

Novel Virus Identification through Metagenomics: A Systematic Review

 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
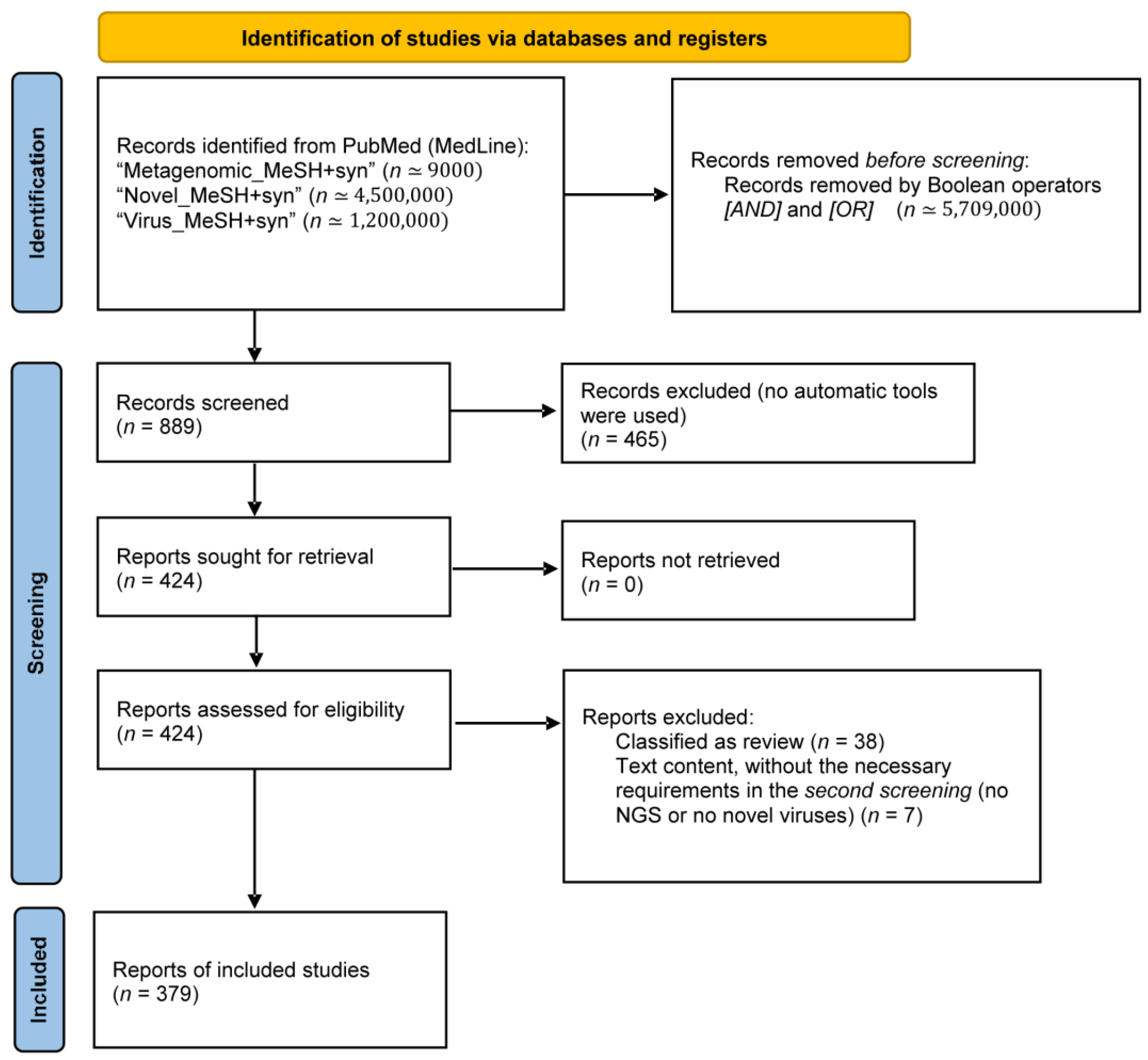
2.1. Search Strategy and Selection Criteria
2.2. Data Extraction and Analysis
3. Results
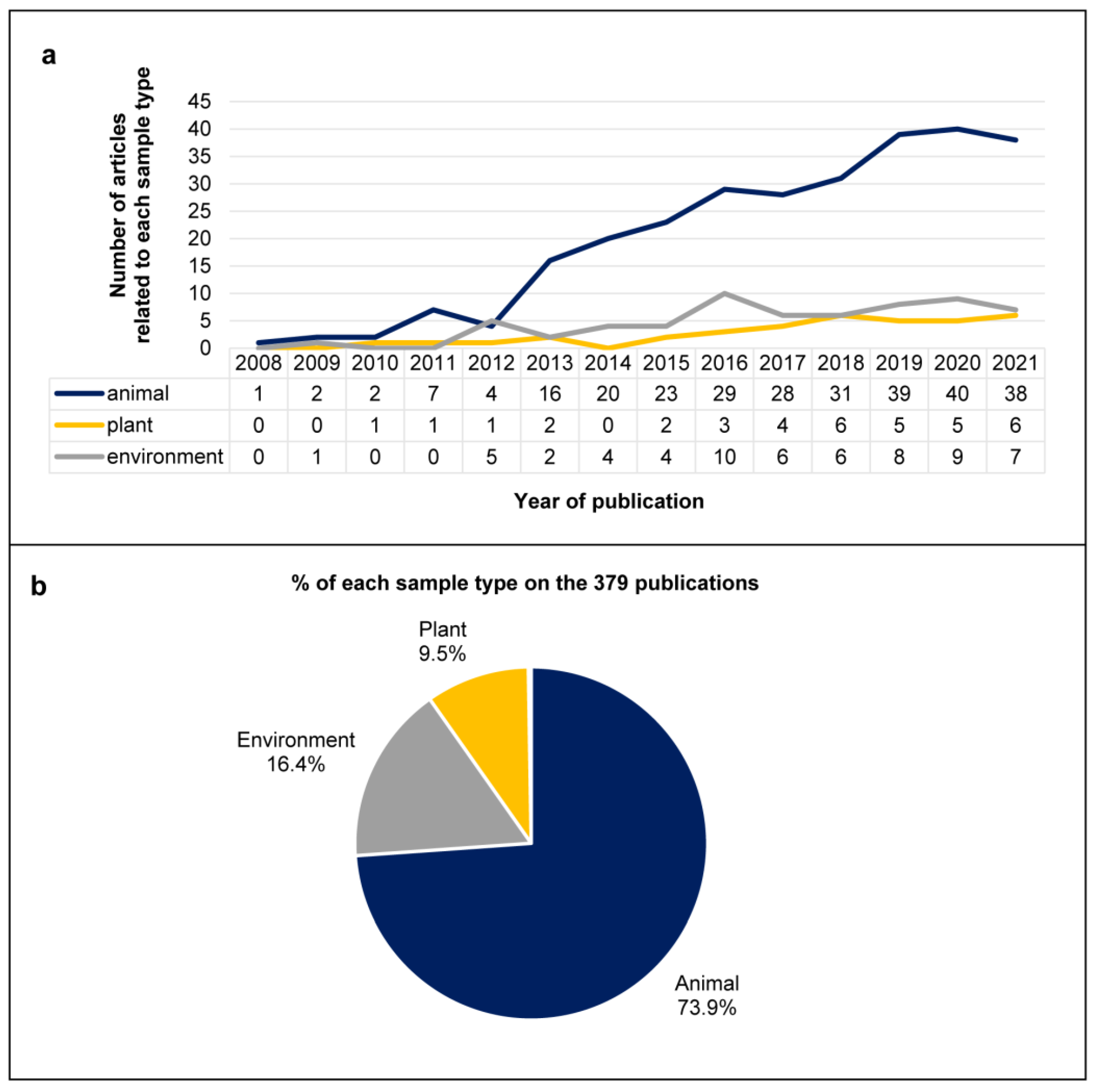
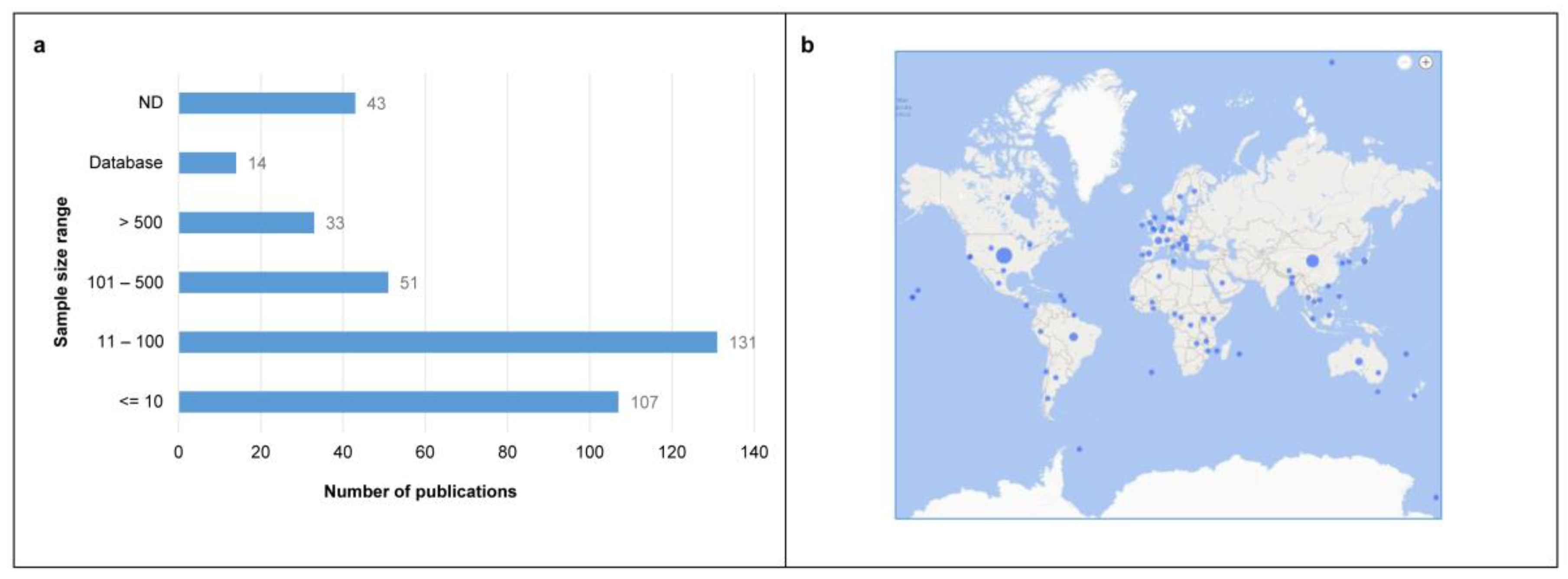
3.1. Literature Search and General Characteristics of the Included Studies
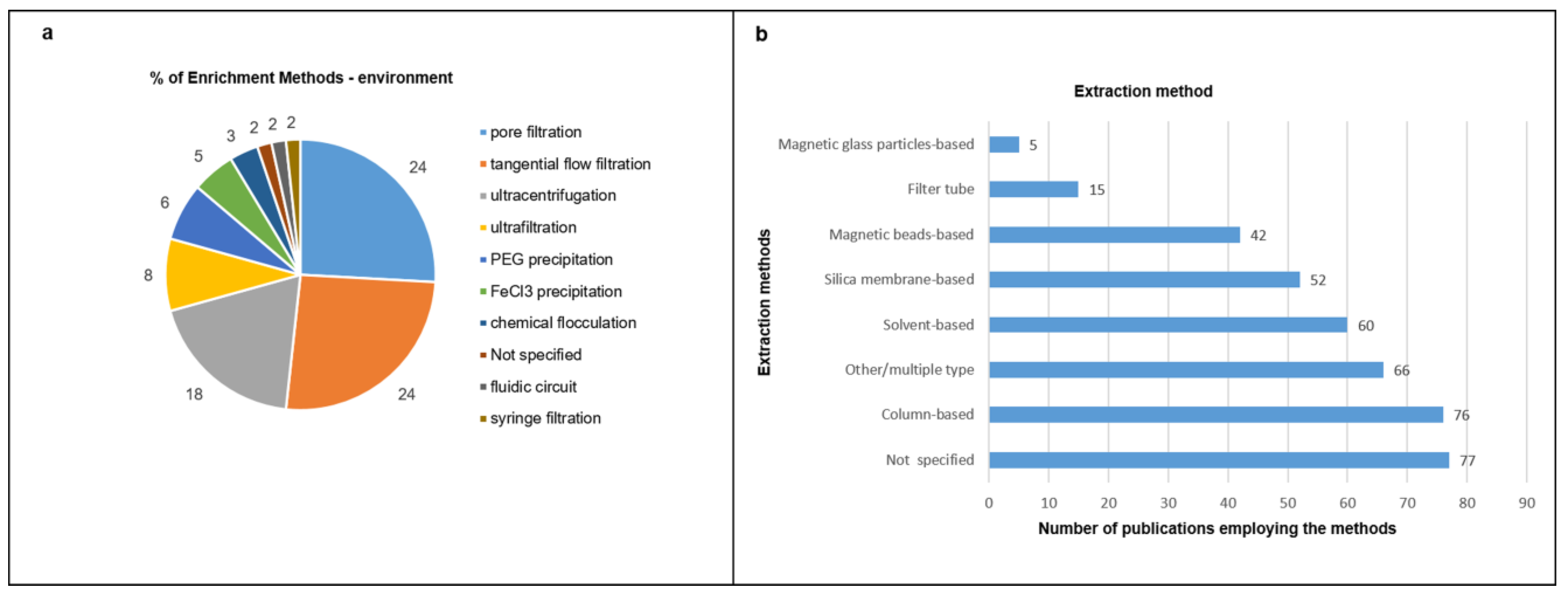
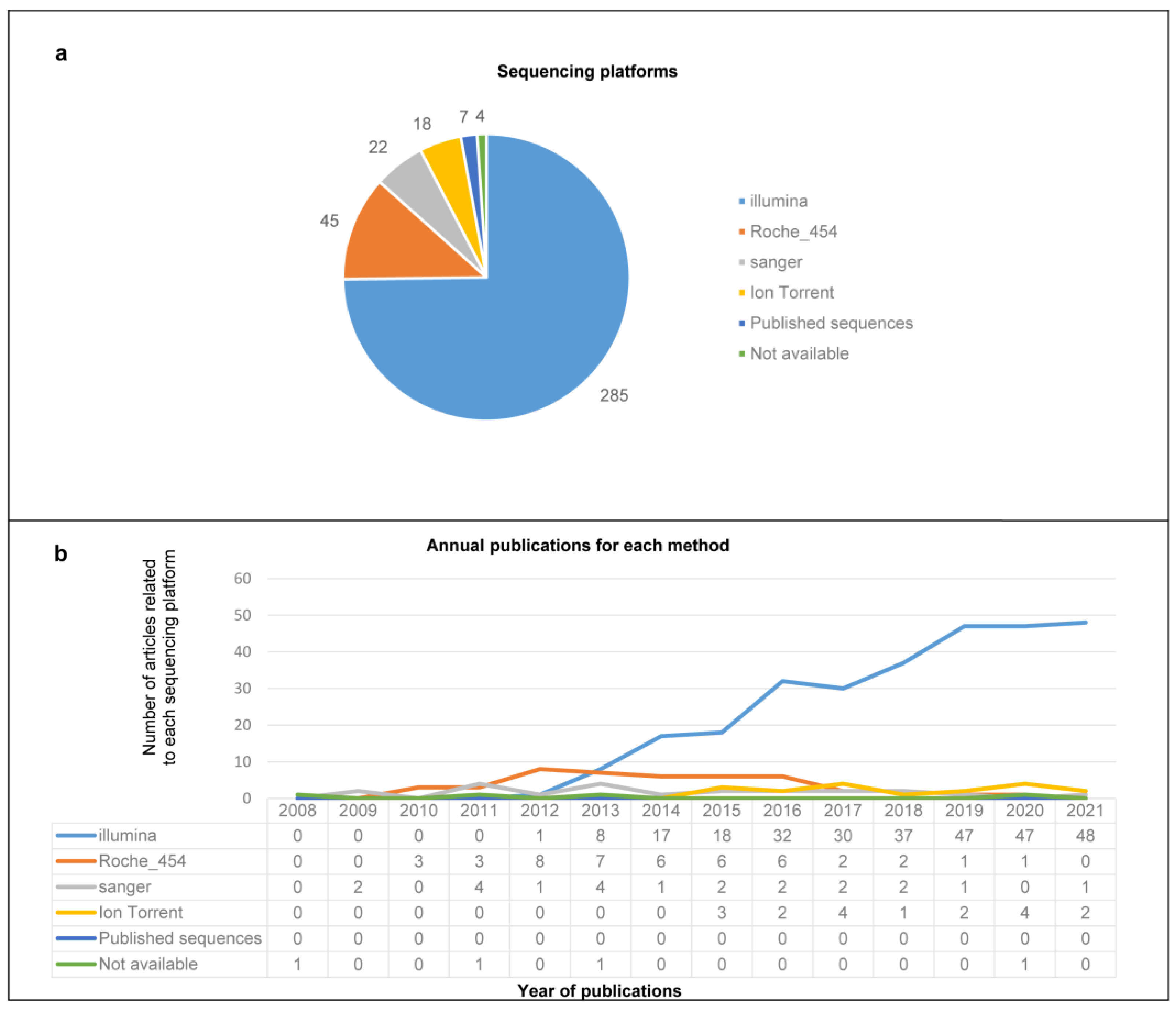
3.2. Enrichment Strategies, Nucleic Acid Purificationand Sequencing Platforms
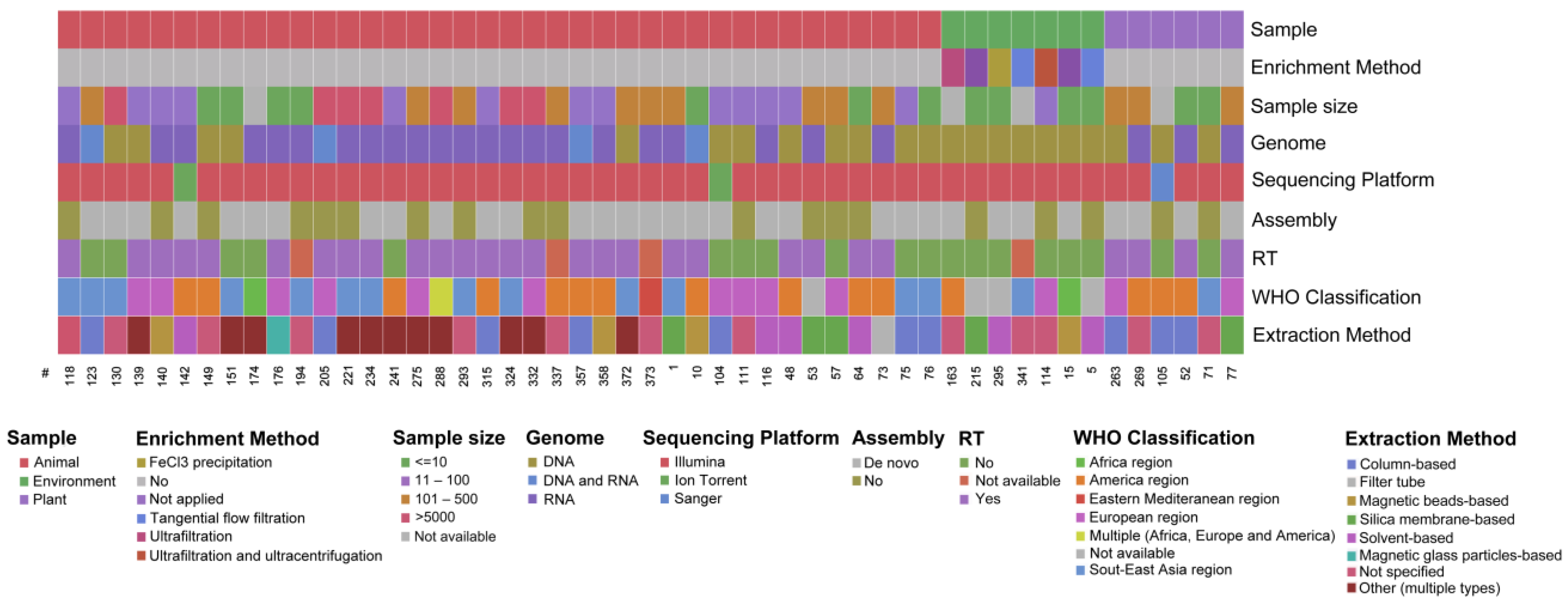
3.3. Overview of the Extracted Characteristics
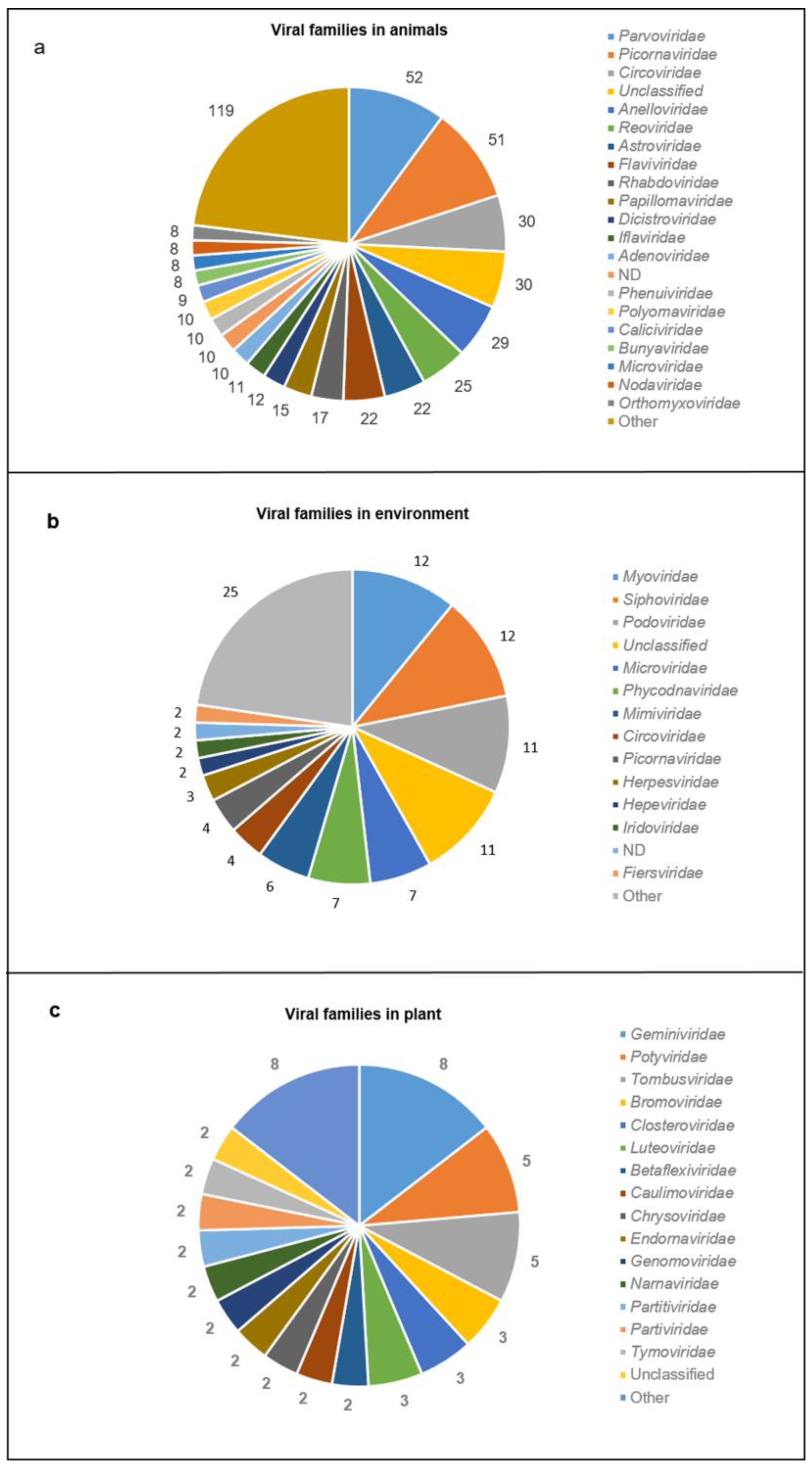
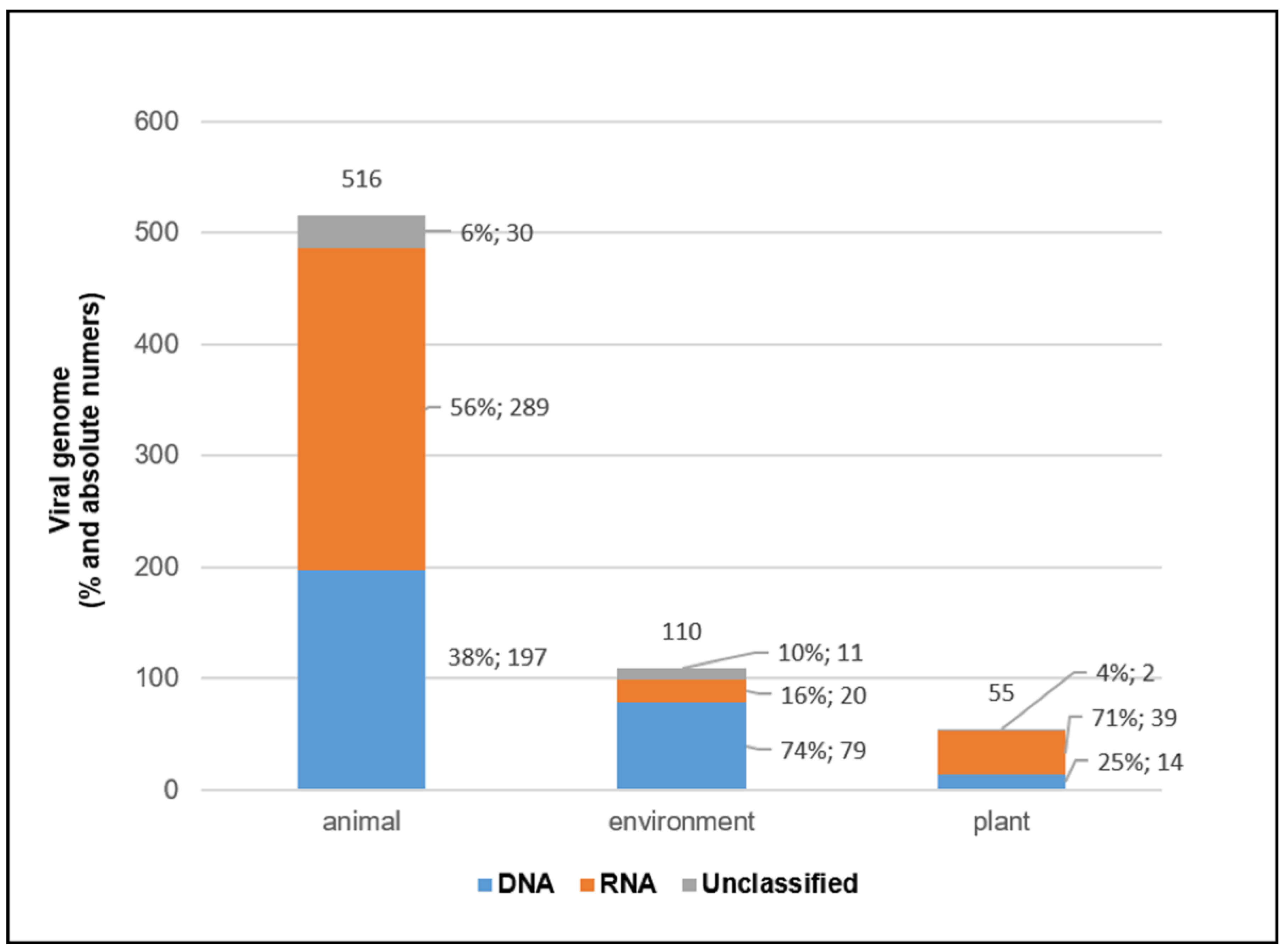
3.4. Viral Genomes and Viral Families in Different Sample Types
3.5. Novel Viruses Found by mNGS Studies
3.6. Bioinformatics Pipelines
3.6.1. Quality Control (QC) Check, Sequence Trimming and Filtering
3.6.2. Viral Genome Identification
3.6.3. Analysis of the Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bar-On, Y.M.; Phillips, R.; Milo, R. The biomass distribution on Earth. Proc. Natl. Acad. Sci. USA 2018, 115, 6506–6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Call, L.; Nayfach, S.; Kyrpides, N.C. Illuminating the Virosphere Through Global Metagenomics. Annu. Rev. Biomed. Data Sci. 2021, 4, 369–391. [Google Scholar] [CrossRef] [PubMed]
- French, R.K.; Holmes, E.C. An Ecosystems Perspective on Virus Evolution and Emergence. Trends Microbiol. 2020, 28, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, A. Food-web structure and ecosystem services: Insights from the Serengeti. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2009, 364, 1665–1682. [Google Scholar] [CrossRef]
- Dobson, A. Population dynamics of pathogens with multiple host species. Am. Nat. 2004, 164 (Suppl. 5), S64–S78. [Google Scholar] [CrossRef]
- Jover, L.F.; Effler, T.C.; Buchan, A.; Wilhelm, S.W.; Weitz, J.S. The elemental composition of virus particles: Implications for marine biogeochemical cycles. Nat. Rev. Microbiol. 2014, 12, 519–528. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Hamilton, R.; Kits, K.D.; Ramonovskaya, V.A.; Rozova, O.N.; Yurimoto, H.; Iguchi, H.; Khmelenina, V.N.; Sakai, Y.; Dunfield, P.F.; Klotz, M.G.; et al. Draft genomes of gammaproteobacterial methanotrophs isolated from terrestrial ecosystems. Genome Announc. 2015, 3, e00515-15. [Google Scholar] [CrossRef] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Westermann, A.J.; Vogel, J. Cross-species RNA-seq for deciphering host-microbe interactions. Nat. Rev. Genet. 2021, 22, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Barba, M.; Czosnek, H.; Hadidi, A. Historical perspective, development and applications of next-generation sequencing in plant virology. Viruses 2014, 6, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knief, C. Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 2014, 5, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maarastawi, S.A.; Frindte, K.; Linnartz, M.; Knief, C. Crop Rotation and Straw Application Impact Microbial Communities in Italian and Philippine Soils and the Rhizosphere of Zea mays. Front. Microbiol. 2018, 9, 1295. [Google Scholar] [CrossRef] [Green Version]
- Manriquez, B.; Muller, D.; Prigent-Combaret, C. Experimental Evolution in Plant-Microbe Systems: A Tool for Deciphering the Functioning and Evolution of Plant-Associated Microbial Communities. Front. Microbiol. 2021, 12, 619122. [Google Scholar] [CrossRef]
- Becker, M.F.; Hellmann, M.; Knief, C. Spatio-temporal variation in the root-associated microbiota of orchard-grown apple trees. Env. Microbiome 2022, 17, 31. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Lapidus, A.L.; Korobeynikov, A.I. Metagenomic Data Assembly—The Way of Decoding Unknown Microorganisms. Front. Microbiol. 2021, 12, 613791. [Google Scholar] [CrossRef]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Kang, Y.; Luo, J.; Pang, K.; Xu, X.; Wu, J.; Li, X.; Jin, S. Next-Generation Sequencing Reveals the Progression of COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 632490. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Mogollón, D.; Kamaliddin, C.; Oberding, L.; Liu, Y.; Mohon, A.N.; Faridi, R.M.; Khan, F.; Pillai, D.R. A metagenomics workflow for SARS-CoV-2 identification, co-pathogen detection, and overall diversity. J. Clin. Virol. 2021, 145, 105025. [Google Scholar] [CrossRef]
- Mackenzie, J.S.; Jeggo, M. The One Health Approach-Why Is It So Important? Trop. Med. Infect. Dis. 2019, 4, 88. [Google Scholar] [CrossRef] [Green Version]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Ponten, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Probst, A.J.; Weinmaier, T.; DeSantis, T.Z.; Santo Domingo, J.W.; Ashbolt, N. New perspectives on microbial community distortion after whole-genome amplification. PLoS ONE 2015, 10, e0124158. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. Meta-IDBA: A de Novo assembler for metagenomic data. Bioinformatics 2011, 27, i94–i101. [Google Scholar] [CrossRef] [Green Version]
- Namiki, T.; Hachiya, T.; Tanaka, H.; Sakakibara, Y. MetaVelvet: An extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012, 40, e155. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampelli, S.; Turroni, S. From Whole-Genome Shotgun Sequencing to Viral Community Profiling: The ViromeScan Tool. Methods Mol. Biol. 2018, 1746, 181–185. [Google Scholar] [CrossRef]
- Garner, E.; Davis, B.C.; Milligan, E.; Blair, M.F.; Keenum, I.; Maile-Moskowitz, A.; Pan, J.; Gnegy, M.; Liguori, K.; Gupta, S.; et al. Next generation sequencing approaches to evaluate water and wastewater quality. Water Res. 2021, 194, 116907. [Google Scholar] [CrossRef] [PubMed]
- Team, T.E. EndNote, EndNote X9; Clarivate: Philadelphia, PA, USA, 2013. [Google Scholar]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Lühken, R.; Jöst, H.; Jansen, S.; Börstler, J.; Rieger, T.; Krüger, A.; Yadouleton, A.; de Mendonça Campos, R.; Cirne-Santos, C.C.; et al. Detection and characterization of a novel rhabdovirus in Aedes cantans mosquitoes and evidence for a mosquito-associated new genus in the family Rhabdoviridae. Infect. Genet. Evol. 2017, 55, 260–268. [Google Scholar] [CrossRef]
- Ge, X.; Wu, Y.; Wang, M.; Wang, J.; Wu, L.; Yang, X.; Zhang, Y.; Shi, Z. Viral metagenomics analysis of planktonic viruses in East Lake, Wuhan, China. Virol. Sin. 2013, 28, 280–290. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, L.; Zhou, Y.; Wang, H.; Xiao, J.; Yan, S.; Wang, Y. Diverse and unique viruses discovered in the surface water of the East China Sea. BMC Genom. 2020, 21, 441. [Google Scholar] [CrossRef]
- Guajardo-Leiva, S.; Chnaiderman, J.; Gaggero, A.; Díez, B. Metagenomic Insights into the Sewage RNA Virosphere of a Large City. Viruses 2020, 12, 1050. [Google Scholar] [CrossRef]
- Kallies, R.; Hölzer, M.; Brizola Toscan, R.; Nunes da Rocha, U.; Anders, J.; Marz, M.; Chatzinotas, A. Evaluation of Sequencing Library Preparation Protocols for Viral Metagenomic Analysis from Pristine Aquifer Groundwaters. Viruses 2019, 11, 484. [Google Scholar] [CrossRef]
