
COVID-19—A Trigger Factor for Severe Immune-Mediated Thrombocytopenia in Active Rheumatoid Arthritis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Causes of Thrombocytopenia
2.1. Decreased Production
2.2. Sequestration
2.3. Increased Destruction
2.4. Drug-Induced Thrombocytopenia
2.5. Pregnancy
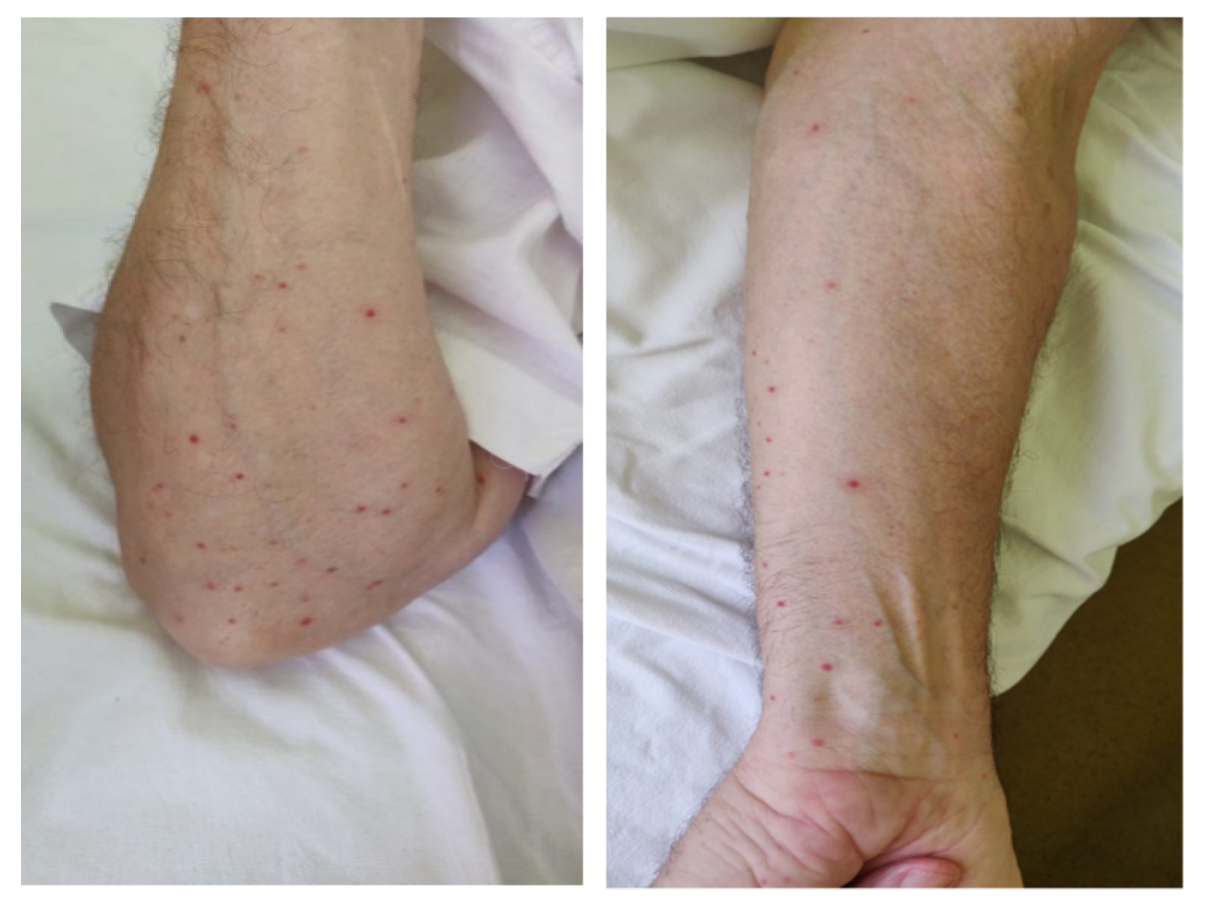
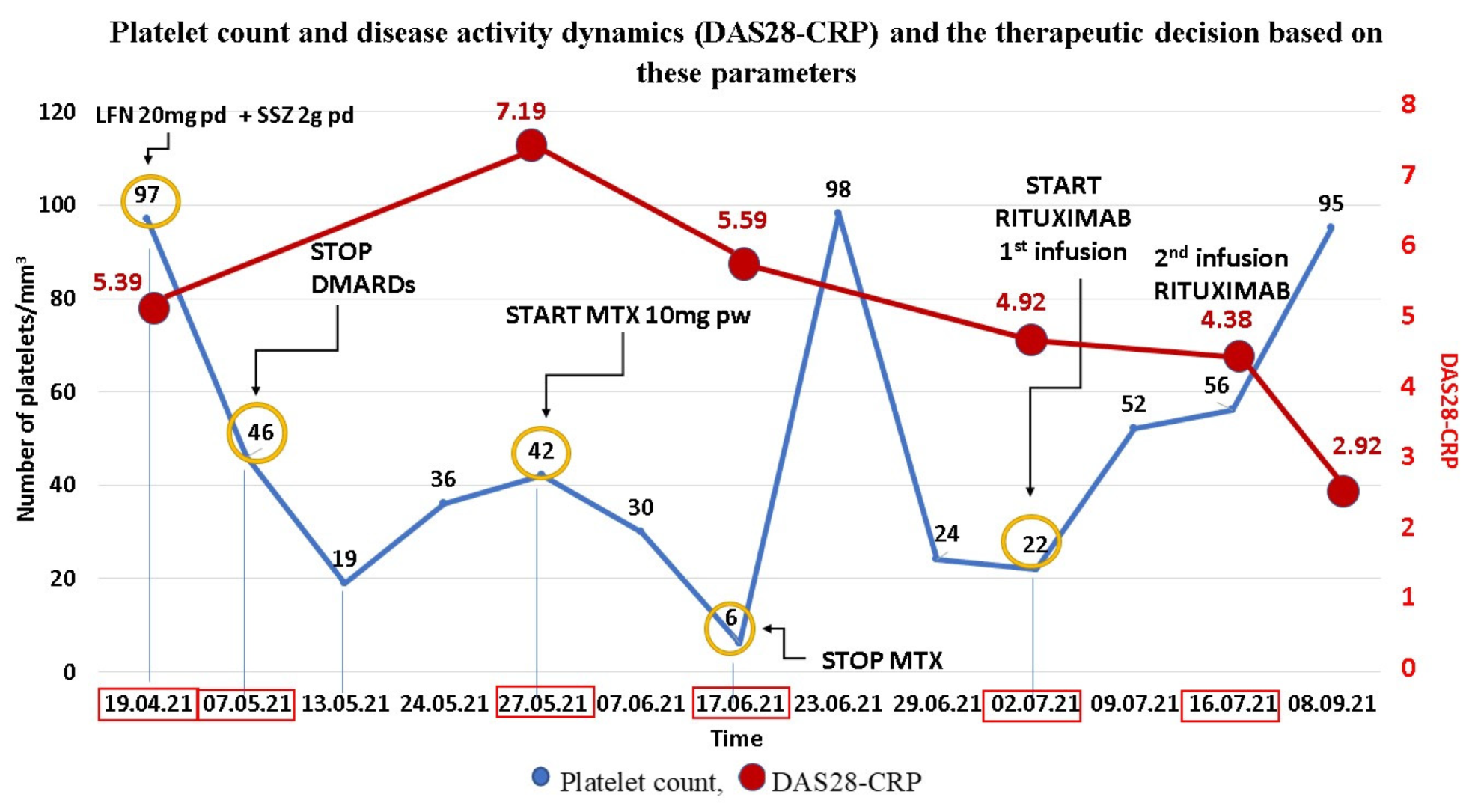
3. Clinical Experience—Case Report
4. Rheumatoid Arthritis-Related Thrombocytopenia
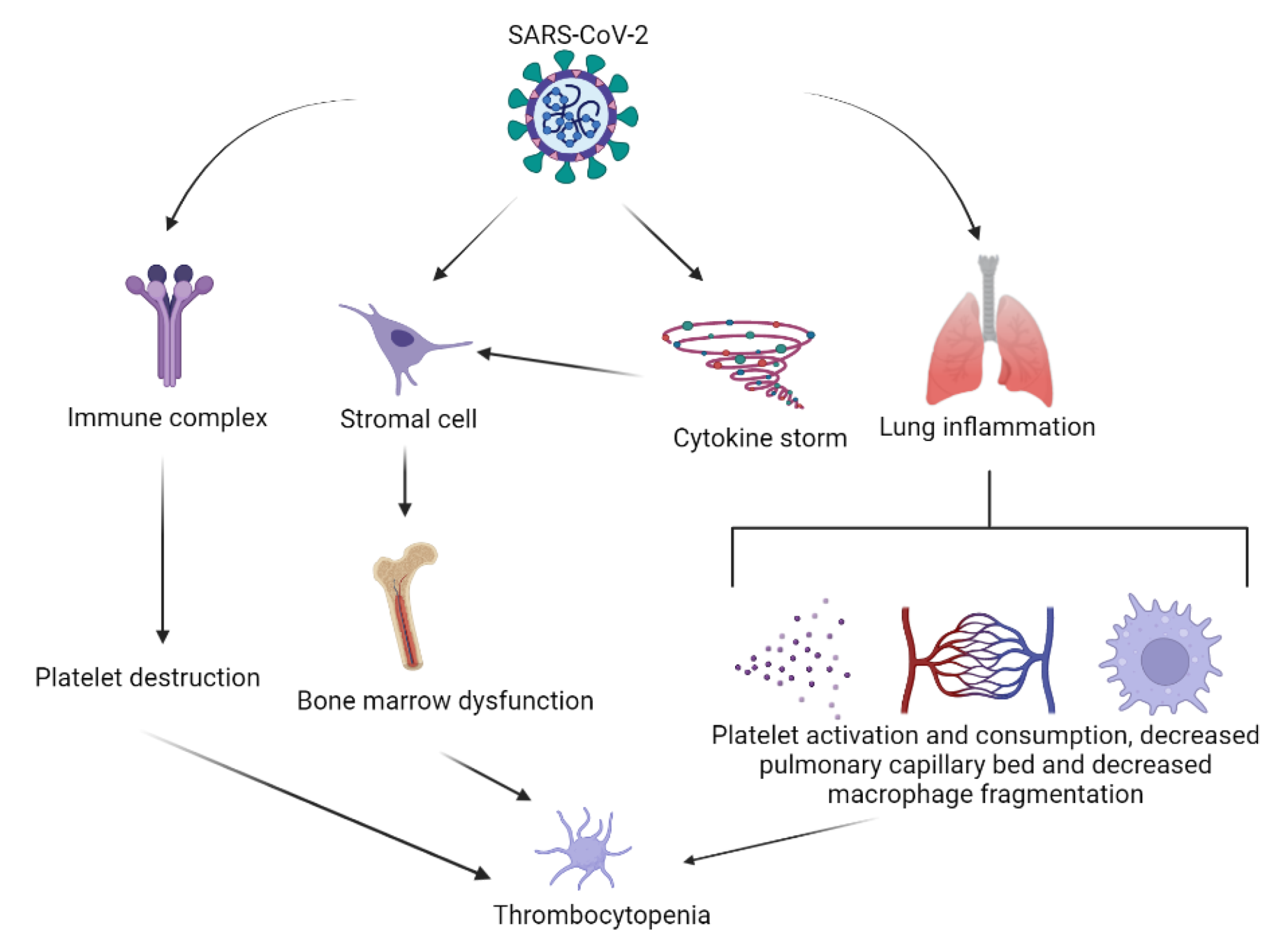
5. SARS-CoV-2 Infection-Induced Thrombocytopenia
- Platelet activation and subsequent clearance by reticuloendothelial system;
- Platelet clearance due to increased endothelial damage;
- Platelet autoantibody formation, with subsequent platelet clearance;
- Splenic/hepatic sequestration;
- Marrow/megakaryocyte suppression;
6. SARS-CoV-2 mRNA Vaccine-Related Thrombosis and Thrombocytopenia
7. Rheumatoid Arthritis and SARS-CoV-2
7.1. The ACE-Dependent Pathway
7.2. The Macrophage-Dependent Pathway
8. Management of RA and Thrombocytopenia in COVID-19 Patients
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenberg, E.M.; Kaled, E.S. Thrombocytopenia. Crit. Care Nurs. Clin. N. Am. 2013, 25, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, A.I. Thrombocytopenia. Prim. Care-Clin. Off. Pract. 2016, 43, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Smock, K.J.; Perkins, S.L. Thrombocytopenia: An update. Int. J. Lab. Hematol. 2014, 36, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Jeon, K.; Kim, M.; Lee, J.; Lee, J.S.; Kim, H.S.; Kang, H.J.; Lee, Y.K. Immature platelet fraction: A useful marker for identifying the cause of thrombocytopenia and predicting platelet recovery. Medicine 2020, 99. [Google Scholar] [CrossRef]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinkels, M.; Rijkers, M.; Voorberg, J.; Vidarsson, G.; Leebeek, F.W.G.; Jansen, A.J.G. Emerging concepts in immune thrombocytopenia. Front. Immunol. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Raadsen, M.; du Toit, J.; Langerak, T.; van Bussel, B.; van Gorp, E.; Goeijenbier, M. Thrombocytopenia in virus infections. J. Clin. Med. 2021, 10, 877. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Jyothi, Y.; Rabbani, S.I. Thrombocytopenia and its causes. J. Chem. Pharm. Res. 2016, 8, 184–189. [Google Scholar]
- Sripati, T.K.; Govindarajan; Ganapathy, H. Evaluation of thrombocytopenia in hematological malignancies. J. Pharm. Res. Int. 2020, 32, 81–85. [Google Scholar] [CrossRef]
