
In Silico Subtractive Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pneumoniae Strain D39
 , , ,
, , ,
Abstract
:1. Introduction
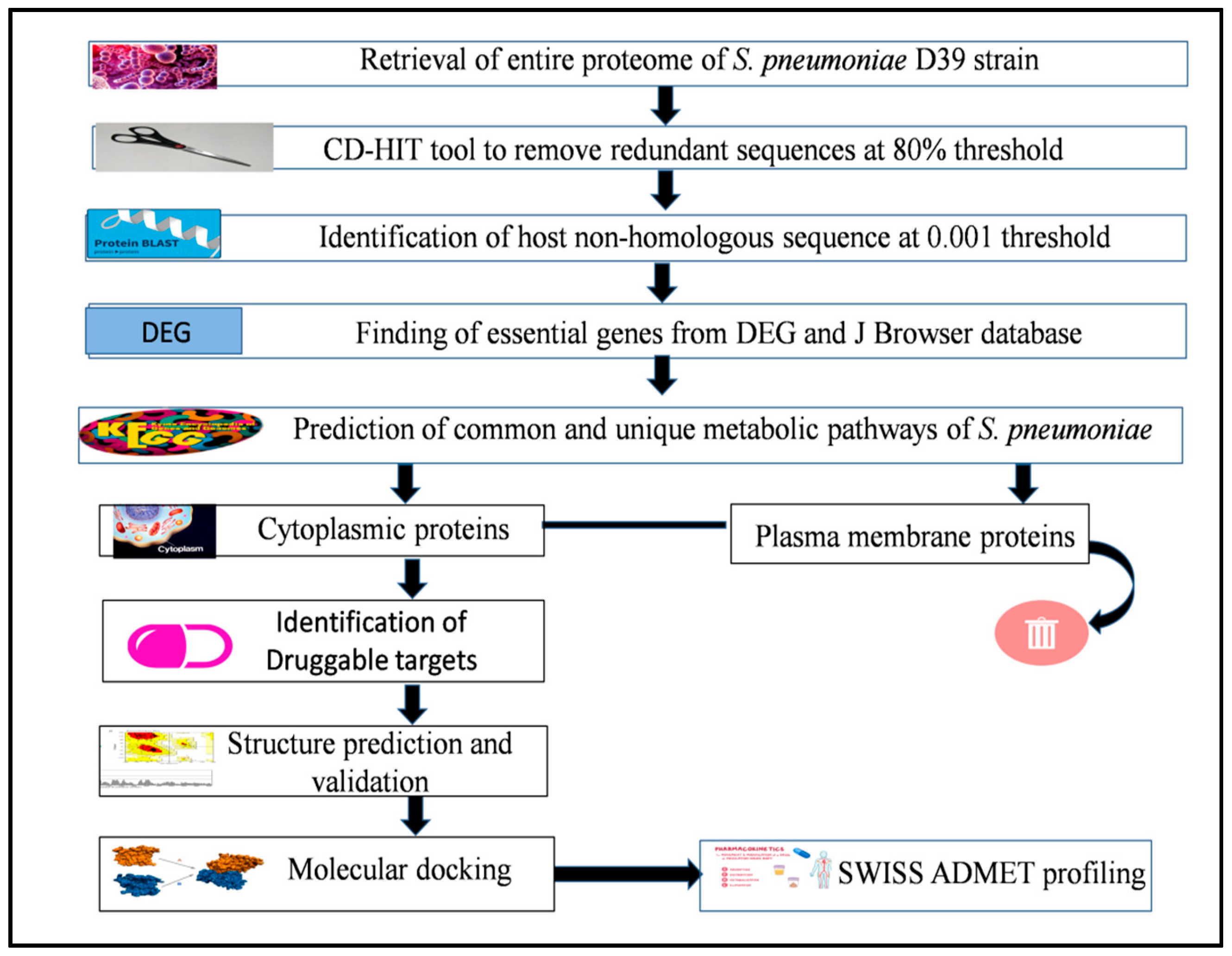
2. Methodology
2.1. Retrieval of Proteome
2.2. Removal of Redundant Sequences
2.3. Elimination of Homologous Protein Sequences
2.4. Determination of Essential Proteins of Pathogen
2.5. Finding of Metabolic Pathways in S. pneumoniae
2.6. Sub-Cellular Localization of Essential Proteins
2.7. Functional Classification of Essential Hypothetical Proteins
2.8. Identification of Druggable Targets
2.9. Structure Prediction and Validation
2.10. Molecular Docking and Visualization
2.11. Druglikness and Toxicity Prediction
2.12. MD Simulation
2.13. Binding Free Energy Calculations
3. Results
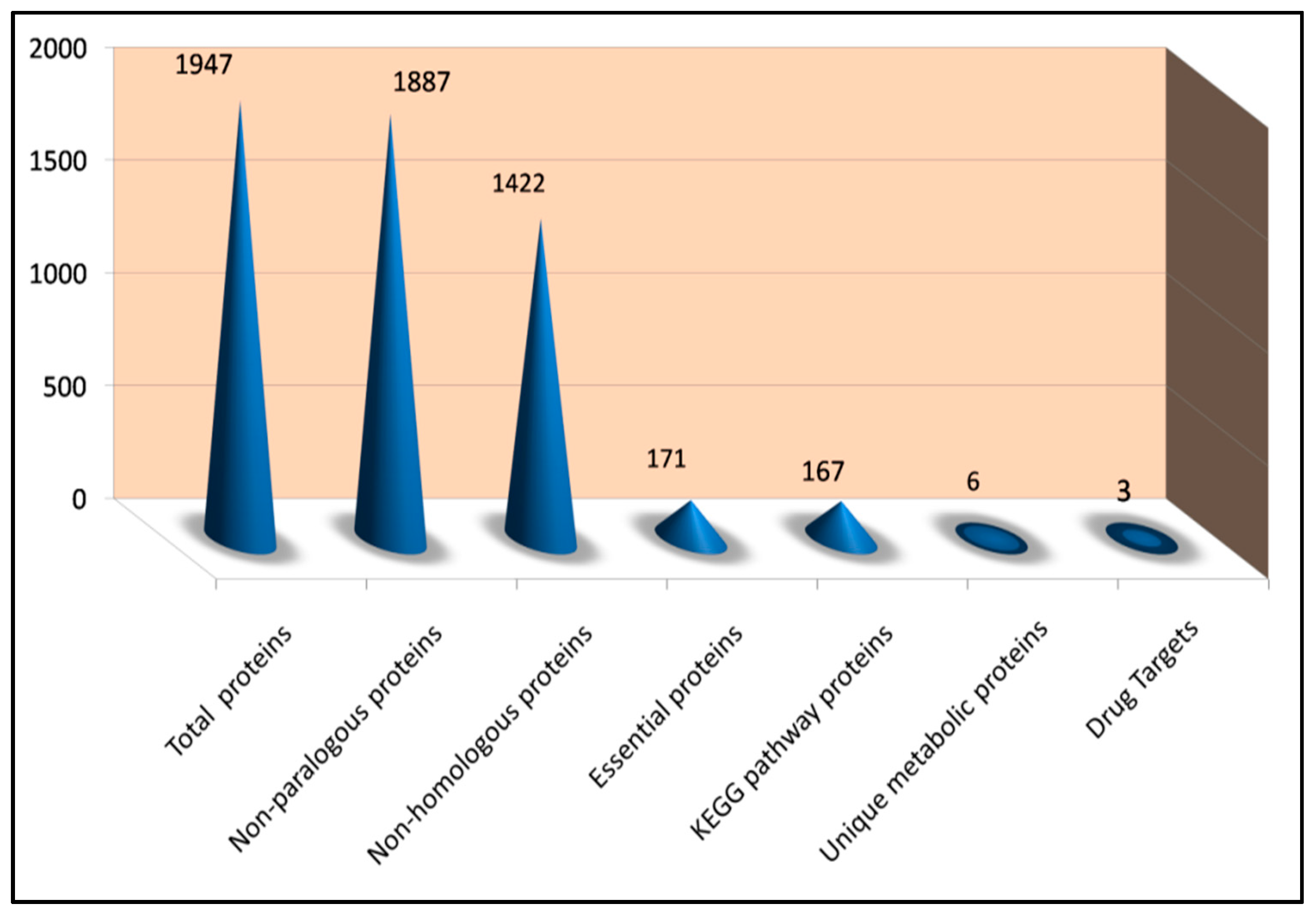
3.1. Detection of Non-Paralogous Sequences
3.2. Identification of Non-Homologous Protein Sequences
3.3. Identification of Essential Proteins
3.4. Analysis of Metabolic Pathways
3.5. Analysis and Exploration of Localization Results
3.6. Drug Ability Analysis
3.7. Analysis of Structure Prediction and Validation
3.8. Protein Multiple Ligands Docking
3.9. ADMET Profiling and Toxicity Prediction
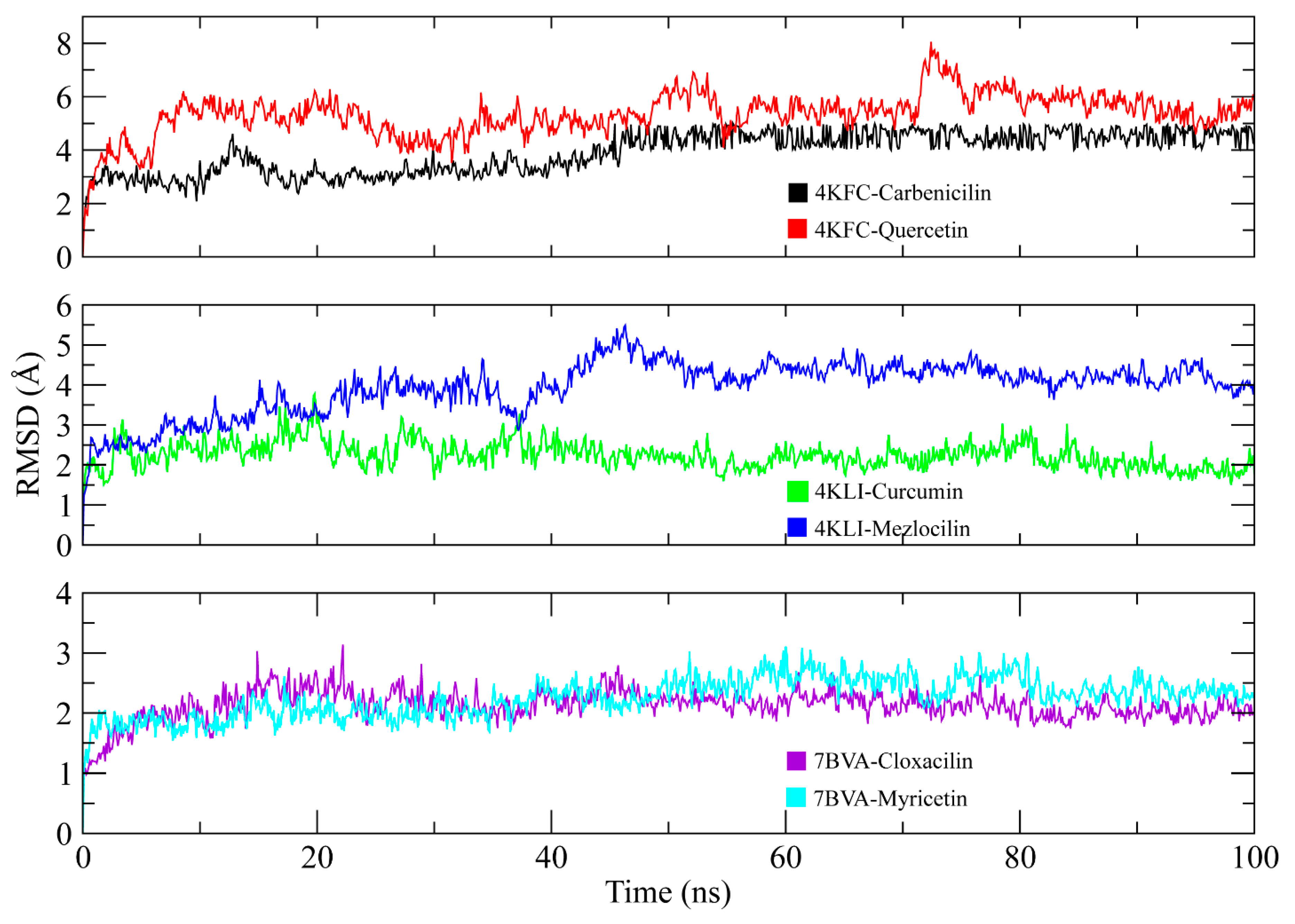
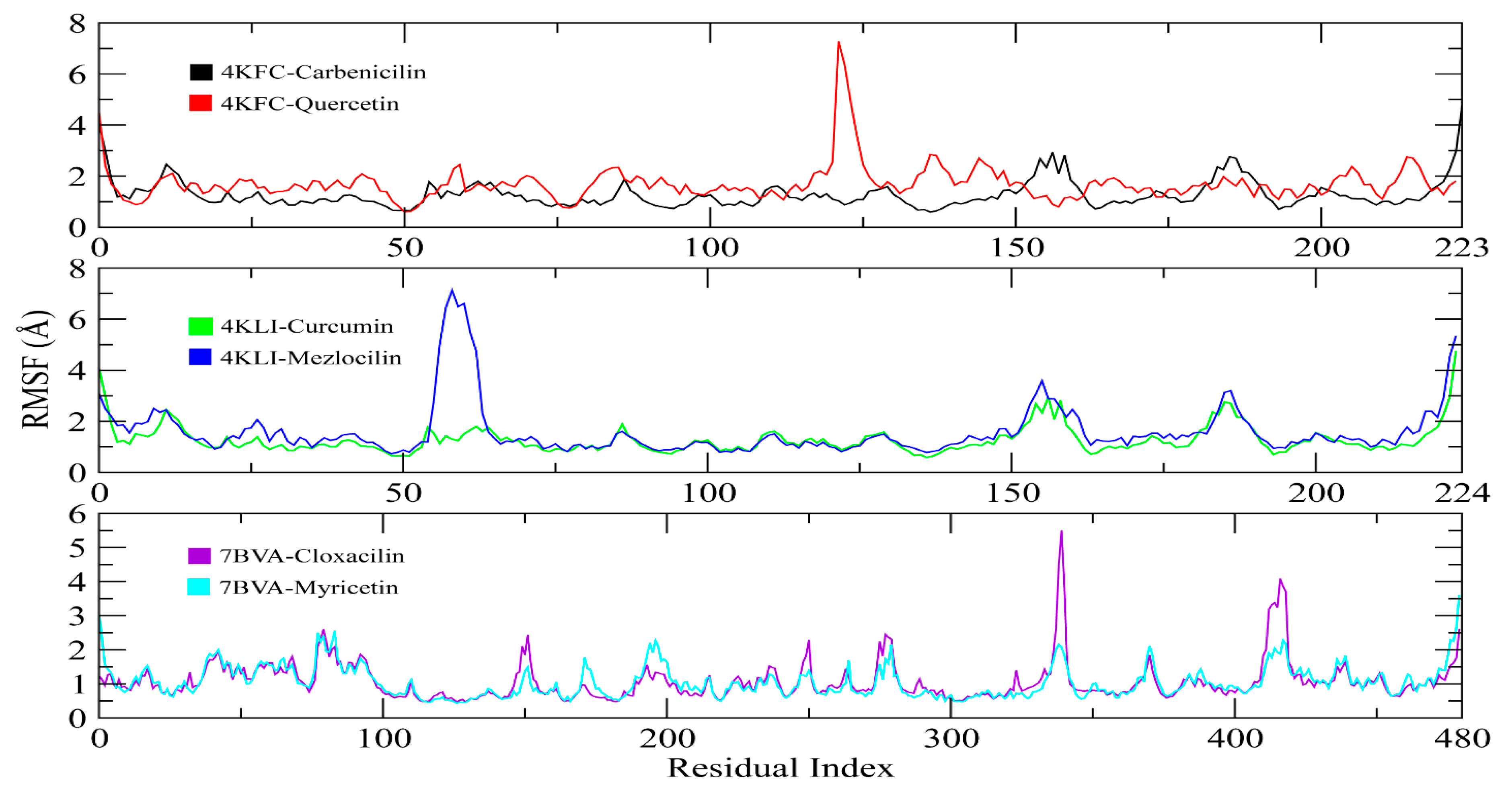
3.10. MD Simulation
3.11. MM/GBSA
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woolhouse, M.E.J. Population biology of emerging and re-emerging pathogens. Trends Microbiol. 2002, 10, s3–s7. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J. Environmental and social influences on emerging infectious diseases: Past, present and future. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 2004, 359, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- McMichael, A.J.; Woodruff, R.E.; Hales, S. Climate change and human health: Present and future risks. Lancet 2006, 367, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M. Infections caused by Streptococcus pneumoniae: Clinical spectrum, pathogenesis, immunity, and treatment. Clin. Infect. Dis. 1992, 14, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Panwhar, S.; Fiedler, B.A. Upstream policy recommendations for pakistan’s child mortality problem. In Translating National Policy to Improve Environmental Conditions Impacting Public Health Through Community Planning; Springer: Berlin/Heidelberg, Germany, 2018; pp. 203–217. [Google Scholar]
- O’Brien, K.L.; Wolfson, L.J.; Watt, J.P.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Mulholland, K.; Levine, O.S.; Cherian, T.; et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 2009, 374, 893–902. [Google Scholar] [CrossRef]
