettere, A.; Portmann, B.; Mowat, A.P. Syndromic paucity of the intrahepatic bile ducts: Diagnostic difficulty; severe morbidity throughout early childhood. J. Pediatr. Gastroenterol. Nutr. 1987, 6, 865–871. [Google Scholar] [CrossRef]
- Hoffenberg, E.J.; Narkewicz, M.R.; Sondheimer, J.M.; Smith, D.J.; Silverman, A.; Sokol, R.J. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J. Pediatr. 1995, 127, 220–224. [Google Scholar] [CrossRef]
- Emerick, K.M.; Rand, E.B.; Goldmuntz, E.; Krantz, I.D.; Spinner, N.B.; Piccoli, D.A. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology 1999, 29, 822–829. [Google Scholar] [CrossRef]
- Quiros-Tejeira, R.E.; Ament, M.E.; Heyman, M.B.; Martin, M.G.; Rosenthal, P.; Hall, T.R.; McDiarmid, S.V.; Vargas, J.H. Variable morbidity in Alagille syndrome: A review of 43 cases. J. Pediatr. Gastroenterol. Nutr. 1999, 29, 431–437. [Google Scholar] [CrossRef]
- Subramaniam, P.; Knisely, A.; Portmann, B.; Qureshi, S.; Aclimandos, W.; Karani, J.; Baker, A. Diagnosis of Alagille syndrome—25 years of experience at King’s College Hospital. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Podkameni, G.; Hutchinson, A.L.; Leonard, L.D.; Gerfen, J.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B.; Loomes, K.M.; Meyers, K. Renal anomalies in Alagille syndrome: A disease-defining feature. Am. J. Med. Genet. A 2012, 158A, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, B.M.; Piccoli, D.A. Alagille syndrome. In Diseases of the Liver in Children; Springer: New York, NY, USA, 2014; pp. 227–246. [Google Scholar]
- Kriegermeier, A.; Wehrman, A.; Kamath, B.M.; Loomes, K.M. Liver disease in alagille syndrome. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 49–65. [Google Scholar]
- Garcia, M.A.; Margarita, R.; Mirta, C.; Hugo, C.; Pablo, L.; Estela, A.; De Davila, M.T. Alagille syndrome: Cutaneous manifestations in 38 children. Pediatr. Dermatol. 2005, 22, 11–14. [Google Scholar] [CrossRef]
- Lykavieris, P.; Hadchouel, M.; Chardot, C.; Bernard, O. Outcome of liver disease in children with Alagille syndrome: A study of 163 patients. Gut. 2001, 49, 431–435. [Google Scholar] [CrossRef]
- Kamath, B.M.; Ye, W.; Goodrich, N.P.; Loomes, K.M.; Romero, R.; Heubi, J.E.; Leung, D.H.; Spinner, N.B.; Piccoli, D.A.; Alonso, E.M. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatol. Commun. 2020, 4, 387–398. [Google Scholar] [CrossRef] [Green Version]
- McElhinney, D.B.; Krantz, I.D.; Bason, L.; Piccoli, D.A.; Emerick, K.M.; Spinner, N.B.; Goldmuntz, E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation 2002, 106, 2567–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferencz, C. Genetic and environmental risk factors of major cardiovascular malformations: The Baltimore-Washington infant study 1981–1989. Perspect. Pediatr. Cardiol. 1997, 5, 346–347. [Google Scholar]
- Wagley, Y.; Mitchell, T.; Ashley, J.; Loomes, K.M.; Hankenson, K. Skeletal involvement in Alagille syndrome. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 121–135. [Google Scholar]
- Hingorani, M.; Nischal, K.K.; Davies, A.; Bentley, C.; Vivian, A.; Baker, A.J.; Mieli-Vergani, G.; Bird, A.C.; Aclimandos, W.A. Ocular abnormalities in Alagille syndrome. Ophthalmology 1999, 106, 330–337. [Google Scholar] [CrossRef]
- Rennie, C.A.; Chowdhury, S.; Khan, J.; Rajan, F.; Jordan, K.; Lamb, R.J.; Vivian, A.J. The prevalence and associated features of posterior embryotoxon in the general ophthalmic clinic. Eye (Lond.) 2005, 19, 396–399. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.; Kirschner, R.; Goldmuntz, E.; Sullivan, K.; Eicher, P.; Gerdes, M.; Moss, E.; Solot, C.; Wang, P.; Jacobs, I. The Philadelphia story: The 22q11. 2 deletion: Report on 250 patients. Genet. Couns. (Geneva, Switzerland) 1999, 10, 11. [Google Scholar]