- Weycker, D.; Hatfield, M.; Grossman, A.; Hanau, A.; Lonshteyn, A.; Sharma, A.; Chandler, D. Risk and consequences of chemotherapy-induced thrombocytopenia in US clinical practice. BMC Cancer 2019, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Silczuk, A.; Habrat, B. Alcohol-induced thrombocytopenia: Current review. Alcohol 2020, 86, 9–16. [Google Scholar] [CrossRef]
- Ostadi, Z.; Shadvar, K.; Sanaie, S.; Mahmoodpoor, A.; Saghaleini, S.H. Thrombocytopenia in the intensive care unit. Pak. J. Med Sci. 2019, 35, 282. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, S.; Sun, Y.; Lin, Q.; Liao, X.; Zhang, J.; Luo, J.; Qian, H.; Duan, L.; Shi, G. Clinical characteristics of immune thrombocytopenia associated with autoimmune disease: A retrospective study. Medicine 2016, 95, e5565. [Google Scholar] [CrossRef]
- Kappler, S.; Ronan-Bentle, S.; Graham, A. Thrombotic microangiopathies (TTP, HUS, HELLP). Hematol. Oncol. Clin. N. Am. 2017, 31, 1081–1103. [Google Scholar] [CrossRef]
- Vayne, C.; Guéry, E.A.; Rollin, J.; Baglo, T.; Petermann, R.; Gruel, Y. Pathophysiology and diagnosis of drug-induced immune thrombocytopenia. J. Clin. Med. 2020, 9, 2212. [Google Scholar] [CrossRef]
- Al-Husban, N.; Al-Kuran, O.; Khadra, M.; Fram, K. Thrombocytopenia in pregnancy; prevalence, causes and fetomaternal outcome. Clin. Exp. Obstet. Gynecol. 2020, 47, 21–26. [Google Scholar] [CrossRef]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Ciurea, A.; Papagiannoulis, E.; Bürki, K.; von Loga, I.; Micheroli, R.; Möller, B.; Rubbert-Roth, A.; Andor, M.; Bräm, R.; Müller, A.; et al. Impact of the COVID-19 pandemic on the disease course of patients with inflammatory rheumatic diseases: Results from the Swiss Clinical Quality Management Cohort. Ann. Rheum. Dis. 2021, 80, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Vanni, K.M.M.; Lyu, H.; Solomon, D.H. Cytopenias among patients with rheumatic diseases using methotrexate: A meta-analysis of randomized controlled clinical trials. Rheumatology 2020, 59, 709–717. [Google Scholar] [CrossRef]
- Hanrahan, P.S.; Scrtvens, G.A.; Russell, A.S. Clinical practice prospective long term follow-up of methotrexate therapy in rheumatoid arthritis: Toxicity, efficacy and radiological progression. Br. J. Rheumatol. 1989, 28, 147–153. [Google Scholar] [CrossRef]
- Alarcóan, G.S.; Tracy, I.C.; Blackburn, W.D. Methotrexate in rheumatoid arthritis. Toxic effects as the major factor in limiting long-term treatment. Arthritis Rheum. 1989, 32, 671–676. [Google Scholar] [CrossRef]