- McIntosh, K. Community-acquired pneumonia in children. N. Engl. J. Med. 2002, 346, 429–437. [Google Scholar] [CrossRef]
- Sharew, B.; Moges, F.; Yismaw, G.; Abebe, W.; Fentaw, S.; Vestrheim, D.; Tessema, B. Antimicrobial resistance profile and multidrug resistance patterns of Streptococcus pneumoniae isolates from patients suspected of pneumococcal infections in Ethiopia. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 26. [Google Scholar] [CrossRef]
- Kaur, R.; Casey, J.R.; Pichichero, M.E. Emerging Streptococcus pneumoniae strains colonizing the nasopharynx in children after 13-valent (PCV13) pneumococcal conjugate vaccination in comparison to the 7-valent (PCV7) era, 2006–2015. Pediatr. Infect. Dis. J. 2016, 35, 901. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, V.C.; Cervi, M.C.; Cintra, O.A.L.; Salathiel, A.S.M.; Gomes, A.C.L.F. Nasopharyngeal colonization with Streptococcus pneumoniae in children infected with human immunodeficiency virus. J. Pediatr. (Rio. J.) 2006, 82, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Chewapreecha, C.; Harris, S.R.; Croucher, N.J.; Turner, C.; Marttinen, P.; Cheng, L.; Pessia, A.; Aanensen, D.M.; Mather, A.E.; Page, A.J.; et al. Dense genomic sampling identifies highways of pneumococcal recombination. Nat. Genet. 2014, 46, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.; Jalal, K.; Khan, A.; Al-Harrasi, A.; Uddin, R. Comparative Metabolic Pathways Analysis and Subtractive genomics Profiling to Prioritize Potential Drug Targets Against Streptococcus pneumoniae. Front. Microbiol. 2021, 12, 4384. [Google Scholar] [CrossRef]
- Saha, S.K.; Al Emran, H.M.; Hossain, B.; Darmstadt, G.L.; Saha, S.; Islam, M.; Chowdhury, A.I.; Foster, D.; Naheed, A.; El Arifeen, S.; et al. Streptococcus pneumoniae serotype-2 childhood meningitis in Bangladesh: A newly recognized pneumococcal infection threat. PLoS ONE 2012, 7, e32134. [Google Scholar] [CrossRef] [Green Version]
- Lanie, J.A.; Ng, W.-L.; Kazmierczak, K.M.; Andrzejewski, T.M.; Davidsen, T.M.; Wayne, K.J.; Tettelin, H.; Glass, J.I.; Winkler, M.E. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 2007, 189, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, M.; Suzuki, Y.; Garcia, I.E.; Fafutis-Morris, M.; Vargas-González, A.; Carreño-Martinez, C.; Fukushima, Y.; Nakajima, C. Possible mode of emergence for drug-resistant leprosy is revealed by an analysis of samples from Mexico. Jpn. J. Infect. Dis. 2010, 63, 412–416. [Google Scholar] [CrossRef]