- Font-Verdera, F.; Liébana, R.; Aldeguer-Riquelme, B.; Gangloff, V.; Santos, F.; Viver, T.; Rosselló-Móra, R. Inverted microbial community stratification and spatial-temporal stability in hypersaline anaerobic sediments from the S’Avall solar salterns. Syst. Appl. Microbiol. 2021, 44, 126231. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Robin, A.; Ravet, V.; Personnic, S.; Theil, S.; Colombet, J.; Sime-Ngando, T.; Debroas, D. Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 2012, 7, e33641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Tang, C.; Yue, H.; Ren, Y.; Song, Z. Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. J. Gen. Virol. 2014, 95, 1603–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baidaliuk, A.; Lequime, S.; Moltini-Conclois, I.; Dabo, S.; Dickson, L.B.; Prot, M.; Duong, V.; Dussart, P.; Boyer, S.; Shi, C.; et al. Novel genome sequences of cell-fusing agent virus allow comparison of virus phylogeny with the genetic structure of Aedes aegypti populations. Virus Evol. 2020, 6, veaa018. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Godinho, R.; Álvares, F.; Yinda, C.K.; Deboutte, W.; Zeller, M.; Laenen, L.; Heylen, E.; Roque, S.; Petrucci-Fonseca, F.; et al. Viral gut metagenomics of sympatric wild and domestic canids, and monitoring of viruses: Insights from an endangered wolf population. Ecol. Evol. 2017, 7, 4135–4146. [Google Scholar] [CrossRef] [Green Version]
- de Souza, W.M.; Fumagalli, M.J.; Martin, M.C.; de Araujo, J.; Orsi, M.A.; Sanfilippo, L.F.; Modha, S.; Durigon, E.L.; Proença-Módena, J.L.; Arns, C.W.; et al. Pingu virus: A new picornavirus in penguins from Antarctica. Virus Evol. 2019, 5, vez047. [Google Scholar] [CrossRef] [PubMed]
- Duraisamy, R.; Akiana, J.; Davoust, B.; Mediannikov, O.; Michelle, C.; Robert, C.; Parra, H.J.; Raoult, D.; Biagini, P.; Desnues, C. Detection of novel RNA viruses from free-living gorillas, Republic of the Congo: Genetic diversity of picobirnaviruses. Virus Genes 2018, 54, 256–271. [Google Scholar] [CrossRef]
- Fernandez-Cassi, X.; Timoneda, N.; Gonzales-Gustavson, E.; Abril, J.F.; Bofill-Mas, S.; Girones, R. A metagenomic assessment of viral contamination on fresh parsley plants irrigated with fecally tainted river water. Int. J. Food Microbiol. 2017, 257, 80–90. [Google Scholar] [CrossRef]
- Frey, K.G.; Biser, T.; Hamilton, T.; Santos, C.J.; Pimentel, G.; Mokashi, V.P.; Bishop-Lilly, K.A. Bioinformatic Characterization of Mosquito Viromes within the Eastern United States and Puerto Rico: Discovery of Novel Viruses. Evol. Bioinform. 2016, 12 (Suppl. 2), 1–12. [Google Scholar] [CrossRef]
- Wang, G.; Huang, Y.; Zhang, W.; Peng, R.; Luo, J.; Liu, S.; Bai, S.; Hu, X.; Wu, Z.; Yang, F.; et al. Identification and genome analysis of a novel picornavirus from captive belugas (Delphinapterus leucas) in China. Sci. Rep. 2021, 11, 21018. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Gustavson, E.; Timoneda, N.; Fernandez-Cassi, X.; Caballero, A.; Abril, J.F.; Buti, M.; Rodriguez-Frias, F.; Girones, R. Identification of sapovirus GV.2, astrovirus VA3 and novel anelloviruses in serum from patients with acute hepatitis of unknown aetiology. PLoS ONE 2017, 12, e0185911. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hua, X.; Zhang, W.; Yang, S.; Shen, Q.; Hu, H.; Li, J.; Liu, Z.; Wang, X.; Wang, H.; et al. Viral metagenomics analysis of feces from coronary heart disease patients reveals the genetic diversity of the Microviridae. Virol. Sin. 2017, 32, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Xiao, J.; Song, Y.; Hong, M.; Dai, G.; Lu, H.; Zhang, M.; Liang, Y.; Yan, D.; Zhu, S.; et al. The Husavirus Posa-Like Viruses in China, and a New Group of Picornavirales. Viruses 2020, 12, 995. [Google Scholar] [CrossRef]
- Hargitai, R.; Boros, Á.; Pankovics, P.; Mátics, R.; Altan, E.; Delwart, E.; Reuter, G. Detection and genetic characterization of a novel parvovirus (family Parvoviridae) in barn owls (Tyto alba) in Hungary. Arch. Virol. 2021, 166, 231–236. [Google Scholar] [CrossRef]
- Yang, J.; Wang, H.; Zhang, X.; Yang, S.; Xu, H.; Zhang, W. Viral metagenomic identification of a novel anellovirus in blood sample of a child with atopic dermatitis. J. Med. Virol. 2021, 93, 4038–4041. [Google Scholar] [CrossRef]
- Kauer, R.V.; Koch, M.C.; Hierweger, M.M.; Werder, S.; Boujon, C.L.; Seuberlich, T. Discovery of novel astrovirus genotype species in small ruminants. PeerJ 2019, 7, e7338. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Giannitti, F.; Low, J.; Keyes, C.; Ullmann, L.S.; Deng, X.; Aleman, M.; Pesavento, P.A.; Pusterla, N.; Delwart, E. Exploring the virome of diseased horses. J. Gen. Virol. 2015, 96, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Pesavento, P.A.; Shan, T.; Leutenegger, C.M.; Wang, C.; Delwart, E. Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. J. Gen. Virol. 2011, 92, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Langat, S.K.; Eyase, F.; Bulimo, W.; Lutomiah, J.; Oyola, S.O.; Imbuga, M.; Sang, R. Profiling of RNA Viruses in Biting Midges (Ceratopogonidae) and Related Diptera from Kenya Using Metagenomics and Metabarcoding Analysis. mSphere 2021, 6, e0055121. [Google Scholar] [CrossRef] [PubMed]
- Law, J.; Jovel, J.; Patterson, J.; Ford, G.; O’Keefe, S.; Wang, W.; Meng, B.; Song, D.; Zhang, Y.; Tian, Z.; et al. Identification of hepatotropic viruses from plasma using deep sequencing: A next generation diagnostic tool. PLoS ONE 2013, 8, e60595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yang, S.; Wang, Y.; Shen, Q.; Yang, Y.; Deng, X.; Zhang, W.; Delwart, E. Identification of a novel human papillomavirus by metagenomic analysis of vaginal swab samples from pregnant women. Virol. J. 2016, 13, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, A.; Shama, S.; Ni, H.; Wang, H.; Ling, Y.; Xu, H.; Yang, S.; Naseer, Q.A.; Zhang, W. Viral Metagenomics Revealed a Novel Cardiovirus in Feces of Wild Rats. Intervirology 2019, 62, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sautu, U.; Wiley, M.R.; Prieto, K.; Chitty, J.A.; Haddow, A.D.; Sanchez-Lockhart, M.; Klein, T.A.; Kim, H.C.; Chong, S.T.; Kim, Y.J.; et al. Novel viruses in hard ticks collected in the Republic of Korea unveiled by metagenomic high-throughput sequencing analysis. Ticks Tick-Borne Dis. 2021, 12, 101820. [Google Scholar] [CrossRef] [PubMed]
- Pyke, A.T.; Shivas, M.A.; Darbro, J.M.; Onn, M.B.; Johnson, P.H.; Crunkhorn, A.; Montgomery, I.; Burtonclay, P.; Jansen, C.C.; van den Hurk, A.F. Uncovering the genetic diversity within the Aedes notoscriptus virome and isolation of new viruses from this highly urbanised and invasive mosquito. Virus Evol. 2021, 7, veab082. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, H.; Ling, Y.; Yang, S.X.; Wang, X.C.; Zhou, R.; Xiao, Y.Q.; Chen, X.; Yang, J.; Fu, W.G.; et al. Viral metagenomics revealed diverse CRESS-DNA virus genomes in faeces of forest musk deer. Virol. J. 2020, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Shan, T.; Liu, Z.; Deng, X.; Zhang, Z.; Bi, W.; Owens, J.R.; Feng, F.; Zheng, L.; Huang, F.; et al. A novel polyomavirus from the nasal cavity of a giant panda (Ailuropoda melanoleuca). Virol. J. 2017, 14, 207. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, A.L.; Colmant, A.M.G.; Warrilow, D.; Huang, B.; Pyke, A.T.; McMahon, J.L.; Meyer, D.B.; Graham, R.M.A.; Jennison, A.V.; Ritchie, S.A.; et al. Metagenomic Analysis of the Virome of Mosquito Excreta. mSphere 2020, 5, e00587-20. [Google Scholar] [CrossRef]
- Ramírez-Martínez, M.M.; Bennett, A.J.; Dunn, C.D.; Yuill, T.M.; Goldberg, T.L. Bat Flies of the Family Streblidae (Diptera: Hippoboscoidea) Host Relatives of Medically and Agriculturally Important “Bat-Associated” Viruses. Viruses 2021, 13, 860. [Google Scholar] [CrossRef]
- Reuter, G.; Pankovics, P.; Gyöngyi, Z.; Delwart, E.; Boros, A. Novel dicistrovirus from bat guano. Arch. Virol. 2014, 159, 3453–3456. [Google Scholar] [CrossRef] [PubMed]