- Gutierrez-Ureña, S.; Molina, J.F.; García, C.O.; Cuéllar, M.L.; Espinoza, L.R. Pancytopenia secondary to methotrexate therapy in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Fraser, P. Elevated mean corpuscular volume as a predictor of hematologic toxicity due to methotrexate therapy. Arthritis Rheum. 1989, 32, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.J. Hematological manifestations of rheumatoid arthritis. Scand. J. Rheumatol. 2002, 31, 251–259. [Google Scholar] [CrossRef]
- Pathare, S.K.; Heycock, C.; Hamilton, J. TNFalpha blocker-induced thrombocytopenia. Rheumatology 2006, 45, 1313–1314. [Google Scholar] [CrossRef] [Green Version]
- Kistangari, G.; McCrae, K.R. Immune thrombocytopenia. Hematol. Oncol. Clin. N. Am. 2013, 27, 495–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Chary, M.; Salehi, I.; Bansal, R. Immune-mediated adalimumab-induced thrombocytopenia for the treatment of ulcerative colitis. Int. J. Pharm. Pharm. Sci. 2015, 7, 456–458. [Google Scholar]
- Ichikawa, N.; Harigai, M.; Nakajima, A.; Hara, M.; Kamatani, N. Immune thrombocytopenic purpura associated with rheumatoid arthritis—A report of five cases and review of the literature. Mod. Rheumatol. 2001, 11, 246–250. [Google Scholar] [CrossRef]
- Han, X.; Li, C.; Zhang, S.; Hou, X.; Chen, Z.; Zhang, J.; Zhang, Y.; Sun, J.; Wang, Y. Why thromboembolism occurs in some patients with thrombocytopenia and treatment strategies. Thromb. Res. 2020, 196, 500–509. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.; Niloofa, R.; Jayarajah, U.; de Mel, S.; Abeysuriya, V.; Seneviratne, S.L. Hematological abnormalities in COVID-19: A narrative review. Am. J. Trop. Med. Hyg. 2021, 104, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhou, Q.; Xu, J. Mechanism of thrombocytopenia in COVID-19 patients. Ann. Hematol. 2020, 99, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Deruelle, E.; ben Hadj Salem, O.; Sep Hieng, S.; Pichereau, C.; Outin, H.; Jamme, M. Immune thrombocytopenia in a patient with COVID-19. Int. J. Hematol. 2020, 112, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Tao, X.; Cui, W.; Yi, B.; Pan, T.; Young, K.H.; Qian, W. SARS-CoV-2 induced thrombocytopenia as an important biomarker significantly correlated with abnormal coagulation function, increased intravascular blood clot risk and mortality in COVID-19 patients. Exp. Hematol. Oncol. 2020, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Xu, W.; Wang, Q.; Wang, C.; Hua, C.; Li, W.; Lu, L.; Jiang, S. Peptide-based membrane fusion inhibitors targeting HCOV-229E spike protein HR1 and HR2 domains. Int. J. Mol. Sci. 2018, 19, 487. [Google Scholar] [CrossRef] [Green Version]