- Wang, C.Y.; Chen, Y.H.; Fang, C.; Zhou, M.M.; Xu, H.M.; Jing, C.M.; Deng, H.L.; Cai, H.J.; Jia, K.; Han, S.Z.; et al. Antibiotic resistance profiles and multidrug resistance patterns of Streptococcus pneumoniae in pediatrics: A multicenter retrospective study in mainland China. Medicine 2019, 98, e15942. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wan, Y.; Lei, Y.; Zobel, J.; Verspoor, K. Evaluation of CD-HIT for constructing non-redundant databases. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 703–706. [Google Scholar]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Liu, X.; Gallay, C.; Kjos, M.; Domenech, A.; Slager, J.; Kessel, S.P.; Knoops, K.; Sorg, R.A.; Zhang, J.; Veening, J. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 2017, 13, 931. [Google Scholar] [CrossRef]
- Zhang, R.; Lin, Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009, 37 (Suppl. S1), D455–D458. [Google Scholar] [CrossRef] [Green Version]
- Slager, J.; Aprianto, R.; Veening, J.-W. Deep genome annotation of the opportunistic human pathogen Streptococcus pneumoniae D39. Nucleic Acids Res. 2018, 46, 9971–9989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36 (Suppl. S1), D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-S.; Cheng, C.-W.; Su, W.-C.; Chang, K.-C.; Huang, S.-W.; Hwang, J.-K.; Lu, C.-H. CELLO2GO: A web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33 (Suppl. S2), W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Sarkar, S.; Banerjee, A.; Chakraborty, N.; Soren, K.; Chakraborty, P.; Bandopadhyay, R. Structural-functional analyses of textile dye degrading azoreductase, laccase and peroxidase: A comparative in silico study. Electron. J. Biotechnol. 2020, 43, 48–54. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37 (Suppl. S2), W623–W633. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC- a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Mani, J.S.; Johnson, J.B.; Hosking, H.; Ashwath, N.; Walsh, K.B.; Neilsen, P.M.; Broszczak, D.A.; Naiker, M. Antioxidative and therapeutic potential of selected Australian plants: A review. J. Ethnopharmacol. 2021, 268, 113580. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Al-Dhahli, A.S.; Al-Hassani, F.A.; Alarjani, K.M.; Yehia, H.M.; Al Lawati, W.M.; Azmi, S.N.H.; Khan, S.A. Essential oil from the rhizomes of the Saudi and Chinese Zingiber officinale cultivars: Comparison of chemical composition, antibacterial and molecular docking studies. J. King Saud Univ. 2020, 32, 3343–3350. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [Green Version]
- Wadhwani, A.; Khanna, V. In silico identification of novel potential vaccine candidates in Streptococcus pneumoniae. Glob. J Technol. Optim. 2016, 7, 2. [Google Scholar]
