
Roles of NAD(P)H:quinone Oxidoreductase 1 in Diverse Diseases

Abstract
:1. Introduction
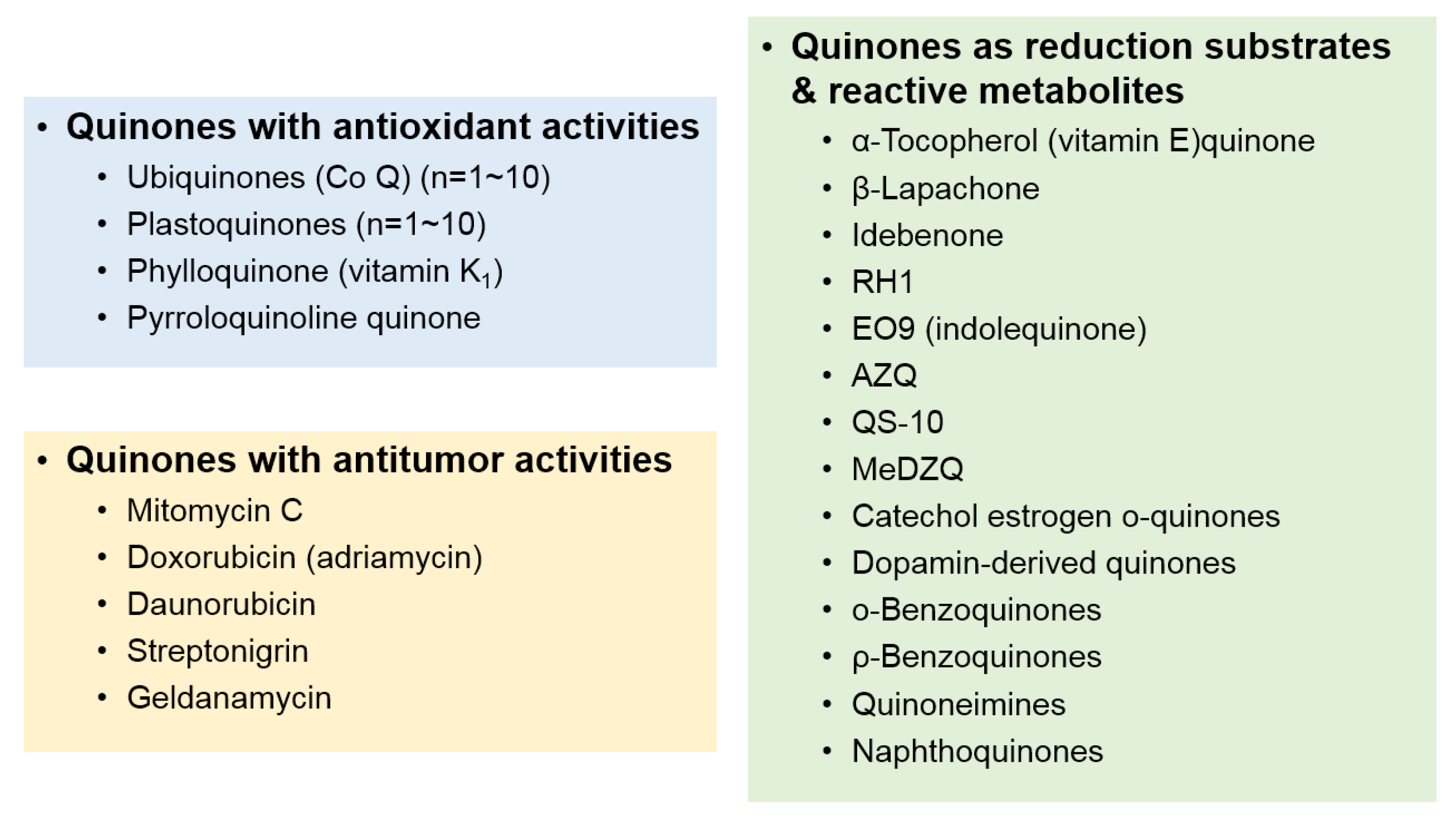
2. Substrates and Induction of NQO1
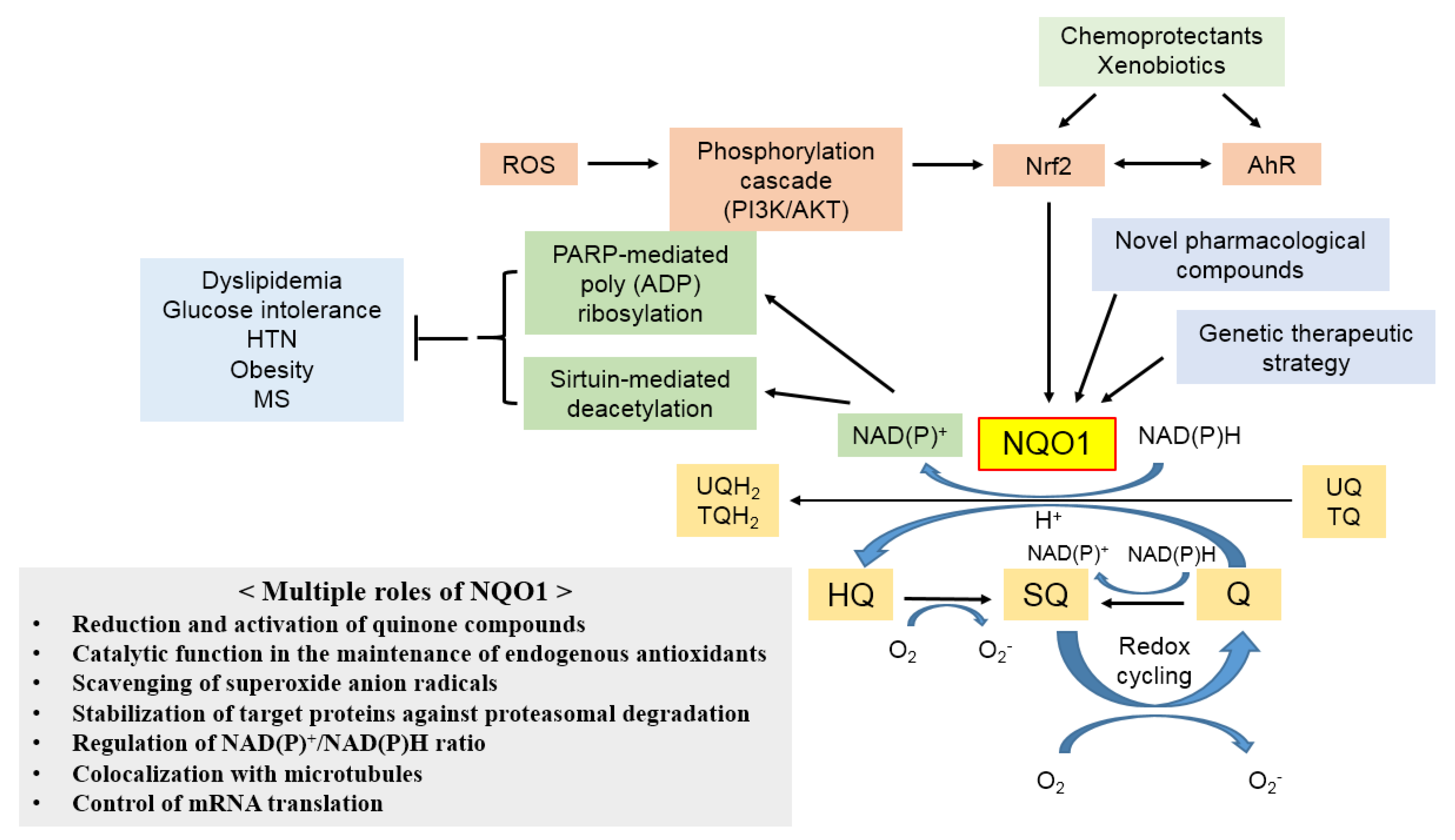
3. Biological Role of NQO1
3.1. Reduction and Activation of Quinone Compounds and Their Derivatives
3.2. Catalytic Function in the Maintenance of Endogenous Antioxidants
3.3. Scavenging of Superoxide Radicals
3.4. Stabilization of Target Proteins
3.5. Generation of NAD+ and β-Lapachone
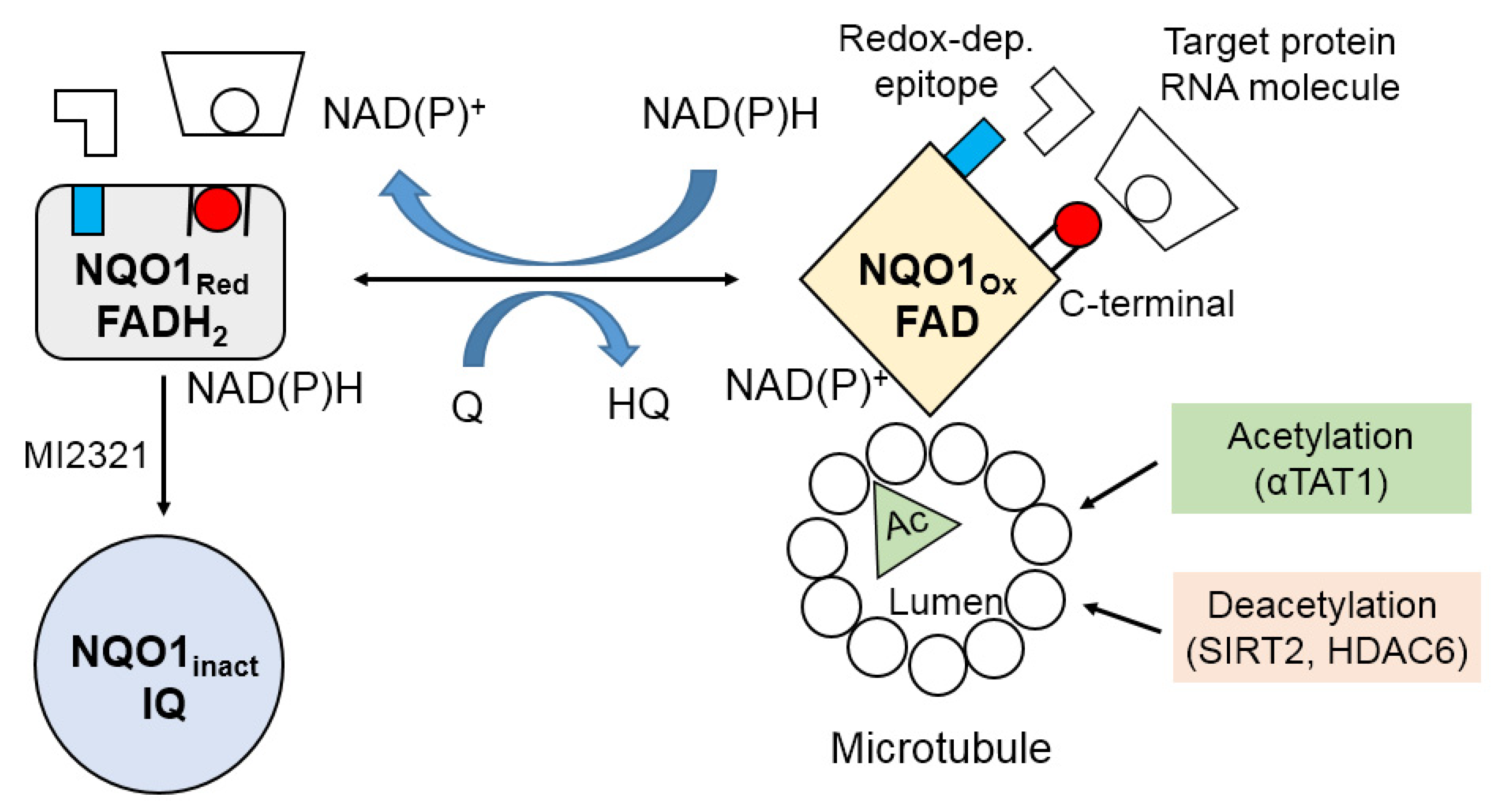
3.6. Colocalization with Microtubules
3.7. Control of mRNA Translation
4. Molecular Redox Switch and Conformational Changes in NQO1
5. Genetic Polymorphisms in NQO1 and Disease
6. NQO1 and Disease
6.1. Atherosclerosis and Cardiovascular Diseases
6.2. Insulin Resistance and Diabetes Mellitus
6.3. Metabolic Syndrome
6.4. Aging
6.5. Alzheimer’s Disease
7. Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, H.; Li, Y. NAD(P)H: Quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc. Toxicol. 2012, 12, 39–45. [Google Scholar] [CrossRef]
- Ernster, L.; Lindberg, O. Animal mitochondria. Annu. Rev. Physiol. 1958, 20, 13–42. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K.; Burnett, P.; Adesnik, M.; McBride, O.W. Nucleotide and deduced amino acid sequence of a human cDNA (NQO2) corresponding to a second member of the NAD(P)H:quinone oxidoreductase gene family. Extensive polymorphism at the NQO2 gene locus on chromosome 6. Biochemistry 1990, 29, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Human NAD(P)H:quinone oxidoreductase2. Gene structure, activity, and tissue-specific expression. J. Biol. Chem. 1994, 269, 14502–14508. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 24, 595. [Google Scholar] [CrossRef]
- Foster, C.E.; Bianchet, M.A.; Talalay, P.; Zhao, Q.; Amzel, L.M. Crystal structure of human quinone reductase type 2, a metalloflavoprotein. Biochemistry 1999, 38, 9881–9886. [Google Scholar] [CrossRef]