- I. Sardi, S.; H. Carvalho, R.; C. Pacheco, L.G.; P. D. Almeida, J.P.; M. D. A. Belitardo, E.M.; S. Pinheiro, C.; S. Campos, G.; R. G. R. Aguiar, E. High-Quality Resolution of the Outbreak-Related Zika Virus Genome and Discovery of New Viruses Using Ion Torrent-Based Metatranscriptomics. Viruses 2020, 12, 782. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.D.; Ziegler, U.; Szillat, K.P.; Szentiks, C.A.; Strobel, B.; Skuballa, J.; Merbach, S.; Grothmann, P.; Tews, B.A.; Beer, M.; et al. In action-an early warning system for the detection of unexpected or novel pathogens. Virus Evol. 2021, 7, veab085. [Google Scholar] [CrossRef]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.B.; Matthijnssens, J. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Simsek, C.; Corman, V.M.; Everling, H.U.; Lukashev, A.N.; Rasche, A.; Maganga, G.D.; Binger, T.; Jansen, D.; Beller, L.; Deboutte, W.; et al. At Least Seven Distinct Rotavirus Genotype Constellations in Bats with Evidence of Reassortment and Zoonotic Transmissions. mBio 2021, 12, e02755-20. [Google Scholar] [CrossRef]
- Siqueira, J.D.; Ng, T.F.; Miller, M.; Li, L.; Deng, X.; Dodd, E.; Batac, F.; Delwart, E. Endemic Infection of Stranded Southern Sea Otters (Enhydra Lutris Nereis) with Novel Parvovirus, Polyomavirus, And Adenovirus. J. Wildl. Dis. 2017, 53, 532–542. [Google Scholar] [CrossRef]
- Souza, W.M.; Fumagalli, M.J.; Torres Carrasco, A.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, T.F.F.; Dill, J.A.; Camus, A.C.; Delwart, E.; Van Meir, E.G. Two new species of betatorqueviruses identified in a human melanoma that metastasized to the brain. Oncotarget 2017, 8, 105800–105808. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.F.; Mesquita, J.R.; Nascimento, M.S.; Kondov, N.O.; Wong, W.; Reuter, G.; Knowles, N.J.; Vega, E.; Esona, M.D.; Deng, X.; et al. Feline fecal virome reveals novel and prevalent enteric viruses. Vet. Microbiol. 2014, 171, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Gong, H.; Shen, X.; Zhang, W.; Shan, T.; Yu, X.; Wang, S.J.; Cui, L. Comparison of Viromes in Ticks from Different Domestic Animals in China. Virol. Sin. 2020, 35, 398–406. [Google Scholar] [CrossRef]
- Theuns, S.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Roukaerts, I.D.; Desmarets, L.M.; Van Ranst, M.; Nauwynck, H.J.; Matthijnssens, J. Characterization of a genetically heterogeneous porcine rotavirus C, and other viruses present in the fecal virome of a non-diarrheic Belgian piglet. Infect. Genet. Evol. 2016, 43, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.; Tsang, A.K.; Tsoi, H.W.; Leung, A.S.; Ho, C.C.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Identification of a novel bat papillomavirus by metagenomics. PLoS ONE 2012, 7, e43986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, K.; Mehmood, A.; Chen, X.; Dar, M.A.; Yang, S.; Zhang, W. Detection and molecular characterization of picobirnaviruses in the wild birds: Identification of a novel picobirnavirus possessing yeast mitochondrial genetic code. Virus Res. 2021, 308, 198624. [Google Scholar] [CrossRef] [PubMed]
- Vandegrift, K.J.; Kumar, A.; Sharma, H.; Murthy, S.; Kramer, L.D.; Ostfeld, S.; Hudson, P.J.; Kapoor, A. Presence of Segmented Flavivirus Infections in North America. Emerg. Infect. Dis. 2020, 26, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Vanmechelen, B.; Merino, M.; Vergote, V.; Laenen, L.; Thijssen, M.; Martí-Carreras, J.; Claerebout, E.; Maes, P. Exploration of the Ixodes ricinus virosphere unveils an extensive virus diversity including novel coltiviruses and other reoviruses. Virus Evol. 2021, 7, veab066. [Google Scholar] [CrossRef] [PubMed]
- Vibin, J.; Chamings, A.; Klaassen, M.; Alexandersen, S. Metagenomic characterisation of additional and novel avian viruses from Australian wild ducks. Sci. Rep. 2020, 10, 22284. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci. Rep. 2020, 10, 12800. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, S.; Shan, T.; Hou, R.; Liu, Z.; Li, W.; Guo, L.; Wang, Y.; Chen, P.; Wang, X.; et al. Virome comparisons in wild-diseased and healthy captive giant pandas. Microbiome 2017, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, B.; Yue, H.; Wang, Y.; Zhou, F.; Zhang, Q.; Tang, C. A novel astrovirus species in the gut of yaks with diarrhoea in the Qinghai–Tibetan Plateau, 2013. J. Gen. Virol. 2015, 96, 3672–3680. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.U.; He, Y.; Li, W.; Kalim, U.; Xiao, Y.; Yang, J.; Wang, X.; Yang, S.; Zhang, W. Identification and Characterization of a Novel Recombinant Porcine Astrovirus from Pigs in Anhui, China. Pol. J. Microbiol. 2020, 69, 471–478. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The fecal virome of red-crowned cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Y.; Ma, J.; Zhou, S.; Fang, W.; Ye, J.; Tan, L.; Ji, J.; Luo, D.; Li, L.; et al. A novel Enterovirus 96 circulating in China causes hand, foot, and mouth disease. Virus Genes 2017, 53, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Zeller, M.; Conceição-Neto, N.; Maes, P.; Deboutte, W.; Beller, L.; Heylen, E.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent reassortant bat rotaviruses in Cameroon, without evidence of zoonosis. Sci. Rep. 2016, 6, 34209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yinda, C.K.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Maes, P.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent sapoviruses detected by metagenomics analysis in straw-colored fruit bats in Cameroon. Emerg. Microbes Infect. 2017, 6, e38. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhang, J.; Yang, S.; Wang, X.; Shen, Q.; Sun, G.; Wang, H.; Zhang, W. Virome analysis of ticks in a forest region of Liaoning, China: Characterization of a novel hepe-like virus sequence. Virol. J. 2021, 18, 163. [Google Scholar] [CrossRef]
- Alex, C.E.; Kubiski, S.V.; Li, L.; Sadeghi, M.; Wack, R.F.; McCarthy, M.A.; Pesavento, J.B.; Delwart, E.; Pesavento, P.A. Amdoparvovirus Infection in Red Pandas (Ailurus fulgens). Vet. Pathol. 2018, 55, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Alex, C.E.; Fahsbender, E.; Altan, E.; Bildfell, R.; Wolff, P.; Jin, L.; Black, W.; Jackson, K.; Woods, L.; Munk, B.; et al. Viruses in unexplained encephalitis cases in American black bears (Ursus americanus). PLoS ONE 2020, 15, e0244056. [Google Scholar] [CrossRef]
- Altan, E.; Delaney, M.A.; Colegrove, K.M.; Spraker, T.R.; Wheeler, E.A.; Deng, X.; Li, Y.; Gulland, F.M.D.; Delwart, E. Complex Virome in a Mesenteric Lymph Node from a Californian Sea Lion (Zalophus Californianus) with Polyserositis and Steatitis. Viruses 2020, 12, 793. [Google Scholar] [CrossRef]
- Altan, E.; Hui, A.; Li, Y.; Pesavento, P.; Asín, J.; Crossley, B.; Deng, X.; Uzal, F.A.; Delwart, E. New Parvoviruses and Picornavirus in Tissues and Feces of Foals with Interstitial Pneumonia. Viruses 2021, 13, 1612. [Google Scholar] [CrossRef]
- Altan, E.; Kubiski, S.V.; Boros, Á.; Reuter, G.; Sadeghi, M.; Deng, X.; Creighton, E.K.; Crim, M.J.; Delwart, E. A Highly Divergent Picornavirus Infecting the Gut Epithelia of Zebrafish (Danio rerio) in Research Institutions Worldwide. Zebrafish 2019, 16, 291–299. [Google Scholar] [CrossRef]
- Altan, E.; Li, Y.; Sabino-Santos, G., Jr.; Sawaswong, V.; Barnum, S.; Pusterla, N.; Deng, X.; Delwart, E. Viruses in Horses with Neurologic and Respiratory Diseases. Viruses 2019, 11, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan, E.; Seguin, M.A.; Leutenegger, C.M.; Phan, T.G.; Deng, X.; Delwart, E. Nasal virome of dogs with respiratory infection signs include novel taupapillomaviruses. Virus Genes 2019, 55, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Eibach, D.; Hogan, B.; Sarpong, N.; Winter, D.; Struck, N.S.; Adu-Sarkodie, Y.; Owusu-Dabo, E.; Schmidt-Chanasit, J.; May, J.; Cadar, D. Viral metagenomics revealed novel betatorquevirus species in pediatric inpatients with encephalitis/meningoencephalitis from Ghana. Sci. Rep. 2019, 9, 2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahsbender, E.; Charlys da-Costa, A.; Elise Gill, D.; Augusto de Padua Milagres, F.; Brustulin, R.; Julio Costa Monteiro, F.; Octavio da Silva Rego, M.; Soares D’Athaide Ribeiro, E.; Cerdeira Sabino, E.; Delwart, E. Plasma virome of 781 Brazilians with unexplained symptoms of arbovirus infection include a novel parvovirus and densovirus. PLoS ONE 2020, 15, e0229993. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Correa, I.; Truchado, D.A.; Gomez-Lucia, E.; Doménech, A.; Pérez-Tris, J.; Schmidt-Chanasit, J.; Cadar, D.; Benítez, L. A novel group of avian astroviruses from Neotropical passerine birds broaden the diversity and host range of Astroviridae. Sci. Rep. 2019, 9, 9513. [Google Scholar] [CrossRef] [Green Version]