- Asalone, K.C.; Nelson, M.M.; Bracht, J.R. Novel sequence discovery by subtractive genomics. J. Vis. Exp. 2019, 143, e58877. [Google Scholar]
- Verhagen, L.M.; De Jonge, M.I.; Burghout, P.; Schraa, K.; Spagnuolo, L.; Mennens, S.; Eleveld, M.J.; Jongh, C.E.V.D.G.-D.; Zomer, A.; Hermans, P.W.M.; et al. Genome-wide identification of genes essential for the survival of Streptococcus pneumoniae in human saliva. PLoS ONE 2014, 9, e89541. [Google Scholar] [CrossRef]
- Elzek, M.A.W.; Christopher, J.A.; Breckels, L.M.; Lilley, K.S. Localization of Organelle Proteins by Isotope Tagging: Current status and potential applications in drug discovery research. Drug Discov. Today Technol. 2021, 39, 57–67. [Google Scholar] [CrossRef]
- Montanaro, N.; Vaccheri, A.; Magrini, N.; Battilana, M. Faimaguida: A databank for the analysis of the italian drug market and drug utilization in general practice. Eur. J. Clin. Pharmacol. 1992, 42, 395–399. [Google Scholar] [CrossRef]
- Uddin, R.; Arif, A.; Zahra, N.-A.; Sufian, M. Comparative proteome-wide study for in-silico identification and characterization of indispensable hypothetical proteins of food bornepathogen Campylobacter jejuni (CJJ) by Subtractive genomics approach. Pak. J. Pharm. Sci. 2021, 34, 1359–1367. [Google Scholar]
- Alamri, M.A.; Mirza, M.U.; Adeel, M.M.; Ashfaq, U.A.; Qamar, M.T.U.; Shahid, F.; Ahmad, S.; Alatawi, E.A.; Albalawi, G.M.; Allemailem, K.S.; et al. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals 2022, 15, 659. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, M.; Adeniji, S.E.; Arthur, D.E.; Haruna, A. Homology modeling and molecular docking simulation of some novel imidazo [1, 2-a] pyridine-3-carboxamide (IPA) series as inhibitors of Mycobacterium tuberculosis. J. Genet. Eng. Biotechnol. 2021, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Durán-Iturbide, N.A.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. In silico ADME/Tox profiling of natural products: A focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef]
- Maurya, S.; Akhtar, S.; Siddiqui, M.H.; Khan, M.K.A. Subtractive Proteomics for Identification of Drug Targets in Bacterial Pathogens: A Review. Int. J. Eng. Res. Technol. 2020, 9, 2278-0181. [Google Scholar]
- Sarangi, A.N.; Lohani, M.; Aggarwal, R. Proteome mining for drug target identification in Listeria monocytogenes strain EGD-e and structure-based virtual screening of a candidate drug target penicillin binding protein 4. J. Microbiol. Methods 2015, 111, 9–18. [Google Scholar] [CrossRef]
- Azam, S.S.; Shamim, A. An insight into the exploration of druggable genome of Streptococcus gordonii for the identification of novel therapeutic candidates. Genomics 2014, 104, 203–214. [Google Scholar] [CrossRef]