- Skelly, J.V.; Sanderson, M.R.; Suter, D.A.; Baumann, U.; Read, M.A.; Gregory, D.S.; Bennet, M.; Hobbs, S.M.; Neidle, S. Crystal structure of human DT-diaphorase: A model for interaction with the cytotoxic prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954). J. Med. Chem. 1999, 42, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yang, X.L.; Holtzclaw, W.D.; Talalay, P. Unexpected genetic and structural relationships of a long-forgotten flavoenzyme to NAD(P)H:quinone reductase (DT-diaphorase). Proc. Natl. Acad. Sci. USA 1997, 94, 1669–1674. [Google Scholar] [CrossRef] [Green Version]
- Bianchet, M.A.; Faig, M.; Amzel, L.M. Structure and mechanism of NAD[P]H:quinone acceptor oxidoreductases (NQO). Methods Enzymol. 2004, 382, 144–174. [Google Scholar]
- Ernster, L. DT-diaphorase. Methods Enzymol. 1967, 10, 309–317. [Google Scholar]
- De Long, M.J.; Prochaska, H.J.; Talalay, P. Induction of NAD(P)H:quinone reductase in murine hepatoma cells by phenolic antioxidants, azo dyes, and other chemoprotectors: A model system for the study of anticarcinogens. Proc. Natl. Acad. Sci. USA 1986, 83, 787–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, C.; Cadenas, E.; Hochstein, P.; Ernster, L. DT-diaphorase: Purification, properties, and function. Methods Enzymol. 1990, 186, 287–301. [Google Scholar]
- Cadenas, E. Antioxidant and prooxidant functions of DTdiaphorase in quinone metabolism. Biochem. Pharmacol. 1995, 49, 127–140. [Google Scholar] [CrossRef]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1): Chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Hosoda, S.; Nakamura, W.; Hayashi, K. Properties and reaction mechanism of DT diaphorase from rat liver. J. Biol. Chem. 1974, 249, 6416–6423. [Google Scholar] [CrossRef]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The threedimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, Y.; Liang, Y.; Naruse, K.; Takahashi, K. Systematic Understanding of Pathophysiological Mechanisms of Oxidative Stress-Related Conditions-Diabetes Mellitus, Cardiovascular Diseases, and Ischemia-Reperfusion Injury. Front. Cardiovasc. Med. 2021, 8, 649785. [Google Scholar] [CrossRef]