- Hayward, A.C.; Aldridge, R.W.; Lewer, D.; Beale, S.; Johnson, A.M.; Zambon, M.; Fragaszy, E.B. Seasonality and immunity to laboratory-confirmed seasonal coronaviruses (HCoV-NL63, HCoV-OC43, and HCoV-229E): Results from the Flu Watch Cohort Study. Wellcome Open Res. 2020, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, X.; Jiao, Y.; Li, Z.; Liu, Q.; Yang, M.; Ye, J. Mechanisms Involved in the Development of Thrombocytopenia in Patients with COVID-19. Thromb. Res. 2020, 193, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Villalba, N.; Zulfiqar, A.A.; Auburtin, M.; Schuhmacher, M.H.; Meyer, A.; Maouche, Y.; Keller, O.; Andres, E. Thrombocytopenia in the course of COVID-19 infection. Eur. J. Case Rep. Intern. Med. 2020, 7, 001702. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, T. Secondary haemophagocytic lymphohistiocytosis: Experience from the Uppsala University Hospital. Upsala J. Med. Sci. 2015, 120, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Chen, L.; Zhang, S.; Dong, X.; Li, W.; Li, R.; Deng, Y.; Wang, T.; Xu, Y.; Liu, J.; et al. Risk factors for secondary hemophagocytic lymphohistiocytosis in severe coronavirus disease 2019 adult patients. BMC Infect. Dis. 2021, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, G.; de Mast, Q.; Markou, N.; Theodorakopoulou, M.; Komnos, A.; Mouktaroudi, M.; Netea, M.G.; Spyridopoulos, T.; Verheggen, R.J.; Hoogerwerf, J.; et al. Favorable anakinra responses in severe COVID-19 patients with secondary hemophagocytic lymphohistiocytosis. Cell Host Microbe 2020, 28, 117–123. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Bashash, D.; Hosseini-Baharanchi, F.S.; Rezaie-Tavirani, M.; Safa, M.; Dilmaghani, N.A.; Dilmaghani, N.A.; Faranoush, M.; Abolghasemi, H. The prognostic value of thrombocytopenia in COVID-19 patients; A systematic review and meta-analysis. Arch. Acad. Emerg. Med. 2020, 8, e75. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Banerjee, M. Immune thrombocytopenia secondary to COVID-19: A systematic review. SN Compr. Clin. Med. 2020, 2, 2048–2058. [Google Scholar] [CrossRef]
- Jayadharani, C.; Gayatri Devi, R.; Lakshmanan, G. Knowledge and awareness about thrombocytopenia associated with COVID-19. Int. J. Pharm. Res. 2020, 12, 419. [Google Scholar] [CrossRef]
- Klhůfek, J. The role of angiotensin-converting enzyme 2 in the pathogenesis of COVID-19: The villain or the hero? Acta Clin. Belg. Int. J. Clin. Lab. Med. 2020, 1–8. [Google Scholar] [CrossRef]