- Umland, T.C.; Schultz, L.W.; MacDonald, U.; Beanan, J.M.; Olson, R.; Russo, T.A. In vivo-validated essential genes identified in Acinetobacter baumannii by using human ascites overlap poorly with essential genes detected on laboratory media. MBio 2012, 3, e00113-12. [Google Scholar] [CrossRef] [Green Version]
- Suresh, A.; Srinivasarao, S.; Khetmalis, Y.M.; Nizalapur, S.; Sankaranarayanan, M.; Sekhar, K.V.G.C. Inhibitors of pantothenate synthetase of Mycobacterium tuberculosis—A medicinal chemist perspective. RSC Adv. 2020, 10, 37098–37115. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Unique Pathways | Common Pathways |
|---|---|---|
| Bifunctional protein GlmU | Spd 00541 O-antigen nucleotide sugar biosynthesis | Spd 01250 Biosynthesis of nucleotide and Spd 00520 Amino sugar and nucleotide sugar metabolism. |
| RNA polymerase sigma factor | Spd 02040 Flagellar assembly | |
| UDP-N-acetylmuramoyl-L-alanyl-D-glutamate—L-lysine ligase | Spd 00550 Peptidoglycan biosynthesis | Spd 01100 Metabolic pathway |
| Single-stranded DNA-binding protein | Spd00550 Peptidoglycan biosynthesis | Spd03030 DNA replication, Spd03430 Mismatch repair, and Spd03440 Homologous recombination |
| Phospho-N-acetylmuramoyl-pentapeptide-transferase | Spd 01502 Vancomycin resistance and Spd00550 Peptidoglycan biosynthesis | Spd 01100 Metabolic pathway |
| Protein translocase subunit SecA | Spd 02024 Quorum sensing, Spd 03070 Bacterial secretion system and Spd 01110 Biosynthesis of secondary metabolites | Spd 03060 Protein export |
| Glycerol-3-phosphate Acyltransferase | Spd 01110 Biosynthesis of secondary metabolites | Spd 00561 Glycerolipid metabolism and Spd 00564 Glycerophospholipid metabolism |
| UDP-N-acetylmuramoyl-tripeptide—D-alanyl-D-alanine ligase | Spd 01502 Vancomycin resistance, Spd 00550 Peptidoglycan biosynthesis and Spd 00300 Lysine biosynthesis | Spd 01100 Metabolic pathway |
| DNA-binding response regulator | Spd 02020 Two-component system | |
| PTS system, IIB component, putative | Spd02060 PhosphoTransferase system (PTS) | |
| Capsular polysaccharide biosynthesis protein | Spd00541 O-antigen nucleotide sugar biosynthesis Spd00552 Teichoic acid biosynthesis | Spd01250 Biosynthesis of nucleotide sugars Spd01100 Metabolic pathways |
| UDP-N-acetylmuramate—L-alanine ligase | Spd 00550 Peptidoglycan biosynthesis | |
| Transcriptional regulator ComX1 (Transcriptional regulator ComX2) | Spd 02020 Two-component system Spd02024 Quorum sensing | |
| Acyl carrier protein (ACP) | Spd 01110 Biosynthesis of secondary metabolites, | Spd 01100 Metabolic pathways |