- García, N.; Zazueta, C.; Aguilera-Aguirre, L. Oxidative Stress and Inflammation in Cardiovascular Disease. Oxid. Med. Cell Longev. 2017, 2017, 5853238. [Google Scholar] [CrossRef] [Green Version]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial reactive oxygen species mediate cardiac structural, functional, and mitochondrial consequences of diet-induced metabolic heart disease. J. Am. Heart Assoc. 2016, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Carlson, D.; Sun, Y.; Ma, L.; Wolf, S.E.; Minei, J.P.; Zang, Q.S. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE 2015, 10, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Tonnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox. Biol. 2021, 41, 101950. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L. DT-Diaphorase: A historical review. Chem. Scripta 1987, 27A, 1–13. [Google Scholar]
- Beyer, R.E.; Segura-Aguilar, J.; Di Bernardo, S.; Cavazzoni, M.; Fato, R.; Fiorentini, D.; Galli, M.; Setti, M.; Landi, L.; Lenaz, G. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA 1996, 93, 2528–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Bolton, E.M.; Burr, J.A.; Liebler, D.C.; Ross, D. The reduction of alphatocopherolquinone by human NAD(P)H: Quinone oxidoreductase: The role of alpha-tocopherol hydroquinone as a cellular antioxidant. Mol. Pharmacol. 1997, 52, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, E.; Chakravarti, D.; Guttenplan, J.; Hart, E.; Ingle, J.; Jankowiak, R.; Muti, P.; Rogan, E.; Russo, J.; Santen, R.; et al. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta 2006, 1766, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Chandrasena, R.E.; Edirisinghe, P.D.; Bolton, J.L.; Thatcher, G.R. Problematic detoxification of estrogen quinones by NAD(P)H-dependent quinone oxidoreductase and glutathione-S-transferase. Chem. Res. Toxicol. 2008, 21, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Kim, H.J.; Shen, A.; Lee, S.B.; Yang, S.H.; Shim, H.; Cho, E.-Y.; Kwon, K.-B.; Kwak, T.-H.; So, H.-S. New Therapeutic Concept of NAD Redox Balance for Cisplatin Nephrotoxicity. Biomed Res. Int. 2016, 2016, 4048390. [Google Scholar] [CrossRef]