- Tiwari, R.; Mishra, A.R.; Mikaeloff, F.; Gupta, S.; Mirazimi, A.; Byrareddy, S.N.; Neogi, U.; Nayak, D. In silico and in vitro studies reveal complement system drives coagulation cascade in SARS-CoV-2 pathogenesis. Comput. Struct. Biotechnol. J. 2020, 18, 3734–3744. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.P.; Berking, C.; Biedermann, T.; Buhl, T.; Erpenbeck, L.; Eyerich, K.; Eyerich, S.; Ghoreschi, K.; Goebeler, M.; Ludwig, R.J.; et al. COVID-19 and immunological regulations—From basic and translational aspects to clinical implications. JDDG J. Ger. Soc. Dermatol. 2020, 18, 795–807. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef]
- Pavord, S.; Thachil, J.; Hunt, B.J.; Murphy, M.; Lowe, G.; Laffan, M.; Makris, M.; Newland, A.C.; Provan, D.; Grainger, J.D.; et al. Practical guidance for the management of adults with immune thrombocytopenia during the COVID-19 pandemic. Br. J. Haematol. 2020, 189, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Hippisley-Cox, J.; Patone, M.; Mei, X.W.; Saatci, D.; Dixon, S.; Khunti, K.; Zaccardi, F.; Watkinson, P.; Shankar-Hari, M.; Doidge, J.; et al. Risk of thrombocytopenia and thromboembolism after Covid-19 vaccination and SARS-CoV-2 positive testing: Self-controlled case series study. BMJ 2021, 374, n1931. [Google Scholar] [CrossRef]
- Al-Mayhani, T.; Saber, S.; Stubbs, M.J.; Losseff, N.A.; Perry, R.J.; Simister, R.J.; Gull, D.; Jäger, H.R.; Scully, M.A.; Werring, D.J. Ischaemic stroke as a presenting feature of ChAdOx1 NCoV-19 vaccine-induced immune thrombotic thrombocytopenia. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1247–1248. [Google Scholar] [CrossRef]
- Jabagi, M.J.; Botton, J.; Bertrand, M.; Weill, A.; Farrington, P.; Zureik, M.; Dray-Spira, R. Myocardial infarction, stroke, and pulmonary embolism after BNT162b2 MRNA COVID-19 vaccine in people aged 75 years or older. JAMA 2022, 327, 80–82. [Google Scholar] [CrossRef]
- Lee, E.J.; Cines, D.B.; Gernsheimer, T.; Kessler, C.; Michel, M.; Tarantino, M.D.; Semple, J.W.; Arnold, D.M.; Godeau, B.; Lambert, M.P.; et al. Thrombocytopenia following Pfizer and Moderna SARS-CoV-2 vaccination. Am. J. Hematol. 2021, 96, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Welsh, K.J.; Baumblatt, J.; Chege, W.; Goud, R.; Nair, N. Thrombocytopenia including immune thrombocytopenia after receipt of MRNA COVID-19 vaccines reported to the vaccine adverse event reporting system (VAERS). Vaccine 2021, 39, 3329–3332. [Google Scholar] [CrossRef]