| Penicillin-binding protein 2B | Spd 00550 Peptidoglycan biosynthesis Spd 01501 beta-Lactam resistance | |
| Nitroreductase family protein | Spd 00541 O-antigen nucleotide sugar biosynthesis | Spd 01100 Metabolic pathways |
| Preprotein translocase, SecE subunit | Spd 02024 Quorum sensing Spd 03070 Bacterial secretion system | Spd 03060 Protein export |
| DNA polymerase III, delta subunit (EC 2.7.7.7) | Spd 03030 DNA replication, Spd 03430 Mismatch repair, and Spd 03440 Homologous recombination | |
| Replicative DNA helicase (EC 3.6.4.12) | Spd 03030 DNA replication | |
| Phosphomevalonate kinase (EC 2.7.4.2) | Spd 01110 Biosynthesis of secondary metabolites | Spd 00900 Terpenoid backbone biosynthesis |
| Lipid II isoglutaminyl synthase | Spd 00550 Peptidoglycan biosynthesis | Spd 00110 metabolic pathway |
| Acyltransferase domain protein | Spd 01110 Biosynthesis of secondary metabolites | Spd 00561Glycerolipid metabolism Spd 00564 Glycerophospholipid metabolism |
| Alanine racemase (EC 5.1.1.1) | Spd 01502 Vancomycin resistance | Spd 00470 D-Amino acid metabolism and Spd 00110 metabolic pathway |
| D-alanine—D-alanine ligase | Spd01502 Vancomycin resistance and Spd00550 Peptidoglycan biosynthesis | Spd01100 metabolic pathway |
| Isopentenyl-diphosphate delta-isomerase | Spd01110Biosynthesis of secondary metabolites | Spd00900Terpenoid backbone biosynthesis |
| S.R Num | Protein Name | Gene Name | Uniprot ID | Drug Bank ID | Location |
|---|---|---|---|---|---|
| 1 | DNA-binding response regulator | SPD_1085 | Q9A515 | DB01972 | Cytoplasm |
| 2 | UDP-N-acetylmuramate—L-alanine ligase | murC SPD_1349 | P45066 | DB01673 | Cytoplasm |
| 3 | RNA polymerase sigma factor SigA | rpoD SigA SPD_0958 | Q18BX5 | DB08874 | Cytoplasm |
| Proteins Name | ERRAT Quality Factor | VERIFY 3D Compatibility Score | PROCHECK Ramachandran | ProsA-Web |
|---|---|---|---|---|
| DNA binding response regulator | 92.82% | 96.87% compatibility score | Core 89.2% | −7.27 |
| Allowed 10.0% | ||||
| General 0.5% | ||||
| Disallowed 0.3% | ||||
| UDP-N-acetylmuramate—L-alanine ligase | 95.92% | 97.91% compatibility score | Core 93.2% | −12.31 |
| Allowed 6.6% | ||||
| General 0.1% | ||||
| Disallowed 0.0% | ||||
| RNA polymerase sigma factor | 81.18% | 66.16% compatibility factor | Core 88.7% | −6.02 |
| Allowed 10.1% | ||||
| General 0.1% | ||||
| Disallowed 0.3% |
| Drug Targets PDB ID | Dimension Angstrom X | Dimension Angstrom Y | Dimension Angstrom Z | Active Site Residues Obtained from Co-Factor Tool |
|---|---|---|---|---|
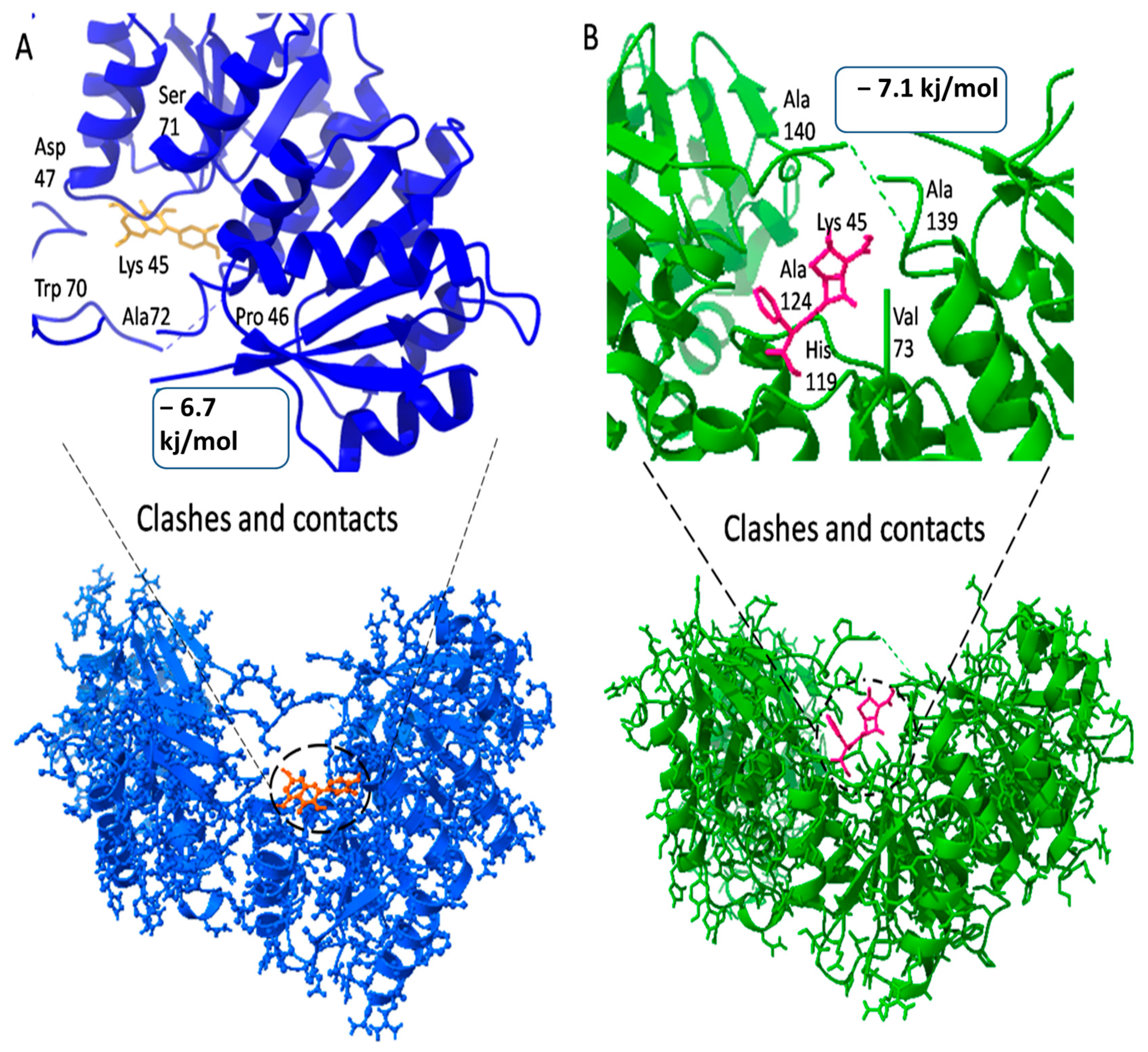