- Herrera, A.; Munoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are dopamine oxidation metabolites involved in the loss of dopaminergic neurone in the nigrstriatal system in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Cassagnes, L.E.; Perio, P.; Ferry, G.; Moulharat, N.; Antoine, M.; Gayon, R.; Boutin, J.A.; Nepveu, F.; Reybier, K. In cellulo monitoring of quinone reductase activity and reactive oxygen species production during the redox cycling of 1,2 and 1,4 quinones. Free Radic. Biol. Med. 2015, 89, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. NQO1 in protection against oxidative stress. Curr. Opin. Toxicol. 2018, 7, 67–72. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Seki, M.; Kawai, T.; Yoshida, K.; Yoshida, M.; Isobe, T.; Hoshino, N.; Shirai, R.; Tanaka, M.; Souzaki, R.; et al. Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis. Oncol. 2020, 4, 20. [Google Scholar] [CrossRef]
- Wu, Y.L.; Wang, D.; Peng, X.E.; Chen, Y.L.; Zheng, D.L.; Chen, W.N.; Lin, X. Epigenetic silencing of NAD(P)H:quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radic. Biol. Med. 2013, 65, 632–644. [Google Scholar] [CrossRef]
- Tada, M.; Yokosuka, O.; Fukai, K.; Chiba, T.; Imazeki, F.; Tokuhisa, T.; Saisho, H. Hypermethylation of NAD(P)H: Quinone oxidoreductase 1 (NQO1) gene in human hepatocellular carcinoma. J. Hepatol. 2005, 42, 511–519. [Google Scholar] [CrossRef]
- Huang, N.; Pei, X.; Lin, W.; Chiu, J.F.; Tao, T.; Li, G. DNA methylation of a non-CpG island promoter represses NQO1 expression in rat arsenic-transformed lung epithelial cells. Acta Biochim. Biophys. Sin. 2018, 50, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Montano, M.M.; Bianco, N.R.; Deng, H.; Wittmann, B.M.; Chaplin, L.C.; Katzenellenbogen, B.S. Estrogen receptor regulation of quinone reductase in breast cancer: Implications for estrogen-induced breast tumor growth and therapeutic uses of tamoxifen. Front. Biosci. 2005, 10, 1440–1461. [Google Scholar] [CrossRef]