- Hargitai, R.; Pankovics, P.; Boros, Á.; Mátics, R.; Altan, E.; Delwart, E.; Reuter, G. Novel picornavirus (family Picornaviridae) from freshwater fishes (Perca fluviatilis, Sander lucioperca, and Ameiurus melas) in Hungary. Arch. Virol. 2021, 166, 2627–2632. [Google Scholar] [CrossRef]
- Phan, T.G.; Luchsinger, V.; Avendaño, L.F.; Deng, X.; Delwart, E. Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. J. Gen. Virol. 2014, 95, 922–927. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Mátics, R.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Detection and complete genome characterization of a novel RNA virus related to members of the Hepe-Virga clade in bird species, hoopoe (Upupa epops). Infect. Genet. Evol. 2020, 81, 104236. [Google Scholar] [CrossRef]
- Sadeghi, M.; Kapusinszky, B.; Yugo, D.M.; Phan, T.G.; Deng, X.; Kanevsky, I.; Opriessnig, T.; Woolums, A.R.; Hurley, D.J.; Meng, X.J.; et al. Virome of US bovine calf serum. Biologicals 2017, 46, 64–67. [Google Scholar] [CrossRef]
- Sadeghi, M.; Popov, V.; Guzman, H.; Phan, T.G.; Vasilakis, N.; Tesh, R.; Delwart, E. Genomes of viral isolates derived from different mosquitos species. Virus Res. 2017, 242, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truchado, D.A.; Diaz-Piqueras, J.M.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; Schmidt-Chanasit, J.; Cadar, D.; Benítez, L. A Novel and Divergent Gyrovirus with Unusual Genomic Features Detected in Wild Passerine Birds from a Remote Rainforest in French Guiana. Viruses 2019, 11, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truchado, D.A.; Llanos-Garrido, A.; Oropesa-Olmedo, D.A.; Cerrada, B.; Cea, P.; Moens, M.A.J.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; et al. Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms 2020, 8, 1869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Delwart, E. What is for dinner? Viral metagenomics of US store bought beef, pork, and chicken. Virology 2014, 468–470, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal virome of cats in an animal shelter. J. Gen. Virol. 2014, 95, 2553–2564. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Idle, A.; Hui, A.; Chan, R.; Seguin, M.A.; Delwart, E. Astrovirus Outbreak in an Animal Shelter Associated with Feline Vomiting. Front. Vet. Sci. 2021, 8, 628082. [Google Scholar] [CrossRef]
- Aw, T.G.; Howe, A.; Rose, J.B. Metagenomic approaches for direct and cell culture evaluation of the virological quality of wastewater. J. Virol. Methods 2014, 210, 15–21. [Google Scholar] [CrossRef]
- Castrignano, S.B.; Nagasse-Sugahara, T.K.; Kisielius, J.J.; Ueda-Ito, M.; Brandão, P.E.; Curti, S.P. Two novel circo-like viruses detected in human feces: Complete genome sequencing and electron microscopy analysis. Virus Res. 2013, 178, 364–373. [Google Scholar] [CrossRef]
- Dai, Z.; Wang, H.; Feng, Z.; Ma, L.; Yang, S.; Shen, Q.; Wang, X.; Zhou, T.; Zhang, W. Identification of a novel circovirus in blood sample of giant pandas (Ailuropoda melanoleuca). Infect. Genet. Evol. 2021, 95, 105077. [Google Scholar] [CrossRef]
- Dai, Z.; Wang, H.; Yang, S.; Shen, Q.; Wang, X.; Zhou, T.; Feng, Z.; Zhang, W. Identification and characterization of a novel bocaparvovirus in tufted deer (Elaphodus cephalophus) in China. Arch. Virol. 2022, 167, 201–206. [Google Scholar] [CrossRef]
- Dunlap, D.S.; Ng, T.F.; Rosario, K.; Barbosa, J.G.; Greco, A.M.; Breitbart, M.; Hewson, I. Molecular and microscopic evidence of viruses in marine copepods. Proc. Natl. Acad. Sci. USA 2013, 110, 1375–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Ling, Y.; Shan, T.; Yang, S.; Xu, H.; Deng, X.; Delwart, E.; Zhang, W. Gut virome of mammals and birds reveals high genetic diversity of the family Microviridae. Virus Evol. 2019, 5, vez013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, N.; Li, Z.; Liu, L.; He, Y.; Meng, J.; Li, S.; Wang, J. Identification of a novel bocaparvovirus in a wild squirrel in Kunming, Yunnan Province, China. Arch. Virol. 2020, 165, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Leigh, B.A.; Bordenstein, S.R.; Brooks, A.W.; Mikaelyan, A.; Bordenstein, S.R. Finer-Scale Phylosymbiosis: Insights from Insect Viromes. mSystems 2018, 3, e00131-18. [Google Scholar] [CrossRef] [Green Version]
- Ling, Y.; Zhang, X.; Qi, G.; Yang, S.; Jingjiao, L.; Shen, Q.; Wang, X.; Cui, L.; Hua, X.; Deng, X.; et al. Viral metagenomics reveals significant viruses in the genital tract of apparently healthy dairy cows. Arch. Virol. 2019, 164, 1059–1067. [Google Scholar] [CrossRef]
- Richard, J.C.; Leis, E.; Dunn, C.D.; Agbalog, R.; Waller, D.; Knowles, S.; Putnam, J.; Goldberg, T.L. Mass mortality in freshwater mussels (Actinonaias pectorosa) in the Clinch River, USA, linked to a novel densovirus. Sci. Rep. 2020, 10, 14498. [Google Scholar] [CrossRef]
- Rosario, K.; Marinov, M.; Stainton, D.; Kraberger, S.; Wiltshire, E.J.; Collings, D.A.; Walters, M.; Martin, D.P.; Breitbart, M.; Varsani, A. Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (Odonata: Anisoptera). J. Gen. Virol. 2011, 92, 1302–1308. [Google Scholar] [CrossRef]
- Rosario, K.; Padilla-Rodriguez, M.; Kraberger, S.; Stainton, D.; Martin, D.P.; Breitbart, M.; Varsani, A. Discovery of a novel mastrevirus and alphasatellite-like circular DNA in dragonflies (Epiprocta) from Puerto Rico. Virus Res. 2013, 171, 231–237. [Google Scholar] [CrossRef]
- Rosario, K.; Schenck, R.O.; Harbeitner, R.C.; Lawler, S.N.; Breitbart, M. Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 2015, 6, 696. [Google Scholar] [CrossRef] [Green Version]
- Rosario, K.; Seah, Y.M.; Marr, C.; Varsani, A.; Kraberger, S.; Stainton, D.; Moriones, E.; Polston, J.E.; Duffy, S.; Breitbart, M. Vector-Enabled Metagenomic (VEM) Surveys Using Whiteflies (Aleyrodidae) Reveal Novel Begomovirus Species in the New and Old Worlds. Viruses 2015, 7, 5553–5570. [Google Scholar] [CrossRef]
- Ng, T.F.; Duffy, S.; Polston, J.E.; Bixby, E.; Vallad, G.E.; Breitbart, M. Exploring the diversity of plant DNA viruses and their satellites using vector-enabled metagenomics on whiteflies. PLoS ONE 2011, 6, e19050. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.F.; Kondov, N.O.; Hayashimoto, N.; Uchida, R.; Cha, Y.; Beyer, A.I.; Wong, W.; Pesavento, P.A.; Suemizu, H.; Muench, M.O.; et al. Identification of an astrovirus commonly infecting laboratory mice in the US and Japan. PLoS ONE 2013, 8, e66937. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.F.; Manire, C.; Borrowman, K.; Langer, T.; Ehrhart, L.; Breitbart, M. Discovery of a novel single-stranded DNA virus from a sea turtle fibropapilloma by using viral metagenomics. J. Virol. 2009, 83, 2500–2509. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.F.F.; Wheeler, E.; Greig, D.; Waltzek, T.B.; Gulland, F.; Breitbart, M. Metagenomic identification of a novel anellovirus in Pacific harbor seal (Phoca vitulina richardsii) lung samples and its detection in samples from multiple years. J. Gen. Virol. 2011, 92, 1318–1323. [Google Scholar] [CrossRef]