- Nischal, K.K.; Hingorani, M.; Bentley, C.R.; Vivian, A.J.; Bird, A.C.; Baker, A.J.; Mowat, A.P.; Mieli-Vergani, G.; Aclimandos, W.A. Ocular Ultrasound in Alagille Syndrome. Ophthalmology 1997, 104, 79–85. [Google Scholar] [CrossRef]
- Chang, M.Y.; Pineles, S.L. Optic disk drusen in children. Surv. Ophthalmol. 2016, 61, 745–758. [Google Scholar] [CrossRef] [Green Version]
- Loomes, K.M.; Spino, C.; Goodrich, N.P.; Hangartner, T.N.; Marker, A.E.; Heubi, J.E.; Kamath, B.M.; Shneider, B.L.; Rosenthal, P.; Hertel, P.M.; et al. Bone Density in Children With Chronic Liver Disease Correlates With Growth and Cholestasis. Hepatology 2019, 69, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bales, C.B.; Kamath, B.M.; Munoz, P.S.; Nguyen, A.; Piccoli, D.A.; Spinner, N.B.; Horn, D.; Shults, J.; Leonard, M.B.; Grimberg, A.; et al. Pathologic lower extremity fractures in children with Alagille syndrome. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Delgado, A.; Mokri, B.; Miller, G.M. Butterfly vertebra. J. Neuroimaging 1996, 6, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Sandal, G.; Aslan, N.; Duman, L.; Ormeci, A. VACTERL association with a rare vertebral anomaly (butterfly vertebra) in a case of monochorionic twin. Genet Couns 2014, 25, 231–235. [Google Scholar]
- Sanderson, E.; Newman, V.; Haigh, S.F.; Baker, A.; Sidhu, P.S. Vertebral anomalies in children with Alagille syndrome: An analysis of 50 consecutive patients. Pediatr. Radiol. 2002, 32, 114–119. [Google Scholar] [CrossRef]
- Kamath, B.M.; Loomes, K.M.; Oakey, R.J.; Krantz, I.D. Supernumerary digital flexion creases: An additional clinical manifestation of Alagille syndrome. Am. J. Med. Genet. 2002, 112, 171–175. [Google Scholar]
- Ryan, R.; Myckatyn, S.; Reid, G.; Munk, P. Alagille syndrome: Case report with bilateral radio-ulnar synostosis and a literature review. Skelet. Radiol. 2003, 32, 489–491. [Google Scholar] [CrossRef]
- Kindler, J.M.; Mitchell, E.L.; Piccoli, D.A.; Grimberg, A.; Leonard, M.B.; Loomes, K.M.; Zemel, B.S. Bone geometry and microarchitecture deficits in children with Alagille syndrome. Bone 2020, 115576. [Google Scholar] [CrossRef]
- Youngstrom, D.; Dishowitz, M.; Bales, C.; Carr, E.; Mutyaba, P.; Kozloff, K.; Shitaye, H.; Hankenson, K.; Loomes, K. Jagged1 expression by osteoblast-lineage cells regulates trabecular bone mass and periosteal expansion in mice. Bone 2016, 91, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Romero, R. The renal sequelae of Alagille Syndrome as a Product of Altered Notch Signaling During Kidney Development. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 103–120. [Google Scholar]
- Kamath, B.M.; Yin, W.; Miller, H.; Anand, R.; Rand, E.B.; Alonso, E.; Bucuvalas, J.; Studies of Pediatric Liver, T. Outcomes of liver transplantation for patients with Alagille syndrome: The studies of pediatric liver transplantation experience. Liver Transpl. 2012, 18, 940–948. [Google Scholar] [CrossRef]
- Kamath, B.M.; Spinner, N.B.; Emerick, K.M.; Chudley, A.E.; Booth, C.; Piccoli, D.A.; Krantz, I.D. Vascular anomalies in Alagille syndrome: A significant cause of morbidity and mortality. Circulation 2004, 109, 1354–1358. [Google Scholar] [CrossRef] [Green Version]