- Okada, Y.; Sakai, R.; Sato-Fitoussi, M.; Nodera, M.; Yoshinaga, S.; Shibata, A.; Kurasawa, T.; Kondo, T.; Amano, K. Potential triggers for thrombocytopenia and/or hemorrhage by the BNT162b2 vaccine, Pfizer-BioNTech. Front. Med. 2021, 8. [Google Scholar] [CrossRef]
- Li, X.; Tong, X.; Yeung, W.W.Y.; Kuan, P.; Yum, S.H.H.; Chui, C.S.L.; Lai, F.T.T.; Wan, E.Y.F.; Wong, C.K.H.; Chan, E.W.Y.; et al. Two-dose COVID-19 vaccination and possible arthritis flare among patients with rheumatoid arthritis in Hong Kong. Ann. Rheum. Dis. 2021. [Google Scholar] [CrossRef]
- Fitzgerald, G.E.; Maguire, S.; Haroon, N. COVID-19: What do rheumatologists need to know? Curr. Rheumatol. Rep. 2021, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Listing, J.; Gerhold, K.; Zink, A. The risk of infections associated with rheumatoid arthritis, with its comorbidity and treatment. Rheumatology 2013, 52, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Hyrich, K.L.; Machado, P.M. Rheumatic disease and COVID-19: Epidemiology and outcomes. Nat. Rev. Rheumatol. 2021, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91. [Google Scholar] [CrossRef]
- Favalli, E.G.; Ingegnoli, F.; de Lucia, O.; Cincinelli, G.; Cimaz, R.; Caporali, R. COVID-19 infection and rheumatoid arthritis: Faraway, so close! Autoimmun. Rev. 2020, 19, 102523. [Google Scholar] [CrossRef] [PubMed]
- Koetz, K.; Bryl, E.; Spickschen, K.; O’Fallon, W.M.; Goronzy, J.J.; Weyand, C.M. T cell homeostasis in patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 9203. [Google Scholar] [CrossRef] [Green Version]
- Contini, C.; di Nuzzo, M.; Barp, N.; Bonazza, A.; de Giorgio, R.; Tognon, M.; Rubino, S. The novel zoonotic COVID-19 pandemic: An expected global health concern. J. Infect. Dev. Ctries 2020, 14, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 1–10. [Google Scholar] [CrossRef]
- Zhu, Z.; Cai, T.; Fan, L.; Lou, K.; Hua, X.; Huang, Z.; Gao, G. The potential role of serum angiotensin-converting enzyme in coronavirus disease 2019. BMC Infect. Dis. 2020, 20, 1–8. [Google Scholar] [CrossRef]
- Walsh, D.A.; Catravas, J.; Wharton, J. Angiotensin converting enzyme in human synovium: Increased stromal [125I] 351A binding in rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 2020, 10, 7448. [Google Scholar] [CrossRef] [PubMed]
- Caso, F.; Costa, L.; Ruscitti, P.; Navarini, L.; del Puente, A.; Giacomelli, R.; Scarpa, R. Could sars-coronavirus-2 trigger autoimmune and/or autoinflammatory mechanisms in genetically predisposed subjects? Autoimmun. Rev. 2020, 19, 102524. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033. [Google Scholar] [CrossRef]