- Wang, X.C.; Wang, H.; Tan, S.D.; Yang, S.X.; Shi, X.F.; Zhang, W. Viral metagenomics reveals diverse anelloviruses in bone marrow specimens from hematologic patients. J. Clin. Virol. 2020, 132, 104643. [Google Scholar] [CrossRef]
- Kim, Y.; Aw, T.G.; Teal, T.K.; Rose, J.B. Metagenomic Investigation of Viral Communities in Ballast Water. Environ. Sci. Technol. 2015, 49, 8396–8407. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gordon, E.; Idle, A.; Altan, E.; Seguin, M.A.; Estrada, M.; Deng, X.; Delwart, E. Virome of a Feline Outbreak of Diarrhea and Vomiting Includes Bocaviruses and a Novel Chapparvovirus. Viruses 2020, 12, 506. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, S.; Quan, R.; Wang, J.; Wei, L.; Yang, B.; Xu, F.; Wang, J.; Chen, F.; Liu, J. Identification and Genome Characterization of the First Sicinivirus Isolate from Chickens in Mainland China by Using Viral Metagenomics. PLoS ONE 2015, 10, e0139668. [Google Scholar] [CrossRef]
- Bodewes, R.; Contreras, G.J.S.; García, A.R.; Hapsari, R.; van de Bildt, M.W.G.; Kuiken, T.; Osterhaus, A. Identification of DNA sequences that imply a novel gammaherpesvirus in seals. J. Gen. Virol. 2015, 96, 1109–1114. [Google Scholar] [CrossRef]
- Castrignano, S.B.; Nagasse-Sugahara, T.K.; Garrafa, P.; Monezi, T.A.; Barrella, K.M.; Mehnert, D.U. Identification of circo-like virus-Brazil genomic sequences in raw sewage from the metropolitan area of São Paulo: Evidence of circulation two and three years after the first detection. Mem. Inst. Oswaldo Cruz 2017, 112, 175–181. [Google Scholar] [CrossRef]
- Crane, A.; Goebel, M.E.; Kraberger, S.; Stone, A.C.; Varsani, A. Novel anelloviruses identified in buccal swabs of Antarctic fur seals. Virus Genes 2018, 54, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, A.; Opong, A.; Jaschke, A.; Hadfield, J.; Baschiera, M.; Dobson, R.C.; Offei, S.K.; Shepherd, D.N.; Martin, D.P.; Varsani, A. Molecular characterisation of a novel cassava associated circular ssDNA virus. Virus Res. 2012, 166, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Cook, C.N.; Schmidlin, K.; Fontenele, R.S.; Bautista, J.; Smith, B.; Varsani, A. Diverse single-stranded DNA viruses associated with honey bees (Apis mellifera). Infect. Genet. Evol. 2019, 71, 179–188. [Google Scholar] [CrossRef]
- Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Walters, M.; Varsani, A. Unravelling the Single-Stranded DNA Virome of the New Zealand Blackfly. Viruses 2019, 11, 532. [Google Scholar] [CrossRef] [Green Version]
- A. Duarte, M.; F. Silva, J.M.; R. Brito, C.; S. Teixeira, D.; L. Melo, F.; M. Ribeiro, B.; Nagata., T.; S. Campos, F. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [Green Version]
- Öncü, C.; Brinkmann, A.; Günay, F.; Kar, S.; Öter, K.; Sarıkaya, Y.; Nitsche, A.; Linton, Y.M.; Alten, B.; Ergünay, K. West Nile virus, Anopheles flavivirus, a novel flavivirus as well as Merida-like rhabdovirus Turkey in field-collected mosquitoes from Thrace and Anatolia. Infect. Genet. Evol. 2018, 57, 36–45. [Google Scholar] [CrossRef]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. [Google Scholar] [CrossRef] [Green Version]
- Popgeorgiev, N.; Boyer, M.; Fancello, L.; Monteil, S.; Robert, C.; Rivet, R.; Nappez, C.; Azza, S.; Chiaroni, J.; Raoult, D.; et al. Marseillevirus-like virus recovered from blood donated by asymptomatic humans. J. Infect. Dis. 2013, 208, 1042–1050. [Google Scholar] [CrossRef]
- Sasaki, M.; Orba, Y.; Ueno, K.; Ishii, A.; Moonga, L.; Hang’ombe, B.M.; Mweene, A.S.; Ito, K.; Sawa, H. Metagenomic analysis of the shrew enteric virome reveals novel viruses related to human stool-associated viruses. J. Gen. Virol. 2015, 96, 440–452. [Google Scholar] [CrossRef]
- Anh, N.T.; Hong, N.T.T.; Nhu, L.N.T.; Thanh, T.T.; Lau, C.Y.; Limmathurotsakul, D.; Deng, X.; Rahman, M.; Chau, N.V.V.; van Doorn, H.R.; et al. Viruses in Vietnamese Patients Presenting with Community-Acquired Sepsis of Unknown Cause. J. Clin. Microbiol. 2019, 57, e00386-19. [Google Scholar] [CrossRef]
- Pan, S.; Yu, T.; Wang, Y.; Lu, R.; Wang, H.; Xie, Y.; Feng, X. Identification of a Torque Teno Mini Virus (TTMV) in Hodgkin’s Lymphoma Patients. Front. Microbiol. 2018, 9, 1680. [Google Scholar] [CrossRef]
- Plyusnin, I.; Kant, R.; Jääskeläinen, A.J.; Sironen, T.; Holm, L.; Vapalahti, O.; Smura, T. Novel NGS pipeline for virus discovery from a wide spectrum of hosts and sample types. Virus Evol. 2020, 6, veaa091. [Google Scholar] [CrossRef]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Mbala-Kingebeni, P.; Naccache, S.N.; Thézé, J.; Bouquet, J.; Federman, S.; Somasekar, S.; Yu, G.; Sanchez-San Martin, C.; Achari, A.; et al. Metagenomic Next-Generation Sequencing of the 2014 Ebola Virus Disease Outbreak in the Democratic Republic of the Congo. J. Clin. Microbiol. 2019, 57, e00827-19. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Wang, Y.; Shi, C.; Atoni, E.; Zhao, L.; Yuan, Z. Comparative Metagenomic Profiling of Viromes Associated with Four Common Mosquito Species in China. Virol. Sin. 2018, 33, 59–66. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Shan, T.L.; Deng, X.; Delwart, E.; Feng, X.P. A novel species of torque teno mini virus (TTMV) in gingival tissue from chronic periodontitis patients. Sci. Rep. 2016, 6, 26739. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shan, T.L.; Li, F.; Yu, T.; Chen, X.; Deng, X.T.; Delwart, E.; Feng, X.P. A novel phage from periodontal pockets associated with chronic periodontitis. Virus Genes 2019, 55, 381–393. [Google Scholar] [CrossRef]
- Blomström, A.L.; Fossum, C.; Wallgren, P.; Berg, M. Viral Metagenomic Analysis Displays the Co-Infection Situation in Healthy and PMWS Affected Pigs. PLoS ONE 2016, 11, e0166863. [Google Scholar] [CrossRef] [Green Version]
- Blomström, A.L.; Ståhl, K.; Masembe, C.; Okoth, E.; Okurut, A.R.; Atmnedi, P.; Kemp, S.; Bishop, R.; Belák, S.; Berg, M. Viral metagenomic analysis of bushpigs (Potamochoerus larvatus) in Uganda identifies novel variants of Porcine parvovirus 4 and Torque teno sus virus 1 and 2. Virol. J. 2012, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Blomström, A.L.; Widén, F.; Hammer, A.S.; Belák, S.; Berg, M. Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J. Clin. Microbiol. 2010, 48, 4392–4396. [Google Scholar] [CrossRef]
- Cebriá-Mendoza, M.; Arbona, C.; Larrea, L.; Díaz, W.; Arnau, V.; Peña, C.; Bou, J.V.; Sanjuán, R.; Cuevas, J.M. Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans. Sci. Rep. 2021, 11, 6921. [Google Scholar] [CrossRef]
- Zhang, D.; You, F.; He, Y.; Te, S.H.; Gin, K.Y. Isolation and Characterization of the First Freshwater Cyanophage Infecting Pseudanabaena. J. Virol. 2020, 94, e00682-20. [Google Scholar] [CrossRef]
- Birnberg, L.; Temmam, S.; Aranda, C.; Correa-Fiz, F.; Talavera, S.; Bigot, T.; Eloit, M.; Busquets, N. Viromics on Honey-Baited FTA Cards as a New Tool for the Detection of Circulating Viruses in Mosquitoes. Viruses 2020, 12, 274. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Yang, F.; Yang, W.; Zhang, Y.; Feng, Y.; Zhou, J.; Xie, J.; Feng, Y.; Bao, X.; Guo, H.; et al. Characterization of a novel G3P[3] rotavirus isolated from a lesser horseshoe bat: A distant relative of feline/canine rotaviruses. J. Virol. 2013, 87, 12357–12366. [Google Scholar] [CrossRef] [Green Version]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543.e524. [Google Scholar] [CrossRef] [Green Version]
- Schlottau, K.; Schulze, C.; Bilk, S.; Hanke, D.; Höper, D.; Beer, M.; Hoffmann, B. Detection of a Novel Bovine Astrovirus in a Cow with Encephalitis. Transbound. Emerg. Dis. 2016, 63, 253–259. [Google Scholar] [CrossRef]
- Dela Cruz, F.N., Jr.; Li, L.; Delwart, E.; Pesavento, P.A. A novel pulmonary polyomavirus in alpacas (Vicugna pacos). Vet. Microbiol. 2017, 201, 49–55. [Google Scholar] [CrossRef]