- Emerick, K.M.; Krantz, I.D.; Kamath, B.M.; Darling, C.; Burrowes, D.M.; Spinner, N.B.; Whitington, P.F.; Piccoli, D.A. Intracranial vascular abnormalities in patients with Alagille syndrome. J. Pediatr. Gastroenterol. Nutr. 2005, 41, 99–107. [Google Scholar] [CrossRef]
- Vandriel, S.M.; Ichord, R.N.; Kamath, B.M. Vascular Manifestations in Alagille Syndrome. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 91–102. [Google Scholar]
- Bérard, E.; Sarles, J.; Triolo, V.; Gagnadoux, M.-F.; Wernert, F.; Hadchouel, M.; Niaudet, P. Renovascular hypertension and vascular anomalies in Alagille syndrome. Pediatr. Nephrol. 1998, 12, 121–124. [Google Scholar] [CrossRef]
- Nishikawa, A.; Mori, H.; Takahashi, M.; Ojima, A.; Shimokawa, K.; Fueuta, T. Alagille’s syndrome: A case with a hamartomatous nodule of the liver. Pathol. Int. 1987, 37, 1319–1326. [Google Scholar] [CrossRef]
- Labrecque, D.R.; Mitros, F.A.; Nathan, R.J.; Romanchuk, K.G.; Judisch, G.F.; El-Khoury, G.H. Four generations of arteriohepatic dysplasia. Hepatology 1982, 2, 467S–474S. [Google Scholar] [CrossRef]
- Kohaut, J.; Pommier, R.; Guerin, F.; Pariente, D.; Jacquemin, E.; Martelli, H.; Branchereau, S. Abdominal Arterial Anomalies in Children With Alagille Syndrome: Surgical Aspects and Outcomes of Liver Transplantation. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 888–891. [Google Scholar] [CrossRef]
- Oda, T.; Elkahloun, A.G.; Pike, B.L.; Okajima, K.; Krantz, I.D.; Genin, A.; Piccoli, D.A.; Meltzer, P.S.; Spinner, N.B.; Collins, F.S. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 1997, 16, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.A.; Bauer, R.C.; Rajagopalan, R.; Grochowski, C.M.; Chao, G.; McEldrew, D.; Nassur, J.A.; Rand, E.B.; Krock, B.L.; Kamath, B.M.; et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum. Mutat. 2019, 40, 2197–2220. [Google Scholar] [CrossRef] [Green Version]
- McDaniell, R.; Warthen, D.M.; Sanchez-Lara, P.A.; Pai, A.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 2006, 79, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Krantz, I.D.; Colliton, R.P.; Genin, A.; Rand, E.B.; Li, L.; Piccoli, D.A.; Spinner, N.B. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am. J. Hum. Genet. 1998, 62, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Spinner, N.B.; Colliton, R.P.; Crosnier, C.; Krantz, I.D.; Hadchouel, M.; Meunier-Rotival, M. Jagged1 mutations in Alagille syndrome. Hum. Mutat. 2001, 17, 18–33. [Google Scholar] [CrossRef]
- Micaglio, E.; Andronache, A.A.; Carrera, P.; Monasky, M.M.; Locati, E.T.; Pirola, B.; Presi, S.; Carminati, M.; Ferrari, M.; Giamberti, A.; et al. Novel JAG1 Deletion Variant in Patient with Atypical Alagille Syndrome. Int. J. Mol. Sci. 2019, 20, 6247. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, X.; Chen, S.; Zhang, J.; Xu, C. Targeted Sequencing and RNA Assay Reveal a Noncanonical JAG1 Splicing Variant Causing Alagille Syndrome. Front. Genet. 2019, 10, 1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar]
- Kamath, B.M.; Bauer, R.C.; Loomes, K.M.; Chao, G.; Gerfen, J.; Hutchinson, A.; Hardikar, W.; Hirschfield, G.; Jara, P.; Krantz, I.D.; et al. NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 2012, 49, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S. Concise review: Notch signaling in stem cell systems. Stem Cells 2006, 24, 2437–2447. [Google Scholar] [CrossRef]
- Huppert, S.S.; Campbell, K.M. Bile Duct Development and the Notch Signaling Pathway. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 11–31. [Google Scholar]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridley, T. Notch signaling in vascular development and physiology. Development 2007, 134, 2709–2718. [Google Scholar] [CrossRef] [Green Version]