- Montano, M.M.; Jaiswal, A.K.; Katzenellenbogen, B.S. Transcriptional regulation of the human quinone reductase gene by antiestrogen-liganded estrogen receptoralpha and estrogen receptor-beta. J. Biol. Chem. 1998, 273, 25443–25449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.; Brüne, B.; Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesis, P.L.; Levin, D.E.; Smith, M.T.; Ernster, L.; Ames, B.N. Mutagenicity of quinones: Pathways of metabolic activation and detoxification. Proc. Natl. Acad. Sci. USA 1984, 81, 1696–1700. [Google Scholar] [CrossRef] [Green Version]
- Lind, C.; Hochstein, P.; Ernster, L. DT-Diaphorase as a quinone reductase: A cellular control device against semiquinone and superoxide radical formation. Arch. Biochem. Biophys. 1982, 216, 178–185. [Google Scholar] [CrossRef]
- Thor, H.; Smith, M.T.; Hartzell, P.; Bellomo, G.; Jewell, S.A.; Orrenius, S. The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes. A study of the implication of oxidative stress in intact cells. J. Biol. Chem. 1982, 257, 12419–12425. [Google Scholar] [CrossRef]
- Prochaska, H.J.; Talalay, P.; Sies, H. Direct protective effect of NAD(P)H:quinone reductase against menadione-induced chemiluminescence of postmitochondrial fractions of mouse liver. J. Biol. Chem. 1987, 262, 1931–1934. [Google Scholar] [CrossRef]
- Wefers, H.; Sies, H. Hepatic low-level chemiluminescence during redox cycling of menadione and the menadione-glutathione conjugate: Relation to glutathione and NAD(P)H:quinone reductase (DT-diaphorase) activity. Arch. Biochem. Biophys. 1983, 224, 568–578. [Google Scholar] [CrossRef]
- Atallah, A.S.; Landolph, J.R.; Ernster, L.; Hochstein, P. DT-diaphorase activity and the cytotoxicity of quinones in C3H/10T1/2 mouse embryo cells. Biochem. Pharmacol. 1988, 37, 2451–2459. [Google Scholar] [CrossRef]
- Iyanagi, T.; Yamazaki, I. Difference in the mechanism of quinone reduction by the NADH dehydrogenase and the NAD(P)H dehydrogenase (DT-diaphorase). Biochim. Biophys. Acta 1970, 216, 282–294. [Google Scholar] [CrossRef]
- Tedeschi, G.; Chen, S.; Massey, V. DT-diaphorase. Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 1995, 270, 1198–1204. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D.; Beall, H.; Prakash, A.S.; Mulcahy, R.T.; Gibson, N.W. DTdiaphorase in activation and detoxification of quinones. Bioreductive activation of mitomycin C. Cancer Metastasis Rev. 1993, 12, 83–101. [Google Scholar] [CrossRef]