| 4KFC | 29.474 | 35.94 | 39.61 | Leu21, Ser35, Arg27, Leu34, Gln35, Leu38, Ala41, Lys45, Pro46, Trp70, Ser71 and Ala72 |
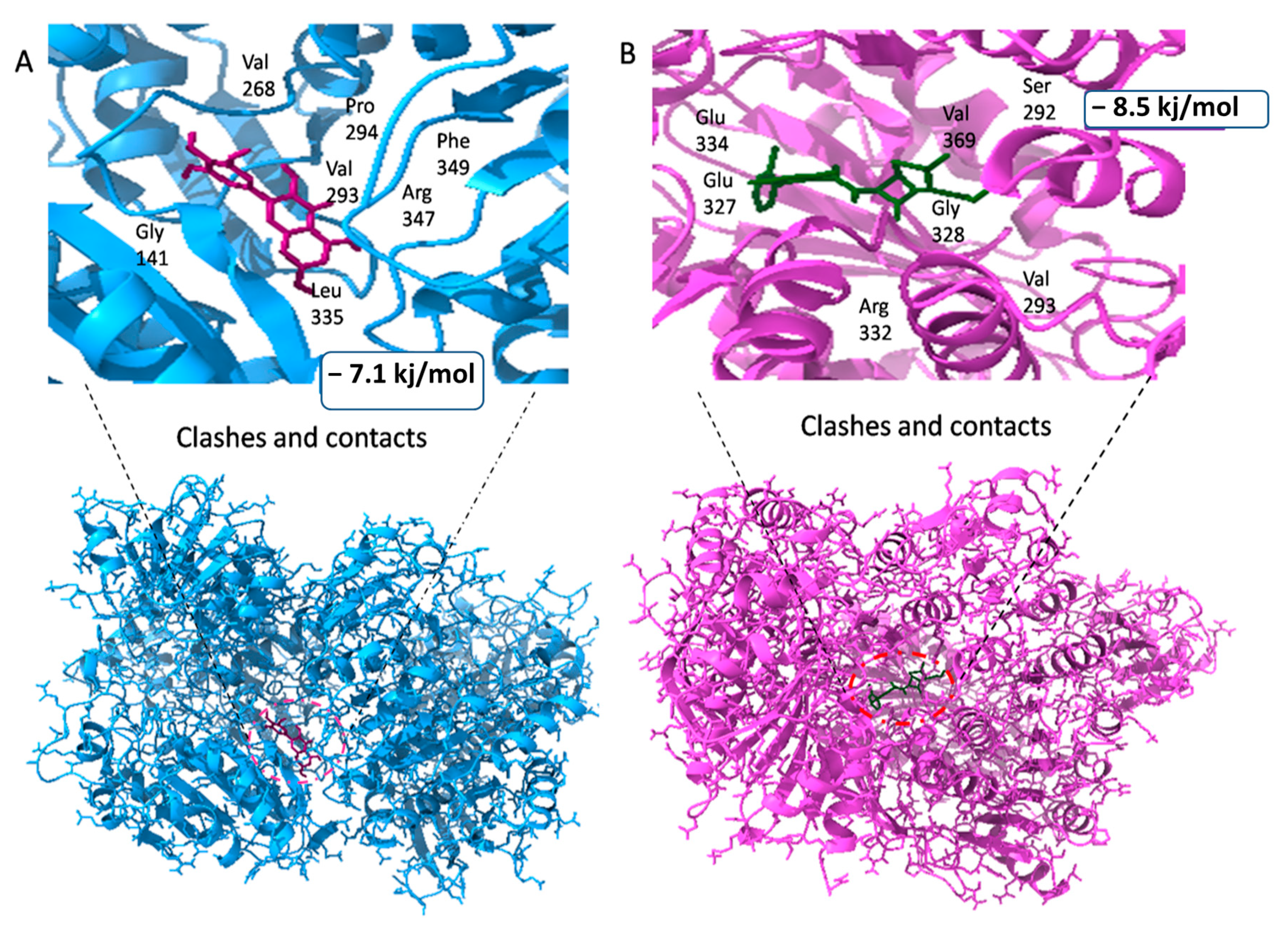
| 7BVA | 29.66 | 25 | 28.42 | Lys126, Thr121, Thr123, His124, Gly125, Lys126, Thr128, Ser131, Ile134, Val135, Gly141, Glu334, Glu327, Ser292 and His160 |
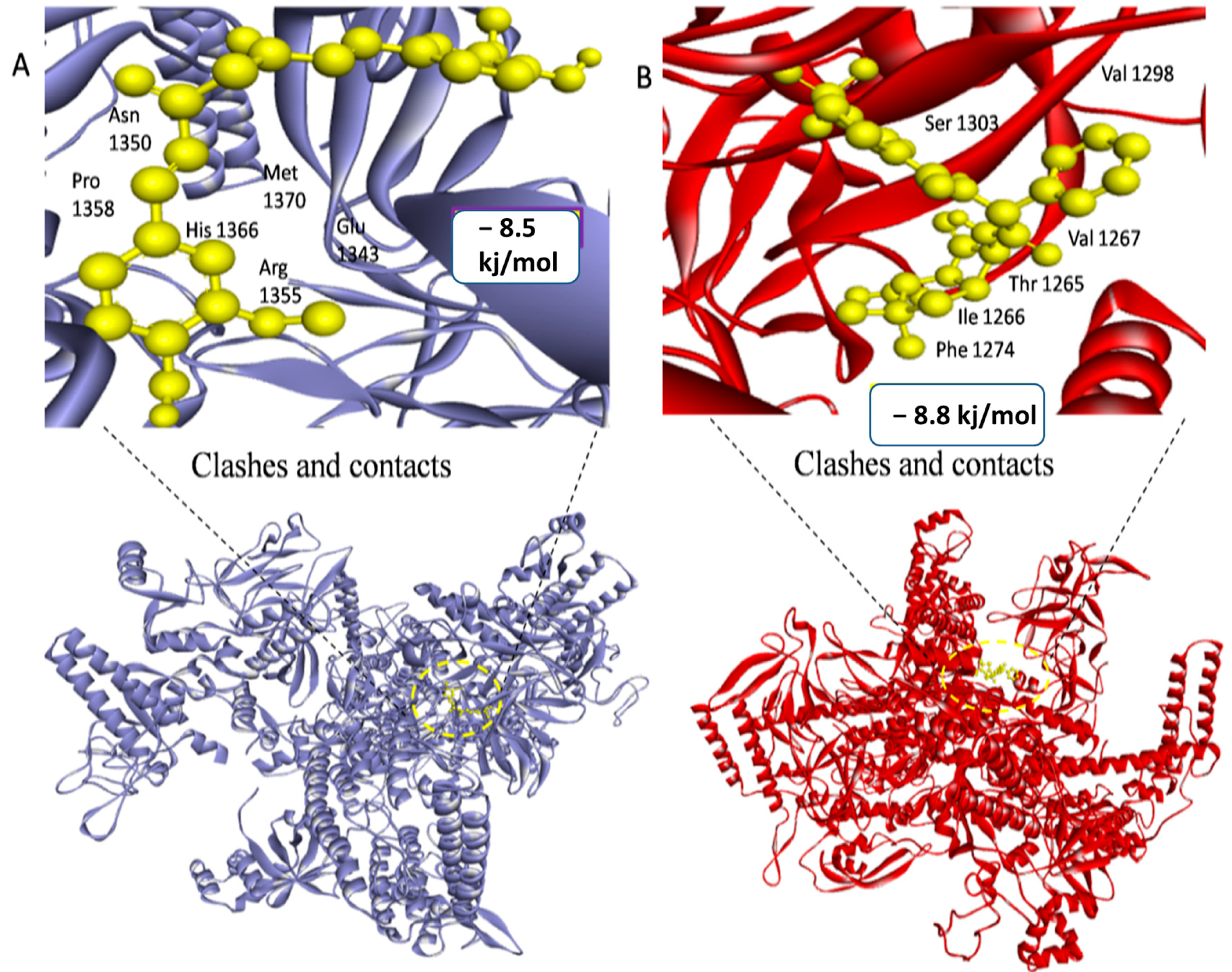
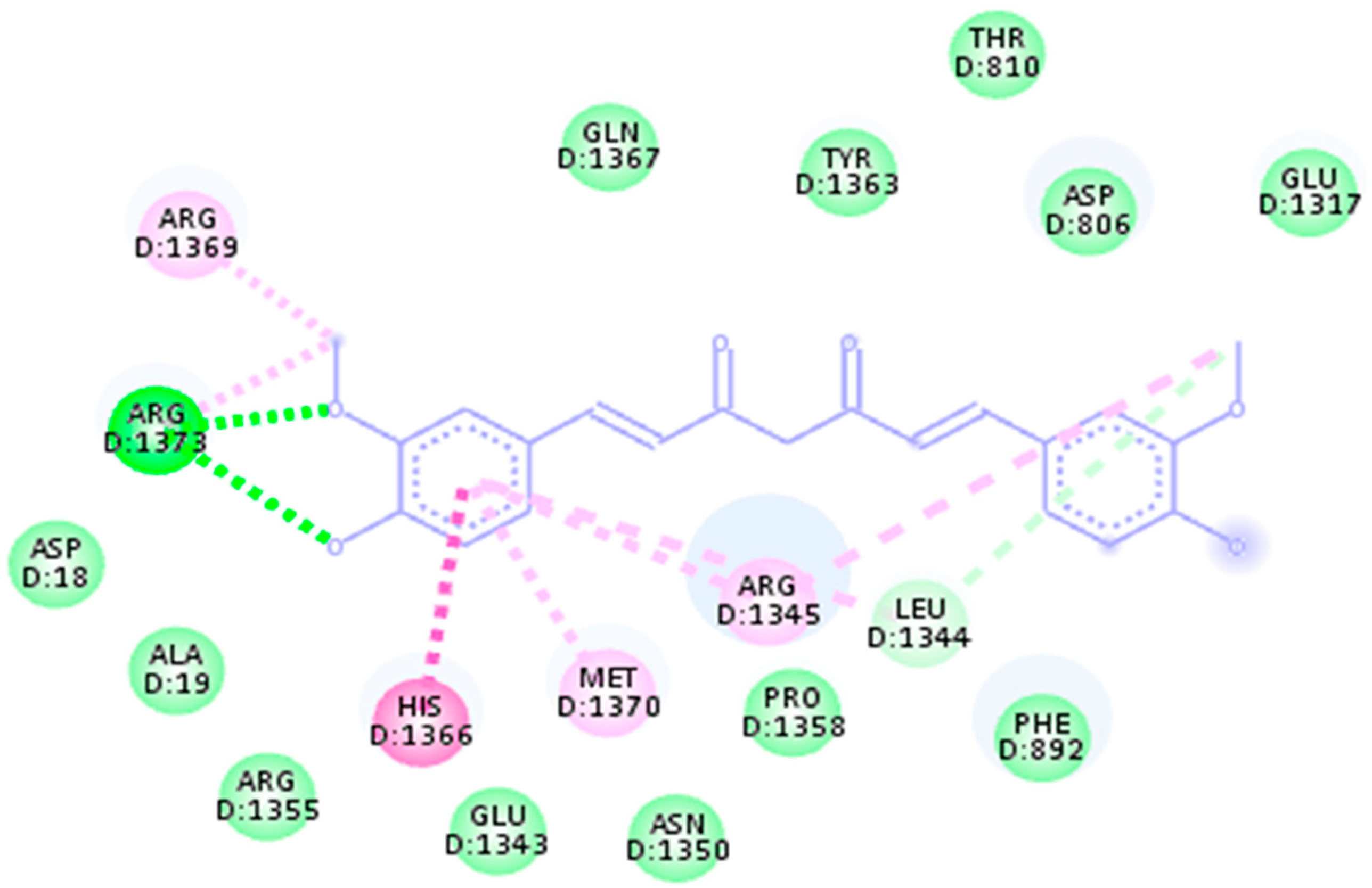
| 4LK1 | 149.36 | 152.76 | 157.24 | Ala42, Arg44, Ile46, leu48, Ser50, Pro52, Gly151, Ser178, Glu181, Ile183, Val185, Asn208, Thr210, Ile217, Arg219, Ala221, leu224, His1366, Met1370, Arg1355, Glu1343, Asn1350 and Pro1358 |
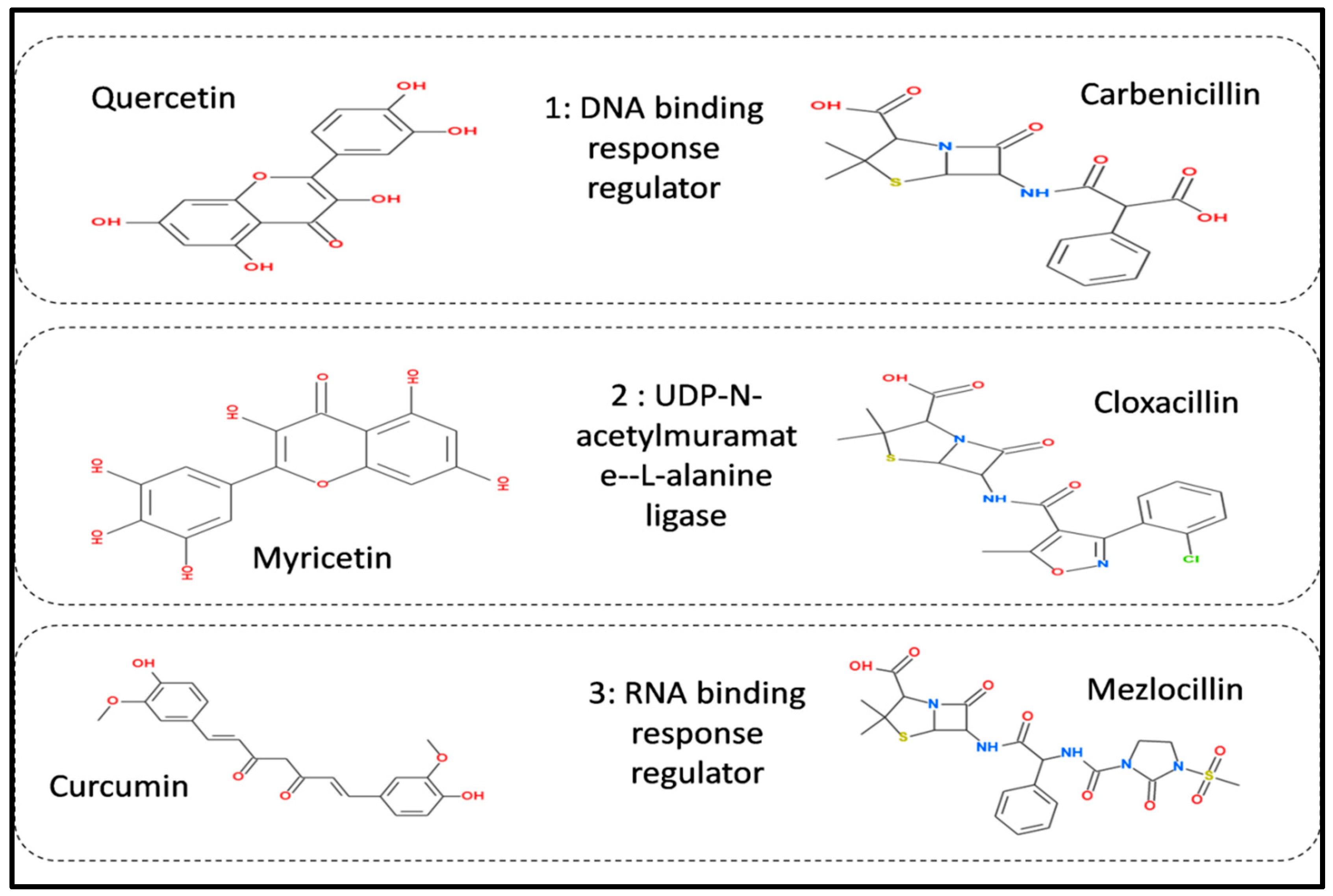
| 1: DNA Binding Response Regulator | ||||
| Compound ID | Compound Name | Binding Affinity | RMSD | Interacting Residues |
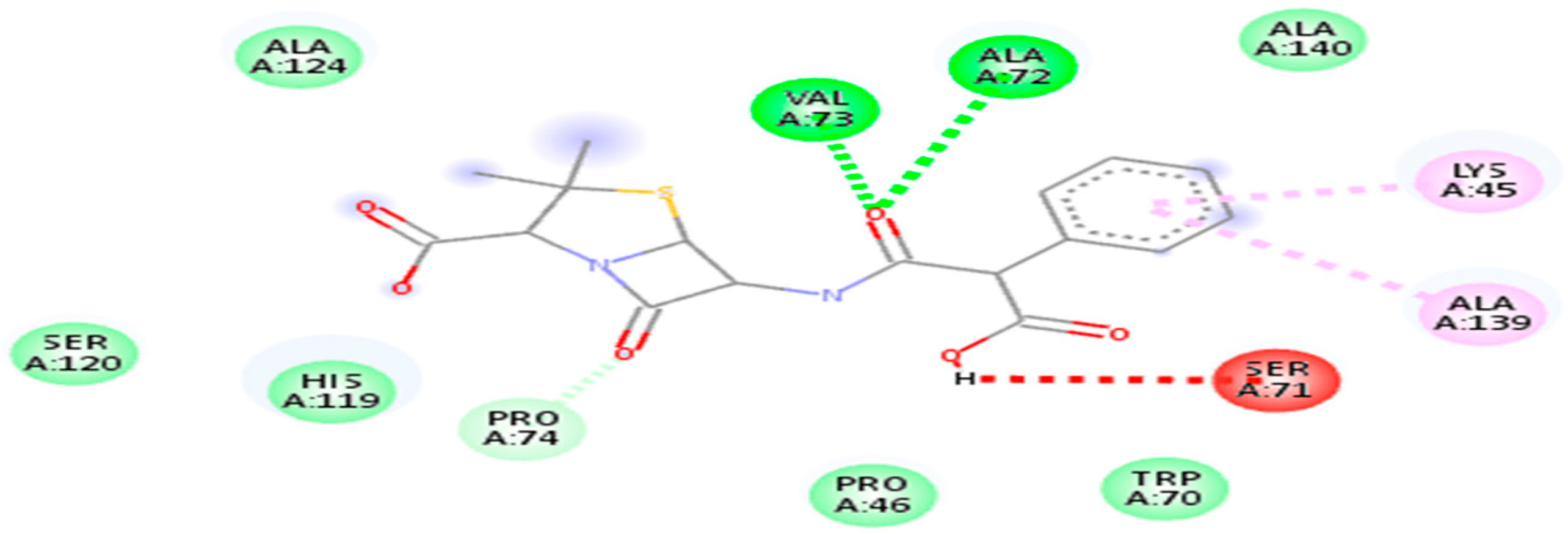
| 20824 | Carbenicillin | −6.7 kj/mol | 1.8 | Lys45, pro46, Trp70, Ser71, Ala72, Val73, pro74, His119, Ser120, Ala 124 and Ala 139 |
| 5280343 | Quercetin | −7.1 kj/mol | 1.72 | Lys45, Pro46, Asp47, Trp70, Ser71, Ala72, Val73, Pro74, Leu116, His119, Ser120, Ala139 and Ala140 |
| 2: UDP-N-acetylmuramate—L-alanine ligase | ||||
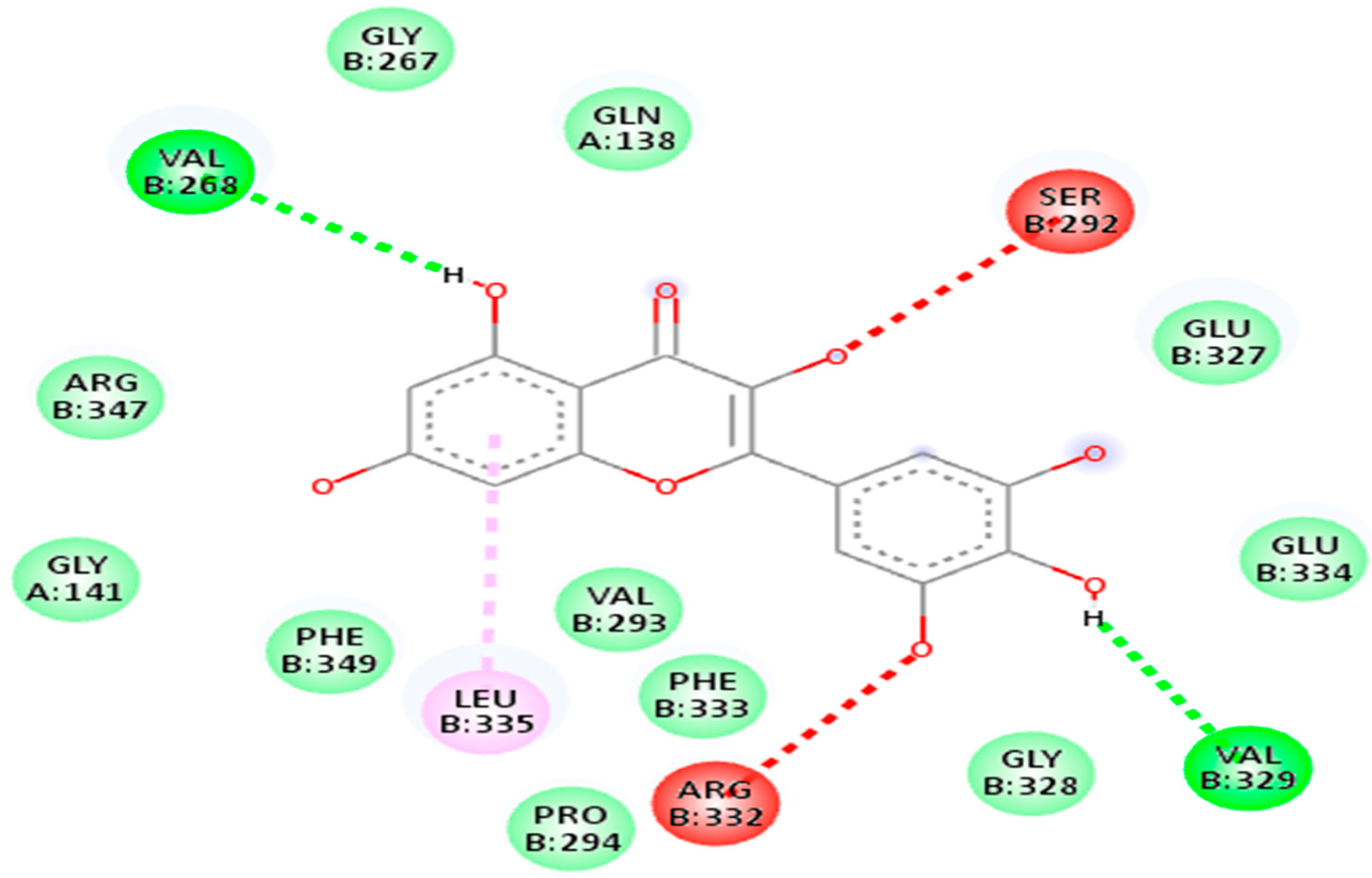
| 5281672 | Myricetin | −7.1 kj/mol | 1.63 | Arg332, Pro294, Phe333, Val293, Leu335, Phe349, Gly141, Arg347, Val268, Gly267, Gln138, Ser292, Glu327, Glu334, Val329 and Gly328 |
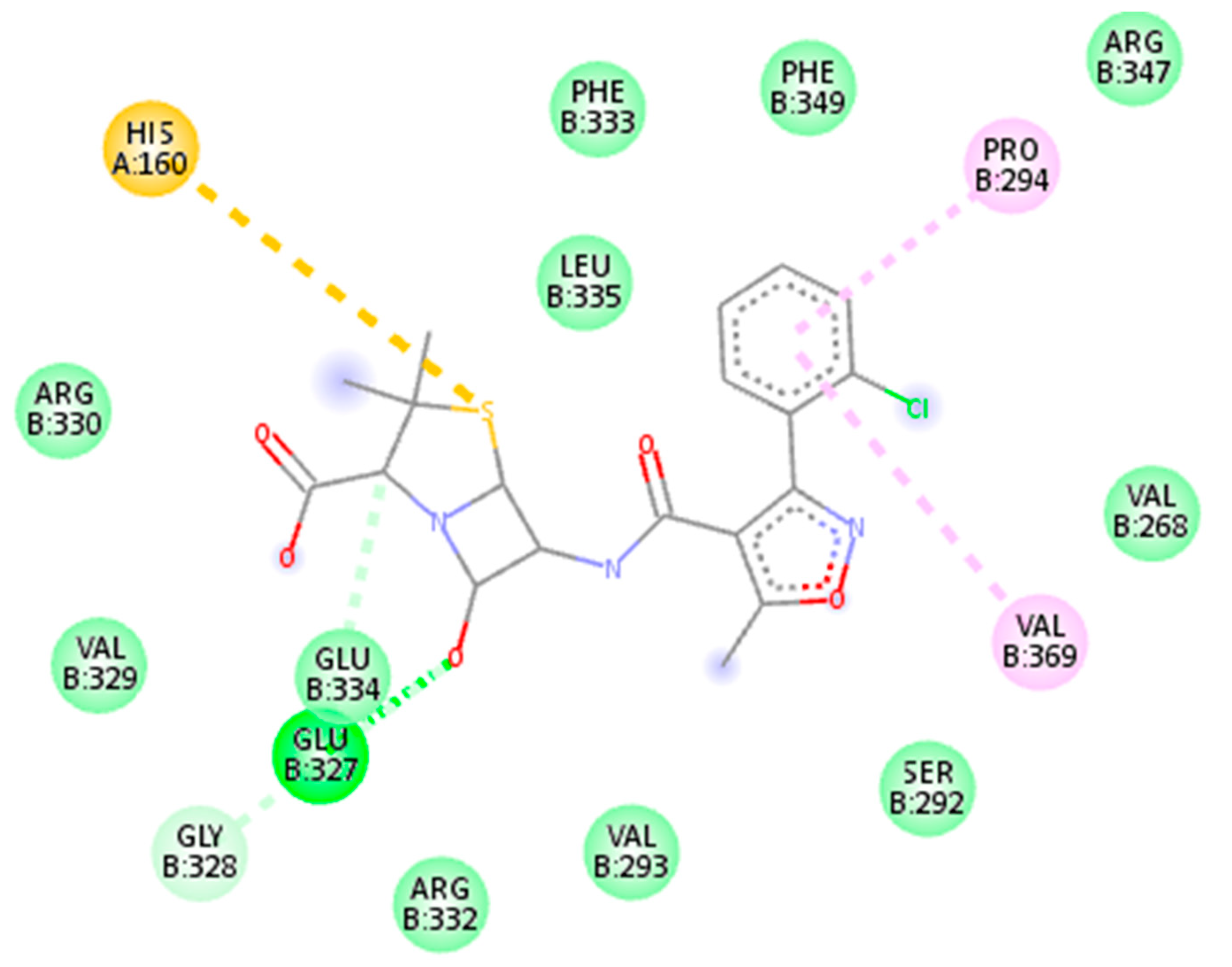
| 6098 | Cloxacillin | −8.5 kj/mol | 1.41 | Glu334, Glu327, Val329, Gly328, Arg332, Val293, Ser292, Val369, Val268, Arg330, His160, Leu335, Phe333, Phe349, Pro294 and Arg347 |
| 3: RNA Polymerase Sigma Factor | ||||
| 969516 | Curcumin | −8.5 kj/mol | 1.15 | His1366, Met1370, Arg1355, Glu1343, Asn1350, Pro1358, Leu1344, Phe892, Asp18, Ala19, Arg1373, Arg1369, Gln1367, Tyr1363, Thr810, Asp806 and Glu1317 |
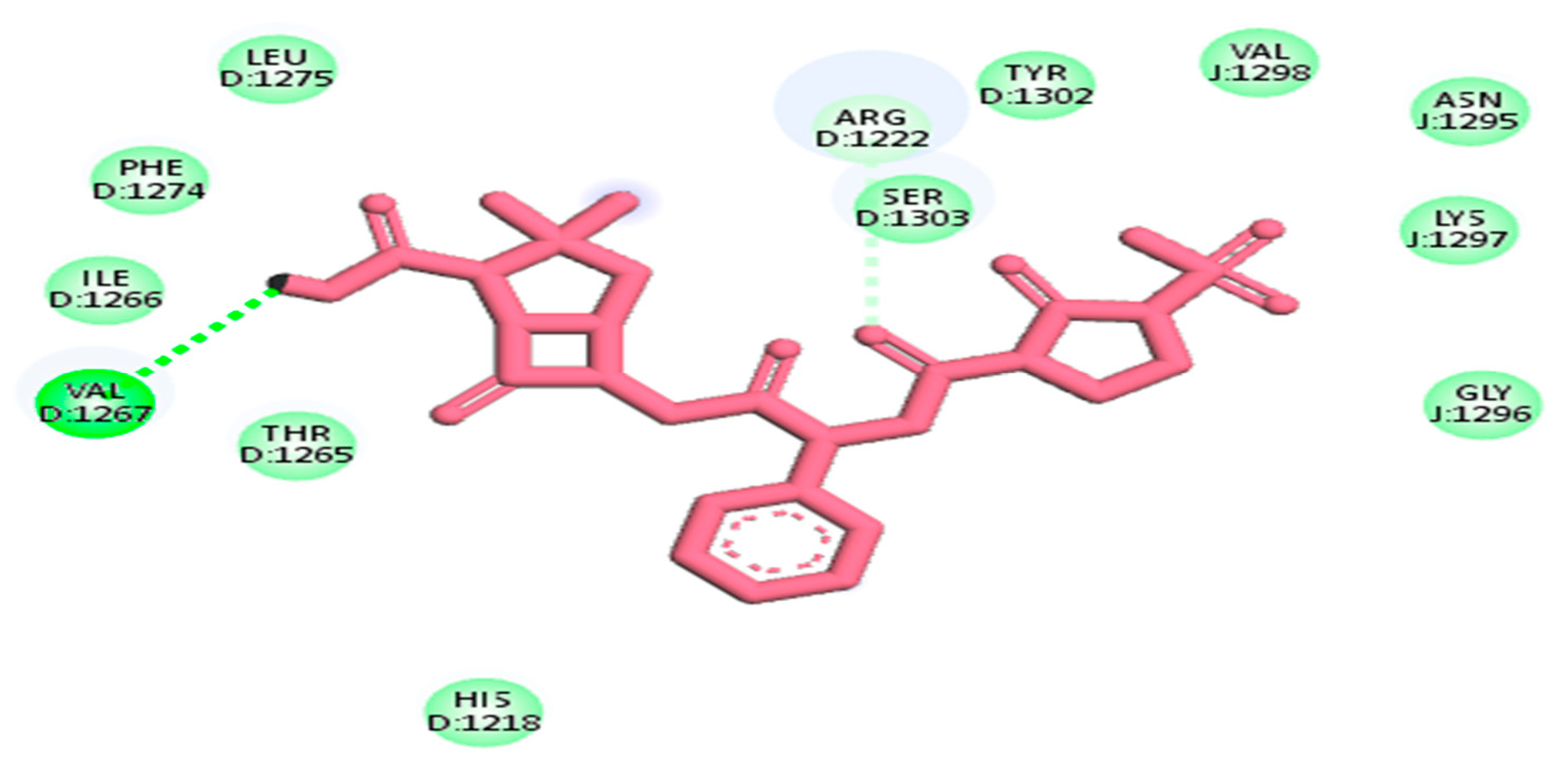
| 656511 | Mezlocillin | −8.8 kj/mol | 2.7 | Val1267, Thr1265, Ile1266, Phe1274, Leu1275, His1218, Arg1222, Ser1303, Tyr1302, Val1298, Asn1295, Lys1297 and Gly1296 |
| 1: DNA Binding Response Regulator | ||||
| Compound Ids | Molecular Weight | Hydrogen Bond Donor | Hydrogen Bond Acceptor | Oral Bio-Availability Score |
| 5280343 | 302.24 | 5 | 7 | 0.55 |
| 20824 | 378.4 | 3 | 6 | 0.56 |
| 2: UDP-N-acetylmuramate—L-alanine ligase | ||||
| 5281672 | 318.24 | 5 | 8 | 0.55 |
| 6098 | 435.88 | 2 | 6 | 0.56 |
| 3: RNA Polymerase Sigma Factor | ||||
| 969516 | 368.38 | 2 | 6 | 0.55 |
| 656511 | 499.5 | 3 | 8 | 0.55 |
| Target Proteins | ||||||
|---|---|---|---|---|---|---|
| Parameters | 1: DNA Binding Response Regulator | 2: UDP-N-acetylmuramate—L-alanine ligase | 3: RNA Polymerase Sigma Factor | |||