- Kirtipal, N.; Bharadwaj, S. Interleukin 6 polymorphisms as an indicator of COVID-19 severity in humans. J. Biomol. Struct. Dyn. 2021, 39, 4563–4565. [Google Scholar] [CrossRef]
- Mikuls, T.R.; Johnson, S.R.; Fraenkel, L.; Arasaratnam, R.J.; Baden, L.R.; Bermas, B.L.; Chatham, W.; Cohen, S.; Costenbader, K.; Gravallese, E.M.; et al. American College of Rheumatology guidance for the management of rheumatic disease in adult patients during the COVID-19 pandemic: Version 1. Arthritis Rheumatol. 2020, 72, 1241–1251. [Google Scholar] [CrossRef]
- Landewé, R.B.; Machado, P.M.; Kroon, F.; Bijlsma, H.W.; Burmester, G.R.; Carmona, L.; Combe, B.; Galli, M.; Gossec, L.; Iagnocco, A.; et al. EULAR provisional recommendations for the management of rheumatic and musculoskeletal diseases in the context of SARS-CoV-2. Ann. Rheum. Dis. 2020, 79, 851–858. [Google Scholar] [CrossRef]
- Strangfeld, A.; Schäfer, M.; Gianfrancesco, M.A.; Lawson-Tovey, S.; Liew, J.W.; Ljung, L.; Mateus, E.F.; Richez, C.; Santos, M.J.; Schmajuk, G.; et al. Factors associated with COVID-19-related death in people with rheumatic diseases: Results from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann. Rheum. Dis. 2021, 80, 930–942. [Google Scholar] [CrossRef]
- Micallef, J.; Soeiro, T.; Jonville-Béra, A.P. Non-steroidal anti-inflammatory drugs, pharmacology, and COVID-19 infection. Therapie 2020, 75, 355–362. [Google Scholar] [CrossRef]
- Reese, J.; Coleman, B.; Chan, L.; Callahan, T.J.; Cappelletti, L.; Fontana, T.; Bradwell, K.R.; Harris, N.L.; Casiraghi, E.; Valentini, G.; et al. Cyclooxygenase inhibitor use is associated with increased COVID-19 severity. medRxiv 2021. [Google Scholar] [CrossRef]
- Rentsch, C.T.; DeVito, N.J.; MacKenna, B.; Morton, C.E.; Bhaskaran, K.; Brown, J.P.; Schultze, A.; Hulme, W.J.; Croker, R.; Walker, A.J.; et al. Effect of pre-exposure use of hydroxychloroquine on COVID-19 mortality: A population-based cohort study in patients with rheumatoid arthritis or systemic lupus erythematosus using the OpenSAFELY platform. Lancet Rheumatol. 2021, 3, e19–e27. [Google Scholar] [CrossRef]
- Sepriano, A.; Kerschbaumer, A.; Smolen, J.S.; van der Heijde, D.; Dougados, M.; van Vollenhoven, R.; McInnes, I.B.; Bijlsma, J.W.; Burmester, G.R.; de Wit, M.; et al. Safety of synthetic and biological DMARDs: A systematic literature review informing the 2019 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2020, 79, S760–S770. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Rajendram, P.; Sacha, G.; Calabrese, L. Practical aspects of targeting IL-6 in COVID-19 disease. Clevel. Clin. J. Med. 2020, 87, 1–5. [Google Scholar] [CrossRef]