- Crosnier, C.; Attie-Bitach, T.; Encha-Razavi, F.; Audollent, S.; Soudy, F.; Hadchouel, M.; Meunier-Rotival, M.; Vekemans, M. JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology 2000, 32, 574–581. [Google Scholar] [CrossRef]
- Lemaigre, F.P. Development of the intrahepatic and extrahepatic biliary tract: A framework for understanding congenital diseases. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Huppert, S.S.; Iwafuchi-Doi, M. Molecular regulation of mammalian hepatic architecture. Curr. Top. Dev. Biol. 2019, 132, 91–136. [Google Scholar]
- Andrews, A.R.; Putra, J. Central Hepatic Regenerative Nodules in Alagille Syndrome: A Clinicopathological Review. Fetal Pediatr. Pathol. 2019, 1–11. [Google Scholar] [CrossRef]
- Rapp, J.B.; Bellah, R.D.; Maya, C.; Pawel, B.R.; Anupindi, S.A. Giant hepatic regenerative nodules in Alagille syndrome. Pediatr. Radiol. 2017, 47, 197–204. [Google Scholar] [CrossRef]
- Alhammad, A.; Kamath, B.M.; Chami, R.; Ng, V.L.; Chavhan, G.B. Solitary Hepatic Nodule Adjacent to the Right Portal Vein: A Common Finding of Alagille Syndrome? J. Pediatr. Gastroenterol. Nutr. 2016, 62, 226–232. [Google Scholar] [CrossRef]
- Treem, W.R.; Krzymowski, G.A.; Cartun, R.W.; Pedersen, C.A.; Hyams, J.S.; Berman, M. Cytokeratin immunohistochemical examination of liver biopsies in infants with Alagille syndrome and biliary atresia. J. Pediatr. Gastroenterol. Nutr. 1992, 15, 73–80. [Google Scholar] [CrossRef]
- Kahn, E. Paucity of interlobular bile ducts. Arteriohepatic dysplasia and nonsyndromic duct paucity. Perspect. Pediatr. Pathol. 1991, 14, 168–215. [Google Scholar]
- Russo, P.; Magee, J.C.; Anders, R.A.; Bove, K.E.; Chung, C.; Cummings, O.W.; Finegold, M.J.; Finn, L.S.; Kim, G.E.; Lovell, M.A. Key histopathological features of liver biopsies that distinguish biliary atresia from other causes of infantile cholestasis and their correlation with outcome: A multicenter study. Am. J. Surg. Pathol. 2016, 40, 1601. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.A.; Spinner, N.B. Genetics of Alagille Syndrome. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 33–48. [Google Scholar]
- Rajagopalan, R.; Grochowski, C.M.; Gilbert, M.A.; Falsey, A.M.; Coleman, K.; Romero, R.; Loomes, K.M.; Piccoli, D.A.; Devoto, M.; Spinner, N.B. Compound heterozygous mutations in NEK8 in siblings with end-stage renal disease with hepatic and cardiac anomalies. Am. J. Med. Genet. Part A 2016, 170, 750–753. [Google Scholar] [CrossRef]
- Grochowski, C.M.; Rajagopalan, R.; Falsey, A.M.; Loomes, K.M.; Piccoli, D.A.; Krantz, I.D.; Devoto, M.; Spinner, N.B. Exome sequencing reveals compound heterozygous mutations in ATP8B1 in a JAG1/NOTCH2 mutation-negative patient with clinically diagnosed Alagille syndrome. Am. J. Med. Genet. Part A 2015, 167, 891–893. [Google Scholar] [CrossRef]
- Kamath, B.M.; Thiel, B.D.; Gai, X.; Conlin, L.K.; Munoz, P.S.; Glessner, J.; Clark, D.; Warthen, D.M.; Shaikh, T.H.; Mihci, E.; et al. SNP array mapping of chromosome 20p deletions: Genotypes, phenotypes, and copy number variation. Hum. Mutat. 2009, 30, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Izumi, K.; Hayashi, D.; Grochowski, C.M.; Kubota, N.; Nishi, E.; Arakawa, M.; Hiroma, T.; Hatata, T.; Ogiso, Y.; Nakamura, T. Discordant clinical phenotype in monozygotic twins with Alagille syndrome: Possible influence of non-genetic factors. Am. J. Med. Genet. Part A 2016, 170, 471–475. [Google Scholar] [CrossRef]