| ID 5280343 | ID 20824 | ID 5281672 | ID 6098 | ID 969516 | ID 656511 | |
| Absorption/Distribution | ||||||
| Blood-Brain Barrier | No | No | No | No | No | No |
| GI Absorption | High | Low | Low | High | High | High |
| p-gp substrate | No | Yes | No | Yes | No | Yes |
| Caco-2 permeability | −5.20 | −6.15 | −5.65 | −5.38 | −4.83 | −6.073 |
| Metabolism | ||||||
| CYP1A2 inhibitor | Yes | No | Yes | No | No | No |
| CYP2C19 inhibitor | No | No | No | Yes | No | No |
| CYP2C9 inhibitor | No | No | No | No | Yes | No |
| CYP2D6 inhibitor | Yes | No | No | No | No | No |
| CYP3A4 inhibitor | Yes | No | Yes | Yes | Yes | No |
| Toxicity | ||||||
| Cytotoxicity | In active | In active | In active | In active | In active | In active |
| Immuno-toxicity | In active | In active | In active | In active | In active | In active |
| AMES toxicity | Non-toxic | Non-toxic | Non-toxic | Non-toxic | Non-toxic | Non-toxic |
| Rat oral acute toxicity | Non-toxic | Non-toxic | Non-toxic | Non-toxic | Non-toxic | Non-toxic |
| Target | Compound | MMGBSA dG Bind | MMGBSA dG Bind Coulomb | MMGBSA dG Bind Covalent | MMGBSA dG Bind Solv GB | MMGBSA dG Bind vdW |
|---|---|---|---|---|---|---|
| 4KFC | Carbenicilin | −0.04115 | −0.5658100 | −0.0147365 | 0.56198971 | −0.0389674 |
| 4KFC | Quercetin | 0.064942 | −0.6541846 | 0.0019109 | 0.73494300 | −0.0163880 |
| 4KLI | Curcumin | −5.46 × 10−12 | 3.64 × 10−12 | 0 | −9.55 × 10−12 | −1.14 × 10−13 |
| 4KLI | Mezlocilin | −2.18 × 10−11 | 8.19 × 10−12 | −4.55 × 10−13 | −2.80 × 10−11 | −1.14 × 10−13 |
| 7BVA | Cloxacilin | −18.7583 | 42.80804 | 6.310527 | −6.20121 | −41.8799 |
| 7BVA | Myricetin | −30.5268 | −4.07858 | 4.33391 | 6.111314 | −17.6194 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shami, A.; Alharbi, N.K.; Al-Saeed, F.A.; Alsaegh, A.A.; Al Syaad, K.M.; Abd El-Rahim, I.H.A.; Mostafa, Y.S.; Ahmed, A.E. In Silico Subtractive Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pneumoniae Strain D39. Life 2023, 13, 1128. https://doi.org/10.3390/life13051128
Shami A, Alharbi NK, Al-Saeed FA, Alsaegh AA, Al Syaad KM, Abd El-Rahim IHA, Mostafa YS, Ahmed AE. In Silico Subtractive Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pneumoniae Strain D39. Life. 2023; 13(5):1128. https://doi.org/10.3390/life13051128
Chicago/Turabian StyleShami, Ashwag, Nada K. Alharbi, Fatimah A. Al-Saeed, Aiman A. Alsaegh, Khalid M. Al Syaad, Ibrahim H. A. Abd El-Rahim, Yasser Sabry Mostafa, and Ahmed Ezzat Ahmed. 2023. "In Silico Subtractive Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pneumoniae Strain D39" Life 13, no. 5: 1128. https://doi.org/10.3390/life13051128