- Aswad, A.; Katzourakis, A. A novel viral lineage distantly related to herpesviruses discovered within fish genome sequence data. Virus Evol. 2017, 3, vex016. [Google Scholar] [CrossRef] [Green Version]
- Carrai, M.; Van Brussel, K.; Shi, M.; Li, C.X.; Chang, W.S.; Munday, J.S.; Voss, K.; McLuckie, A.; Taylor, D.; Laws, A.; et al. Identification of A Novel Papillomavirus Associated with Squamous Cell Carcinoma in A Domestic Cat. Viruses 2020, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Neyvaldt, J.; Enache, L.; Sikora, P.; Mattsson, A.; Johansson, A.; Lindh, M.; Bergstedt, O.; Norder, H. Variations among Viruses in Influent Water and Effluent Water at a Wastewater Plant over One Year as Assessed by Quantitative PCR and Metagenomics. Appl. Environ. Microbiol. 2020, 86, e02073-20. [Google Scholar] [CrossRef]
- Ng, T.F.; Alavandi, S.; Varsani, A.; Burghart, S.; Breitbart, M. Metagenomic identification of a nodavirus and a circular ssDNA virus in semi-purified viral nucleic acids from the hepatopancreas of healthy Farfantepenaeus duorarum shrimp. Dis. Aquat. Org. 2013, 105, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.J.; Ashley, W.; Gil-Fernandez, M.; Newsome, T.M.; Di Giallonardo, F.; Ortiz-Baez, A.S.; Mahar, J.E.; Towerton, A.L.; Gillings, M.; Holmes, E.C.; et al. Red fox viromes in urban and rural landscapes. Virus Evol. 2020, 6, veaa065. [Google Scholar] [CrossRef] [PubMed]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics. J. Virol. 2019, 93, e00205-19. [Google Scholar] [CrossRef] [Green Version]
- Geoghegan, J.L.; Di Giallonardo, F.; Wille, M.; Ortiz-Baez, A.S.; Costa, V.A.; Ghaly, T.; Mifsud, J.C.O.; Turnbull, O.M.H.; Bellwood, D.R.; Williamson, J.E.; et al. Virome composition in marine fish revealed by meta-transcriptomics. Virus Evol. 2021, 7, veab005. [Google Scholar] [CrossRef] [PubMed]
- Harvey, E.; Rose, K.; Eden, J.S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive Diversity of RNA Viruses in Australian Ticks. J. Virol. 2019, 93, e01358-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, A.F.; Pettersson, J.H.; Chang, W.S.; Harvey, E.; Rose, K.; Shi, M.; Eden, J.S.; Buchmann, J.; Moritz, C.; Holmes, E.C. Novel hepaci- and pegi-like viruses in native Australian wildlife and non-human primates. Virus Evol. 2020, 6, veaa064. [Google Scholar] [CrossRef]
- Scavizzi, F.; Bassi, C.; Lupini, L.; Guerriero, P.; Raspa, M.; Sabbioni, S. A comprehensive approach for microbiota and health monitoring in mouse colonies using metagenomic shotgun sequencing. Anim. Microbiome 2021, 3, 53. [Google Scholar] [CrossRef]
- Coutinho, F.H.; Edwards, R.A.; Rodríguez-Valera, F. Charting the diversity of uncultured viruses of Archaea and Bacteria. BMC Biol. 2019, 17, 109. [Google Scholar] [CrossRef] [Green Version]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; You, X.; Mai, G.; Tokuyasu, T.; Liu, C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 2018, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Mushegian, A.; Shipunov, A.; Elena, S.F. Changes in the composition of the RNA virome mark evolutionary transitions in green plants. BMC Biol. 2016, 14, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, K.; Duffy, S.; Breitbart, M. Diverse circovirus-like genome architectures revealed by environmental metagenomics. J. Gen. Virol. 2009, 90, 2418–2424. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Kapitonov, V.V.; Koonin, E.V. A new family of hybrid virophages from an animal gut metagenome. Biol. Direct 2015, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Zaragoza-Solas, A.; Rodriguez-Valera, F.; Lopez-Perez, M. Metagenome Mining Reveals Hidden Genomic Diversity of Pelagimyophages in Aquatic Environments. mSystems 2020, 5, e00905-19. [Google Scholar] [CrossRef] [Green Version]
- Victoria, J.G.; Kapoor, A.; Dupuis, K.; Schnurr, D.P.; Delwart, E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008, 4, e1000163. [Google Scholar] [CrossRef] [Green Version]
- Dayaram, A.; Galatowitsch, M.; Harding, J.S.; Argüello-Astorga, G.R.; Varsani, A. Novel circular DNA viruses identified in Procordulia grayi and Xanthocnemis zealandica larvae using metagenomic approaches. Infect. Genet. Evol. 2014, 22, 134–141. [Google Scholar] [CrossRef]
- Fehér, E.; Mihalov-Kovács, E.; Kaszab, E.; Malik, Y.S.; Marton, S.; Bányai, K. Genomic Diversity of CRESS DNA Viruses in the Eukaryotic Virome of Swine Feces. Microorganisms 2021, 9, 1426. [Google Scholar] [CrossRef]
- Jackson, E.W.; Bistolas, K.S.; Button, J.B.; Hewson, I. Novel Circular Single-Stranded DNA Viruses among an Asteroid, Echinoid and Holothurian (Phylum: Echinodermata). PLoS ONE 2016, 11, e0166093. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Nishijima, S.; Furuta, Y.; Yoshimura, J.; Suda, W.; Oshima, K.; Hattori, M.; Morishita, S. Long-read metagenomic exploration of extrachromosomal mobile genetic elements in the human gut. Microbiome 2019, 7, 119. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Dupont, C.L.; Jones, M.B.; Pham, K.M.; Jiang, Z.D.; DuPont, H.L.; Highlander, S.K. Visualization-assisted binning of metagenome assemblies reveals potential new pathogenic profiles in idiopathic travelers’ diarrhea. Microbiome 2018, 6, 201. [Google Scholar] [CrossRef]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of Bat Guano from Nine Northern California Roosts. J. Virol. 2021, 95, e01713-20. [Google Scholar] [CrossRef] [PubMed]
- Gulino, K.; Rahman, J.; Badri, M.; Morton, J.; Bonneau, R.; Ghedin, E. Initial Mapping of the New York City Wastewater Virome. mSystems 2020, 5, e00876-19. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhou, H.; Huang, Y.; Xie, Z.; Zhang, M.; Wei, Y.; Li, J.; Ma, Y.; Luo, M.; Ding, W.; et al. Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep. Genome Biol. 2021, 22, 207. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, C.B.; Shkoporov, A.N.; Upadrasta, A.; Khokhlova, E.V.; Ross, R.P.; Hill, C. Probing the “Dark Matter” of the Human Gut Phageome: Culture Assisted Metagenomics Enables Rapid Discovery and Host-Linking for Novel Bacteriophages. Front. Cell. Infect. Microbiol. 2021, 11, 616918. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.M.; Kelly, L.; Muscatt, G.; Box, J.D.; Hargraves, N.; Lilley, D.; Jameson, E. The Human Gut Phageome: Origins and Roles in the Human Gut Microbiome. Front. Cell. Infect. Microbiol. 2021, 11, 643214. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.S.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 68. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral communities of the human gut: Metagenomic analysis of composition and dynamics. Mob. DNA 2017, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Chaudhary, S.; Lu, N.; Duff, M.; Heffel, M.; McKinney, C.A.; Bedenice, D.; Marthaler, D. Metagenomic Next-Generation Sequencing Reveal Presence of a Novel Ungulate Bocaparvovirus in Alpacas. Viruses 2019, 11, 701. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Ahmed, S.S.; Yeung, H.C.; Li, K.S.M.; Fan, R.Y.Y.; Cheng, T.Y.C.; Cai, J.P.; Wang, M.; Zheng, B.J.; Wong, S.S.Y.; et al. Identification and interspecies transmission of a novel bocaparvovirus among different bat species in China. J. Gen. Virol. 2016, 97, 3345–3358. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Tsoi, H.W.; Patteril, N.G.; Yeung, H.C.; Joseph, S.; Wong, E.Y.M.; Muhammed, R.; Chow, F.W.N.; Wernery, U.; et al. Two novel dromedary camel bocaparvoviruses from dromedaries in the Middle East with unique genomic features. J. Gen. Virol. 2017, 98, 1349–1359. [Google Scholar] [CrossRef]