- Jafar-Nejad, H.; Leonardi, J.; Fernandez-Valdivia, R. Role of glycans and glycosyltransferases in the regulation of Notch signaling. Glycobiology 2010, 20, 931–949. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Valdivia, R.; Takeuchi, H.; Samarghandi, A.; Lopez, M.; Leonardi, J.; Haltiwanger, R.S.; Jafar-Nejad, H. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 2011, 138, 1925–1934. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.J.; Bales, C.; Nelson, A.; Gonzalez, D.M.; Underkoffler, L.; Segalov, M.; Wilson-Rawls, J.; Cole, S.E.; Moran, J.L.; Russo, P.; et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology 2008, 48, 1989–1997. [Google Scholar] [CrossRef]
- Thakurdas, S.M.; Lopez, M.F.; Kakuda, S.; Fernandez-Valdivia, R.; Zarrin-Khameh, N.; Haltiwanger, R.S.; Jafar-Nejad, H. Jagged1 heterozygosity in mice results in a congenital cholangiopathy which is reversed by concomitant deletion of one copy of Poglut1 (Rumi). Hepatology 2016, 63, 550–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, E.A.; Gilbert, M.A.; Grochowski, C.M.; Underkoffler, L.A.; Meng, H.; Zhang, X.; Wang, M.M.; Shitaye, H.; Hankenson, K.D.; Piccoli, D.; et al. THBS2 Is a Candidate Modifier of Liver Disease Severity in Alagille Syndrome. Cell. Mol. Gastroenterol Hepatol 2016, 2, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier-Torres, L.; Beraza, N.; Fernández-Tussy, P.; Lopitz-Otsoa, F.; Fernández-Ramos, D.; Zubiete-Franco, I.; Varela-Rey, M.; Delgado, T.C.; Gutiérrez, V.; Anguita, J. Histone deacetylase 4 promotes cholestatic liver injury in the absence of prohibitin-1. Hepatology 2015, 62, 1237–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, P.; Ruchelli, E.D.; Piccoli, D.A. Pathology of Pediatric Gastrointestinal and Liver Disease; Springer: Berlin, Heidelberg, 2014. [Google Scholar]
- Kahn, E.; Markowitz, J.; Aiges, H.; Daum, F. Human ontogeny of the bile duct to portal space ratio. Hepatology 1989, 10, 21–23. [Google Scholar] [CrossRef]
- Sergi, C.; Bahitham, W.; Al-Bahrani, R. Bile duct paucity in infancy. In Liver Biopsy in Modern Medicine; InTech: Rijeka, Croatia, 2011; pp. 295–304. [Google Scholar]
- Lally, K.P.; Kanegaye, J.; Matsumura, M.; Rosenthal, P.; Sinatra, F.; Atkinson, J.B. Perioperative factors affecting the outcome following repair of biliary atresia. Pediatrics 1989, 83, 723–726. [Google Scholar] [PubMed]
- Kaye, A.J.; Rand, E.B.; Munoz, P.S.; Spinner, N.B.; Flake, A.W.; Kamath, B.M. Effect of Kasai procedure on hepatic outcome in Alagille syndrome. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, J.; Suzuki, K.; Watanabe, M.; Uotani, C.; Takezoe, T.; Takamoto, N.; Hayashi, K. Outcomes of Alagille syndrome following the Kasai operation: A systematic review and meta-analysis. Pediatr. Surg. Int. 2018, 34, 1073–1077. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, J.; Shen, Z.; Lu, X.; Chen, G.; Huang, Y.; Dong, R.; Zheng, S. Serum MMP-7 in the diagnosis of biliary atresia. Pediatrics 2019, 144. [Google Scholar] [CrossRef]
- Rovner, A.J.; Schall, J.I.; Jawad, A.F.; Piccoli, D.A.; Stallings, V.A.; Mulberg, A.E.; Zemel, B.S. Rethinking growth failure in Alagille syndrome: The role of dietary intake and steatorrhea. J. Pediatr. Gastroenterol. Nutr. 2002, 35, 495–502. [Google Scholar] [CrossRef]
- Wasserman, D.; Zemel, B.S.; Mulberg, A.E.; John, H.A.; Emerick, K.M.; Barden, E.M.; Piccoli, D.A.; Stallings, V.A. Growth, nutritional status, body composition, and energy expenditure in prepubertal children with Alagille syndrome. J. Pediatr. 1999, 134, 172–177. [Google Scholar] [CrossRef]