- Powis, G. Metabolism and reactions of quinoid anticancer agents. Pharmacol. Ther. 1987, 35, 57–162. [Google Scholar] [CrossRef]
- Ross, D.; Beall, H.D.; Siegel, D.; Traver, R.D.; Gustafson, D.L. Enzymology of bioreductive drug activation. Br. J. Cancer 1996, 74 (Suppl. XXVII), S1–S8. [Google Scholar]
- Beall, H.D.; Mulcahy, R.T.; Siegel, D.; Traver, R.D.; Gibson, N.W.; Ross, D. Metabolism of bioreductive antitumor compounds by purified rat and human DTdiaphorases. Cancer Res. 1994, 54, 3196–3201. [Google Scholar]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H: Quinone oxidoreductase activity is the principal determinant of betalapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Gibson, N.W.; Preusch, P.C.; Ross, D. Metabolism of mitomycin C by DTdiaphorase: Role in mitomycin C- induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990, 50, 7483–7489. [Google Scholar]
- Bair, J.S.; Palchaudhuri, R.; Hergenrother, P.J. Chemistry and biology of deoxynyboquinone, a potent inducer of cancer cell death. J. Am. Chem. Soc. 2010, 132, 5469–5478. [Google Scholar] [CrossRef]
- Bailey, S.M.; Lewis, A.D.; Knox, R.J.; Patterson, L.H.; Fisher, G.R.; Workman, P. Reduction of the indoloquinone anticancer drug EO9 by purified DT-diaphorase: A detailed kinetic study and analysis of metabolites. Biochem. Pharmacol. 1998, 56, 613–621. [Google Scholar] [CrossRef]
- Choudry, G.A.; Stewart, P.A.; Double, J.A.; Krul, M.R.; Naylor, B.; Flannigan, G.M.; Shah, T.K.; Brown, J.E.; Phillips, R.M. A novel strategy for NQO1 (NAD(P)H:quinone oxidoreductase, EC 1.6.99.2) mediated therapy of bladder cancer based on the pharmacological properties of EO9. Br. J. Cancer 2001, 85, 1137–1146. [Google Scholar] [CrossRef]
- Guo, W.; Reigan, P.; Siegel, D.; Zirrolli, J.; Gustafson, D.; Ross, D. Formation of 17- allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H: Quinone oxidoreductase 1: Role of 17-AAG hydroquinone in heat shock protein 90 inhibition. Cancer Res. 2005, 65, 10006–10015. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Gibson, N.W.; Preusch, P.C.; Ross, D. Metabolism of diaziquone by NAD(P)H:(quinone acceptor) oxidoreductase (DT-diaphorase): Role in diaziquone-induced DNA damage and cytotoxicity in human colon carcinoma cells. Cancer Res. 1990, 50, 7293–7300. [Google Scholar]