- Kapoor, A.; Mehta, N.; Esper, F.; Poljsak-Prijatelj, M.; Quan, P.L.; Qaisar, N.; Delwart, E.; Lipkin, W.I. Identification and characterization of a new bocavirus species in gorillas. PLoS ONE 2010, 5, e11948. [Google Scholar] [CrossRef] [Green Version]
- Ao, Y.; Li, X.; Li, L.; Xie, X.; Jin, D.; Yu, J.; Lu, S.; Duan, Z. Two novel bocaparvovirus species identified in wild Himalayan marmots. Sci. China. Life Sci. 2017, 60, 1348–1356. [Google Scholar] [CrossRef]
- Gunn, L.; Collins, P.J.; Fanning, S.; McKillen, J.; Morgan, J.; Staines, A.; O’Shea, H. Detection and characterisation of novel bocavirus (genus Bocaparvovirus) and gastroenteritis viruses from asymptomatic pigs in Ireland. Infect. Ecol. Epidemiol. 2015, 5, 27270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Song, F.; Xiu, L.; Liu, Y.; Yang, J.; Yao, L.; Peng, J. Identification and characterization of a novel rodent bocavirus from different rodent species in China. Emerg. Microbes Infect. 2018, 7, 48. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Alabi, O.J.; Westrick, N.M.; Golino, D. Prunus geminivirus A: A Novel Grablovirus Infecting Prunus spp. Plant Dis. 2018, 102, 1246–1253. [Google Scholar] [CrossRef] [Green Version]
- Voorburg, C.M.; Yan, Z.; Bergua-Vidal, M.; Wolters, A.A.; Bai, Y.; Kormelink, R. Ty-1, a universal resistance gene against geminiviruses that is compromised by co-replication of a betasatellite. Mol. Plant Pathol. 2020, 21, 160–172. [Google Scholar] [CrossRef] [Green Version]
- De Nazare Almeida Dos Reis, L.; Fonseca, M.E.N.; Ribeiro, S.G.; Naito, F.Y.B.; Boiteux, L.S.; Pereira-Carvalho, R.C. Metagenomics of Neotropical Single-Stranded DNA Viruses in Tomato Cultivars with and without the Ty-1 Gene. Viruses 2020, 12, 819. [Google Scholar] [CrossRef]
- Alarcon-Schumacher, T.; Guajardo-Leiva, S.; Anton, J.; Diez, B. Elucidating Viral Communities During a Phytoplankton Bloom on the West Antarctic Peninsula. Front. Microbiol. 2019, 10, 1014. [Google Scholar] [CrossRef]
- Hendry, K.R.; Meredith, M.P.; Ducklow, H.W. The marine system of the West Antarctic Peninsula: Status and strategy for progress. Philos. Trans. A Math Phys Eng Sci. 2018, 376, 20170179. [Google Scholar] [CrossRef] [PubMed]
- Faizah, A.N.; Kobayashi, D.; Isawa, H.; Amoa-Bosompem, M.; Murota, K.; Higa, Y.; Futami, K.; Shimada, S.; Kim, K.S.; Itokawa, K.; et al. Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan. Viruses 2020, 12, 264. [Google Scholar] [CrossRef] [Green Version]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S., 3rd; Bolay, F.K.; Diclaro, J.W., 2nd; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Ducatez, M.F.; Guérin, J.L. Identification of a novel coronavirus from guinea fowl using metagenomics. Methods Mol. Biol. 2015, 1282, 27–31. [Google Scholar] [CrossRef]
- Liais, E.; Croville, G.; Mariette, J.; Delverdier, M.; Lucas, M.N.; Klopp, C.; Lluch, J.; Donnadieu, C.; Guy, J.S.; Corrand, L.; et al. Novel avian coronavirus and fulminating disease in guinea fowl, France. Emerg. Infect. Dis. 2014, 20, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Dennis, T.P.W.; Flynn, P.J.; de Souza, W.M.; Singer, J.B.; Moreau, C.S.; Wilson, S.J.; Gifford, R.J. Insights into Circovirus Host Range from the Genomic Fossil Record. J. Virol. 2018, 92, e00145-18. [Google Scholar] [CrossRef] [Green Version]
- Hui, A.; Altan, E.; Slovis, N.; Fletcher, C.; Deng, X.; Delwart, E. Circovirus in Blood of a Febrile Horse with Hepatitis. Viruses 2021, 13, 944. [Google Scholar] [CrossRef]
- Katsuta, R.; Sunaga, F.; Oi, T.; Doan, Y.H.; Tsuzuku, S.; Suzuki, Y.; Sano, K.; Katayama, Y.; Omatsu, T.; Oba, M.; et al. First identification of Sapoviruses in wild boar. Virus Res. 2019, 271, 197680. [Google Scholar] [CrossRef]
- Boukari, W.; Alcalá-Briseño, R.I.; Kraberger, S.; Fernandez, E.; Filloux, D.; Daugrois, J.H.; Comstock, J.C.; Lett, J.M.; Martin, D.P.; Varsani, A.; et al. Occurrence of a novel mastrevirus in sugarcane germplasm collections in Florida, Guadeloupe and Réunion. Virol. J. 2017, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Fontenele, R.S.; Alves-Freitas, D.M.T.; Silva, P.I.T.; Foresti, J.; Silva, P.R.; Godinho, M.T.; Varsani, A.; Ribeiro, S.G. Discovery of the first maize-infecting mastrevirus in the Americas using a vector-enabled metagenomics approach. Arch. Virol. 2018, 163, 263–267. [Google Scholar] [CrossRef]
- Claverie, S.; Ouattara, A.; Hoareau, M.; Filloux, D.; Varsani, A.; Roumagnac, P.; Martin, D.P.; Lett, J.M.; Lefeuvre, P. Exploring the diversity of Poaceae-infecting mastreviruses on Reunion Island using a viral metagenomics-based approach. Sci. Rep. 2019, 9, 12716. [Google Scholar] [CrossRef]
- Feng, C.; Feng, J.; Wang, Z.; Pedersen, C.; Wang, X.; Saleem, H.; Domier, L.; Marzano, S.L. Identification of the Viral Determinant of Hypovirulence and Host Range in Sclerotiniaceae of a Genomovirus Reconstructed from the Plant Metagenome. J. Virol. 2021, 95, e0026421. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Padilla, A.; Rodriguez-Romero, J.; Gomez-Cid, I.; Pacifico, D.; Ayllon, M.A. Novel Mycoviruses Discovered in the Mycovirome of a Necrotrophic Fungus. mBio 2021, 12, e03705-20. [Google Scholar] [CrossRef]
- Saad, N.; Olmstead, J.W.; Varsani, A.; Polston, J.E.; Jones, J.B.; Folimonova, S.Y.; Harmon, P.F. Discovery of Known and Novel Viruses in Wild and Cultivated Blueberry in Florida through Viral Metagenomic Approaches. Viruses 2021, 13, 1165. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Yoo, D.; Liu, W.T. Metagenomics Reveals a Novel Virophage Population in a Tibetan Mountain Lake. Microbes Environ. 2016, 31, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Miranda, J.A.; Culley, A.I.; Schvarcz, C.R.; Steward, G.F. RNA viruses as major contributors to Antarctic virioplankton. Environ. Microbiol. 2016, 18, 3714–3727. [Google Scholar] [CrossRef]
- Molnar, J.; Magyar, B.; Schneider, G.; Laczi, K.; Valappil, S.K.; Kovacs, A.L.; Nagy, I.K.; Rakhely, G.; Kovacs, T. Identification of a novel archaea virus, detected in hydrocarbon polluted Hungarian and Canadian samples. PLoS ONE 2020, 15, e0231864. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Watai, H.; Honda, T.; Mihara, T.; Omae, K.; Roux, S.; Blanc-Mathieu, R.; Yamamoto, K.; Hingamp, P.; Sako, Y.; et al. Environmental Viral Genomes Shed New Light on Virus-Host Interactions in the Ocean. mSphere 2017, 2, e00359-16. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, Y.; Ermini, L.; Wang, H.; Carty, K.; Cheung, M.S. LongQC: A Quality Control Tool for Third Generation Sequencing Long Read Data. G3 (Bethesda) 2020, 10, 1193–1196. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 June 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F. Trim Galore! Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 28 June 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Insights, Q.D. QIAGEN CLC Genomics Workbench, Online Resource. Available online: https://digitalinsights.qiagen.com (accessed on 28 June 2022).
- BBMap–Bushnell B. Available online: sourceforge.net/projects/bbmap/ (accessed on 28 June 2022).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [Green Version]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019, 20, 1125–1136. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef] [Green Version]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Muller, W.E.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Ruby, J.G.; Bellare, P.; Derisi, J.L. PRICE: Software for the targeted assembly of components of (Meta) genomic sequence data. G3 (Bethesda) 2013, 3, 865–880. [Google Scholar] [CrossRef] [Green Version]
- CodonCode Aligner-DNA Sequence Assembly and Alignment on Windows and Mac OS X. CodonCode Corporation, Dedham, MA, USA. Online Resource. Available online: http://www.codoncode.com/ (accessed on 28 June 2022).