- Feranchak, A.P.; Sokol, R. Medical and nutritional management of cholestasis in infants and children. Liver Dis. Child. 2007, 3, 190–231. [Google Scholar]
- Kronsten, V.; Fitzpatrick, E.; Baker, A. Management of cholestatic pruritus in paediatric patients with alagille syndrome: The King’s College Hospital experience. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 149–154. [Google Scholar] [CrossRef]
- Kamath, B.M.; Loomes, K.M.; Piccoli, D.A. Medical management of Alagille syndrome. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 580–586. [Google Scholar] [CrossRef]
- Hofmann, A. Rifampicin and treatment of cholestatic pruritus. Gut 2002, 51, 756–757. [Google Scholar] [CrossRef] [Green Version]
- Wietholtz, H.; Marschall, H.-U.; Jan, S.; Matern, S. Stimulation of bile acid 6α-hydroxylation by rifampin. J. Hepatol. 1996, 24, 713–718. [Google Scholar] [CrossRef]
- Ständer, S.; Steinhoff, M.; Schmelz, M.; Weisshaar, E.; Metze, D.; Luger, T. Neurophysiology of pruritus: Cutaneous elicitation of itch. Arch. Dermatol. 2003, 139, 1463–1470. [Google Scholar] [CrossRef]
- Mayo, M.J.; Handem, I.; Saldana, S.; Jacobe, H.; Getachew, Y.; Rush, A.J. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007, 45, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Thebaut, A.; Habes, D.; Gottrand, F.; Rivet, C.; Cohen, J.; Debray, D.; Jacquemin, E.; Gonzales, E. Sertraline as an Additional Treatment for Cholestatic Pruritus in Children. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 431–435. [Google Scholar] [CrossRef]
- Whitington, P.F.; Whitington, G.L. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology 1988, 95, 130–136. [Google Scholar] [CrossRef]
- Wang, K.S.; Tiao, G.; Bass, L.M.; Hertel, P.M.; Mogul, D.; Kerkar, N.; Clifton, M.; Azen, C.; Bull, L.; Rosenthal, P.; et al. Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology 2017, 65, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Hsu, E.; Rand, E. Transplant Considerations in Alagille Syndrome. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 67–76. [Google Scholar]
- Arnon, R.; Annunziato, R.; Miloh, T.; Suchy, F.; Sakworawich, A.; Sogawa, H.; Kishore, I.; Kerkar, N. Orthotopic liver transplantation for children with Alagille syndrome. Pediatr. Transplant. 2010, 14, 622–628. [Google Scholar] [CrossRef]
- Karpen, S.J.; Kelly, D.; Mack, C.; Stein, P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol. Int. 2020. [Google Scholar] [CrossRef]
- Dawson, P.A.; Haywood, J.; Craddock, A.L.; Wilson, M.; Tietjen, M.; Kluckman, K.; Maeda, N.; Parks, J.S. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J. Biol. Chem. 2003, 278, 33920–33927. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Apte, U. Bile acid metabolism and signaling in cholestasis, inflammation, and cancer. Adv. Pharmacol. 2015, 74, 263–302. [Google Scholar] [CrossRef] [Green Version]
- Shneider, B.L.; Spino, C.; Kamath, B.M.; Magee, J.C.; Bass, L.M.; Setchell, K.D.; Miethke, A.; Molleston, J.P.; Mack, C.L.; Squires, R.H.; et al. Placebo-Controlled Randomized Trial of an Intestinal Bile Salt Transport Inhibitor for Pruritus in Alagille Syndrome. Hepatol. Commun. 2018, 2, 1184–1198. [Google Scholar] [CrossRef]
- Gonzales, E.; Sturm, E.; Stormon, M.; Sokal, E.; Hardikar, W.; Lacaille, F.; Gliwicz, D.; Hierro, L.; Jaecklin, T.; Gu, J. PS-193-Phase 2 open-label study with a placebo-controlled drug withdrawal period of the apical sodium-dependent bile acid transporter inhibitor maralixibat in children with Alagille Syndrome: 48-week interim efficacy analysis. J. Hepatol. Suppl. 2019, 70, e119. [Google Scholar] [CrossRef]