- Godeau, B.; Chevret, S.; Varet, B.; Lefrère, F.; Zini, J.M.; Bassompierre, F.; Chèze, S.; Legouffe, E.; Hulin, C.; Grange, M.J.; et al. Intravenous immunoglobulin or high-dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: A randomised, multicentre trial. Lancet 2002, 359, 23–29. [Google Scholar] [CrossRef]
- Mazzucconi, M.G.; Fazi, P.; Bernasconi, S.; de Rossi, G.; Leone, G.; Gugliotta, L.; Vianelli, N.; Avvisati, G.; Rodeghiero, F.; Amendola, A.; et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: A GIMEMA experience. Blood 2007, 109, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Neunert, C.; Lim, W.; Crowther, M.; Cohen, A.; Solberg, L.; Crowther, M.A. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef] [Green Version]
- Ghanima, W.; Cooper, N.; Rodeghiero, F.; Godeau, B.; Bussel, J.B. Thrombopoietin receptor agonists: Ten years later. Haematologica 2019, 104, 1112. [Google Scholar] [CrossRef] [Green Version]
- Godeau, B.; Porcher, R.; Fain, O.; Lefrère, F.; Fenaux, P.; Cheze, S.; Vekhoff, A.; Chauveheid, M.P.; Stirnemann, J.; Galicier, L.; et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: Results of a prospective multicenter phase 2 study. Blood 2008, 112, 999–1004. [Google Scholar] [CrossRef]
- Zaja, F.; Baccarani, M.; Mazza, P.; Bocchia, M.; Gugliotta, L.; Zaccaria, A.; Vianelli, N.; Defina, M.; Tieghi, A.; Amadori, S.; et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010, 115, 2755–2762. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobircă, A.; Bobircă, F.; Ancuța, I.; Florescu, A.; Bojincă, M.; Muscă, A.; Florescu, D.N.; Florescu, L.M.; Sima, R.M.; Florescu, A.; et al. COVID-19—A Trigger Factor for Severe Immune-Mediated Thrombocytopenia in Active Rheumatoid Arthritis. Life 2022, 12, 77. https://doi.org/10.3390/life12010077
Bobircă A, Bobircă F, Ancuța I, Florescu A, Bojincă M, Muscă A, Florescu DN, Florescu LM, Sima RM, Florescu A, et al. COVID-19—A Trigger Factor for Severe Immune-Mediated Thrombocytopenia in Active Rheumatoid Arthritis. Life. 2022; 12(1):77. https://doi.org/10.3390/life12010077
Chicago/Turabian StyleBobircă, Anca, Florin Bobircă, Ioan Ancuța, Anca Florescu, Mihai Bojincă, Alice Muscă, Dan Nicolae Florescu, Lucian Mihai Florescu, Romina Marina Sima, Alesandra Florescu, and et al. 2022. "COVID-19—A Trigger Factor for Severe Immune-Mediated Thrombocytopenia in Active Rheumatoid Arthritis" Life 12, no. 1: 77. https://doi.org/10.3390/life12010077