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, S.; Raymond, F.; Godzaridis, E.; Laviolette, F.; Corbeil, J. Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2013, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S.L.; Phillippy, A.M.; Zimin, A.; Puiu, D.; Magoc, T.; Koren, S.; Treangen, T.J.; Schatz, M.C.; Delcher, A.L.; Roberts, M.; et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012, 22, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- EBI. Multiple Sequence Alignment. Online Resource. Available online: https://www.ebi.ac.uk/Tools/msa/ (accessed on 28 June 2022).
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Taking variation of evolutionary rates between sites into account in inferring phylogenies. J. Mol. Evol. 2001, 53, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G.; Pikis, A.; Thompson, J. Evolution and biochemistry of family 4 glycosidases: Implications for assigning enzyme function in sequence annotations. Mol. Biol. Evol. 2009, 26, 2487–2497. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temmam, S.; Davoust, B.; Berenger, J.M.; Raoult, D.; Desnues, C. Viral metagenomics on animals as a tool for the detection of zoonoses prior to human infection? Int. J. Mol. Sci. 2014, 15, 10377–10397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarestrup, F.M.; Koopmans, M.G. Sharing Data for Global Infectious Disease Surveillance and Outbreak Detection. Trends Microbiol. 2016, 24, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Destoumieux-Garzon, D.; Mavingui, P.; Boetsch, G.; Boissier, J.; Darriet, F.; Duboz, P.; Fritsch, C.; Giraudoux, P.; Le Roux, F.; Morand, S.; et al. The One Health Concept: 10 Years Old and a Long Road Ahead. Front. Vet. Sci. 2018, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Andreani, J.; Schulz, F.; Di Pinto, F.; Levasseur, A.; Woyke, T.; La Scola, B. Morphological and Genomic Features of the New Klosneuvirinae Isolate Fadolivirus IHUMI-VV54. Front. Microbiol. 2021, 12, 719703. [Google Scholar] [CrossRef]
- Zakham, F.; Albalawi, A.E.; Alanazi, A.D.; Truong Nguyen, P.; Alouffi, A.S.; Alaoui, A.; Sironen, T.; Smura, T.; Vapalahti, O. Viral RNA Metagenomics of Hyalomma Ticks Collected from Dromedary Camels in Makkah Province, Saudi Arabia. Viruses 2021, 13, 1396. [Google Scholar] [CrossRef]
- Guajardo-Leiva, S.; Pedrós-Alió, C.; Salgado, O.; Pinto, F.; Díez, B. Active Crossfire Between Cyanobacteria and Cyanophages in Phototrophic Mat Communities Within Hot Springs. Front. Microbiol. 2018, 9, 2039. [Google Scholar] [CrossRef]
- Muhamad Rizal, N.S.; Neoh, H.M.; Ramli, R.; PR, A.L.K.P.; Hanafiah, A.; Abdul Samat, M.N.; Tan, T.L.; Wong, K.K.; Nathan, S.; Chieng, S.; et al. Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics 2020, 10, 816. [Google Scholar] [CrossRef]
- Harvey, E.; Holmes, E.C. Diversity and evolution of the animal virome. Nat. Rev. Microbiol. 2022, 20, 321–334. [Google Scholar] [CrossRef]
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses by Use of RNA Sequencing-Based Metagenomics: A Systematic Comparison to a Commercial PCR Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddaway, A.P.; Wood, A.M.; Hedges, L.V. How to Do a Systematic Review: A Best Practice Guide for Conducting and Reporting Narrative Reviews, Meta-Analyses, and Meta-Syntheses. Annu. Rev. Psychol. 2019, 70, 747–770. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H. Literature review as a research methodology: An overview and guidelines. J. Bus. Res. 2019, 104, 333–339. [Google Scholar] [CrossRef]
- Walden, C.; Carbonero, F.; Zhang, W. Assessing impacts of DNA extraction methods on next generation sequencing of water and wastewater samples. J. Microbiol. Methods 2017, 141, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Klenner, J.; Kohl, C.; Dabrowski, P.W.; Nitsche, A. Comparing Viral Metagenomic Extraction Methods. Curr. Issues Mol. Biol. 2017, 24, 59–70. [Google Scholar] [CrossRef]
- Hjelmso, M.H.; Hellmer, M.; Fernandez-Cassi, X.; Timoneda, N.; Lukjancenko, O.; Seidel, M.; Elsasser, D.; Aarestrup, F.M.; Lofstrom, C.; Bofill-Mas, S.; et al. Evaluation of Methods for the Concentration and Extraction of Viruses from Sewage in the Context of Metagenomic Sequencing. PLoS ONE 2017, 12, e0170199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Chen, Y.; Zhang, S.; Lyu, Y.; Zou, Y.; Li, J. DNA Enrichment Methods for Microbial Symbionts in Marine Bivalves. Microorganisms 2022, 10, 393. [Google Scholar] [CrossRef]
- Parras-Molto, M.; Rodriguez-Galet, A.; Suarez-Rodriguez, P.; Lopez-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Conceicao-Neto, N.; Zeller, M.; Lefrere, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Shi, Y.; Tong, H.I.; Kang, W.; Wang, Z.; Allmann, E.; Lu, Y. Effective concentration, recovery, and detection of infectious adenoviruses from environmental waters. J. Virol. Methods 2016, 229, 78–85. [Google Scholar] [CrossRef]
- Shi, X.; Shao, C.; Luo, C.; Chu, Y.; Wang, J.; Meng, Q.; Yu, J.; Gao, Z.; Kang, Y. Microfluidics-Based Enrichment and Whole-Genome Amplification Enable Strain-Level Resolution for Airway Metagenomics. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotz, C.A.; Sanders, J.G.; Zuniga, C.; Zaramela, L.S.; Knight, R.; Zengler, K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.T.; Pope, C.E.; Marsh, R.L.; Wolter, D.J.; Weiss, E.J.; Hager, K.R.; Vo, A.T.; Brittnacher, M.J.; Radey, M.C.; Hayden, H.S.; et al. Human and Extracellular DNA Depletion for Metagenomic Analysis of Complex Clinical Infection Samples Yields Optimized Viable Microbiome Profiles. Cell Rep. 2019, 26, 2227–2240.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoendel, M.; Jeraldo, P.R.; Greenwood-Quaintance, K.E.; Yao, J.Z.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Comparison of microbial DNA enrichment tools for metagenomic whole genome sequencing. J. Microbiol. Methods 2016, 127, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, M.; Feehily, C.; Walsh, C.J.; Fenelon, M.; Murphy, E.F.; McAuliffe, F.M.; van Sinderen, D.; O’Toole, P.W.; O’Sullivan, O.; Cotter, P.D. Evaluation of methods for the reduction of contaminating host reads when performing shotgun metagenomic sequencing of the milk microbiome. Sci. Rep. 2020, 10, 21665. [Google Scholar] [CrossRef] [PubMed]
- Ahannach, S.; Delanghe, L.; Spacova, I.; Wittouck, S.; Van Beeck, W.; De Boeck, I.; Lebeer, S. Microbial enrichment and storage for metagenomics of vaginal, skin, and saliva samples. iScience 2021, 24, 103306. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; Sanz, T.K.C. Next-generation sequencing and waste/wastewater treatment: A comprehensive overview. Rev. Env. Sci. Biotechnol. 2019, 18, 635–680. [Google Scholar] [CrossRef]
- Ayling, M.; Clark, M.D.; Leggett, R.M. New approaches for metagenome assembly with short reads. Brief. Bioinform. 2020, 21, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Arroyo Muhr, L.S.; Lagheden, C.; Hassan, S.S.; Kleppe, S.N.; Hultin, E.; Dillner, J. De novo sequence assembly requires bioinformatic checking of chimeric sequences. PLoS ONE 2020, 15, e0237455. [Google Scholar] [CrossRef]
- Somerville, V.; Lutz, S.; Schmid, M.; Frei, D.; Moser, A.; Irmler, S.; Frey, J.E.; Ahrens, C.H. Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiol. 2019, 19, 143. [Google Scholar] [CrossRef]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Jehmlich, N.; Palatinszky, M.; Vierheilig, J.; Bulaev, A.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Frank, J.A.; Pan, Y.; Tooming-Klunderud, A.; Eijsink, V.G.H.; McHardy, A.C.; Nederbragt, A.J.; Pope, P.B. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci. Rep. 2016, 6, 25373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porubsky, D.; Garg, S.; Sanders, A.D.; Korbel, J.O.; Guryev, V.; Lansdorp, P.M.; Marschall, T. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun. 2017, 8, 1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favale, N.; Costa, S.; Scapoli, C.; Carrieri, A.; Sabbioni, S.; Tamburini, E.; Benazzo, A.; Bernacchia, G. Reconstruction of Acinetobacter johnsonii ICE_NC genome using hybrid de novo genome assemblies and identification of the 12alpha-hydroxysteroid dehydrogenase gene. J. Appl. Microbiol. 2022, 133, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.G.; Herrera-Galeano, J.E.; Redden, C.L.; Luu, T.V.; Servetas, S.L.; Mateczun, A.J.; Mokashi, V.P.; Bishop-Lilly, K.A. Comparison of three next-generation sequencing platforms for metagenomic sequencing and identification of pathogens in blood. BMC Genom. 2014, 15, 96. [Google Scholar] [CrossRef] [Green Version]
- Be, N.A.; Thissen, J.B.; Gardner, S.N.; McLoughlin, K.S.; Fofanov, V.Y.; Koshinsky, H.; Ellingson, S.R.; Brettin, T.S.; Jackson, P.J.; Jaing, C.J. Detection of Bacillus anthracis DNA in complex soil and air samples using next-generation sequencing. PLoS ONE 2013, 8, e73455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowska-Osko, I.; Perlejewski, K.; Nakamura, S.; Motooka, D.; Stokowy, T.; Kosinska, J.; Popiel, M.; Ploski, R.; Horban, A.; Lipowski, D.; et al. Sensitivity of Next-Generation Sequencing Metagenomic Analysis for Detection of RNA and DNA Viruses in Cerebrospinal Fluid: The Confounding Effect of Background Contamination. Adv. Exp. Med. Biol. 2016, 944, 53–62. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Novel Viruses Found in mNGS Studies | References |
|---|---|---|
| Animal | Flavivirus | [45,67,85,147,212,213] |
| Coronanvirus | [25,214,215] | |
| Circovirus | [63,120,159,216,217] | |
| Bocaparvovirus | [121,124,200] | |
| Siphoviridae, Myoviridae, Podoviridae, crAss-like viruses | [195] | |
| Sapovirus | [44,95,218]. | |
| Plant | Prunus Geminivirus | [207] |
| Mastrevirus | [219,220,221] | |
| Begomovirus | [132,209] | |
| Genomovirus | [222] | |
| Narnavirus | [223] | |
| Tepovirus | [224] | |
| Environment | Phycodnavirus | [210,225] |
| Picornavirus | [50,226] | |
| PA-SR01 | [163] | |
| Picobirnaviridae | [40] | |
| Epatitis E virus | [171] | |
| Methanosarcina virus MV (MetMV) | [227] | |
| Halovirus | [42,228] | |
| SAR11 phage | [185] |
| Software | Reference | Available at |
|---|---|---|
| FastQC | [230] | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 June 2022) |
| Trimmomatic | [231] | |
| Trim Galore | [232] | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 28 June 2022) |
| Cutadapt | [233] | https://journal.embnet.org/index.php/embnetjournal/article/view/200/479 (accessed on 28 June 2022) |
| CLC Genomics Workbench (Qiagen) | [234] | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/qiagen-clc-genomics/ (accessed on 28 June 2022) |
| BBDuk (part of BBTools/BBMap package) | [235] | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/ (accessed on 28 June 2022) |
| Software | Reference |
|---|---|
| SPAdes | [31] |
| MetaSPAdes | [244] |
| Megahit | [245] |
| Velvet | [246] |
| Trinity | [247] |
| SOAPdenovo2 | [248] |
| Ensemble Assembler | [249] |
| MIRA | [250] |
| PRICE | [251] |
| Codon Code Aligner | [252]; https://www.codoncode.com/aligner/ (accessed on 28 June 2022) |
| ABySS | [253] |
| Ray Meta | [254] |
| CAP3 | [255] |
| CLC Genomics Workbench (Qiagen) | [234] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassi, C.; Guerriero, P.; Pierantoni, M.; Callegari, E.; Sabbioni, S. Novel Virus Identification through Metagenomics: A Systematic Review. Life 2022, 12, 2048. https://doi.org/10.3390/life12122048
Bassi C, Guerriero P, Pierantoni M, Callegari E, Sabbioni S. Novel Virus Identification through Metagenomics: A Systematic Review. Life. 2022; 12(12):2048. https://doi.org/10.3390/life12122048
Chicago/Turabian StyleBassi, Cristian, Paola Guerriero, Marina Pierantoni, Elisa Callegari, and Silvia Sabbioni. 2022. "Novel Virus Identification through Metagenomics: A Systematic Review" Life 12, no. 12: 2048. https://doi.org/10.3390/life12122048