- Schaub, J.R.; Huppert, K.A.; Kurial, S.N.; Hsu, B.Y.; Cast, A.E.; Donnelly, B.; Karns, R.A.; Chen, F.; Rezvani, M.; Luu, H.Y. De novo formation of the biliary system by TGFβ-mediated hepatocyte transdifferentiation. Nature 2018, 557, 247–251. [Google Scholar] [CrossRef]
- Pollheimer, M.J.; Trauner, M.; Fickert, P. Will we ever model PSC?—“It’s hard to be a PSC model!”. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 792–804. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, M.; Ogawa, S.; Bear, C.E.; Ahmadi, S.; Chin, S.; Li, B.; Grompe, M.; Keller, G.; Kamath, B.M.; Ghanekar, A. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 853–861. [Google Scholar] [CrossRef]
- Sampaziotis, F.; De Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Xu, D.; Garfin, P.M.; Ehmer, U.; Hurwitz, M.; Enns, G.; Michie, S.; Wu, M.; Zheng, M.; Nishimura, T. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.E.; Loring, J.F. Genomic instability in pluripotent stem cells: Implications for clinical applications. J. Biol. Chem. 2014, 289, 4578–4584. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.R. Future Therapeutic Approaches for Alagille Syndrome. In Alagille Syndrome; Springer: Cham, Switzerland, 2018; pp. 167–193. [Google Scholar]
- Vandriel, S.; Wang, J.-S.; Li, L.; Piccoli, D.A.; Loomes, K.M.; Sokal, E.; Demaret, T. Clinical features and outcomes in an international cohort of 731 Alagille syndrome patients from 19 countries. Hepatology 2019, 70, 55A–56A. [Google Scholar] [CrossRef]


{kind=link}
| Disease Type | Cause |
|---|---|
| Genetic | Alagille syndrome |
| Trisomy 21 | |
| Williams syndrome | |
| Peroxisomal disorders | |
| Metabolic | α1-antitrypsin deficiency |
| Cystic fibrosis | |
| Panhypopituitarism | |
| Infections | Congenital cytomegalovirus |
| Congenital rubella | |
| Congenital syphilis | |
| Immune/Inflammatory disorders | Hemophagocytic lymphohistiocytosis |
| Sclerosing cholangitis | |
| Graft-versus-host disease | |
| Chronic allograft rejection | |
| Other | Drug-associated vanishing bile duct syndrome |
| Biliary atresia (late finding) |
| Medication Class | Medication | Side Effect Profile |
|---|---|---|
| 1st line: Choleretics | Ursodeoxycholic acid | Generally safe; Diarrhea, abdominal pain, vomiting |
| Bile salt-binding agents | Cholestyramine | Constipation, abdominal pain, worsening FSVD, poor palatability |
| 2nd line: Bile acid hydroxylation | Rifampin | Red discoloration of bodily fluids (sweat, tears), vomiting, hepatitis, idiosyncratic hypersensitivity reaction |
| 3rd line: Opioid antagonists | Naltrexone | Limited data; abdominal pain, nausea, irritability, diarrhea |
| 4th line (not yet approved by regulators): Intestinal bile acid transport (IBAT) inhibitors | Maralixibat Odevixibat | Limited data; vomiting, diarrhea, abdominal pain, rash, hepatitis, FSV deficiencies |
| Adjunctive therapy: | ||
| Antihistamines | Diphenhydramine | Drowsiness |
| SSRI | Sertraline | Limited data; agitation, alopecia and drug eruption, vomiting, hypertension |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayoub, M.D.; Kamath, B.M. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics 2020, 10, 907. https://doi.org/10.3390/diagnostics10110907
Ayoub MD, Kamath BM. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics. 2020; 10(11):907. https://doi.org/10.3390/diagnostics10110907
Chicago/Turabian StyleAyoub, Mohammed D., and Binita M. Kamath. 2020. "Alagille Syndrome: Diagnostic Challenges and Advances in Management" Diagnostics 10, no. 11: 907. https://doi.org/10.3390/diagnostics10110907