- Dehn, D.L.; Winski, S.L.; Ross, D. Development of a new isogenic cell-xenograft system for evaluation of NAD(P)H: Quinone oxidoreductase-directed antitumor quinones: Evaluation of the activity of RH1. Clin. Cancer Res. 2004, 10, 3147–3155. [Google Scholar] [CrossRef] [Green Version]
- Dehn, D.L.; Inayat-Hussain, S.H.; Ross, D. RH1 induces cellular damage in an NAD(P)H: Quinone oxidoreductase 1-dependent manner: Relationship between DNA cross-linking, cell cycle perturbations, and apoptosis. J. Pharmacol. Exp. Ther. 2005, 313, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Ross, D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic. Biol. Med. 2000, 29, 246–253. [Google Scholar] [CrossRef]
- Awadallah, N.S.; Dehn, D.; Shah, R.J.; Russell Nash, S.; Chen, Y.K.; Ross, D.; Bentz, J.S.; Shroyer, K.R. NQO1 expression in pancreatic cancer and its potential use as a biomarker. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Volpato, M.; Abou-Zeid, N.; Tanner, R.W.; Glassbrook, L.T.; Taylor, J.; Stratford, I.; Loadman, P.M.; Jaffar, M.; Phillips, R.M. Chemical synthesis and biological evaluation of a NAD(P)H:quinone oxidoreductase-1 targeted tripartite quinone drug delivery system. Mol. Cancer Ther. 2007, 6, 3122–3130. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Desai, A.; Tang, S.; Thomas, T.P.; Baker, J.R., Jr. The synthesis of a c (RGDyK) targeted SN38 prodrug with an indolequinone structure for bioreductive drug release. Org. Lett. 2010, 12, 1384–1387. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Iyer, A.; Sengupta, K.; Chakrapani, H. INDQ/NO, a bioreductively activated nitric oxide prodrug. Org. Lett. 2013, 15, 2636–2639. [Google Scholar] [CrossRef]
- Liu, P.; Xu, J.; Yan, D.; Zhang, P.; Zeng, F.; Li, B.; Wu, S. A DT-diaphorase responsive theranostic prodrug for diagnosis, drug release monitoring and therapy. Chem. Commun. 2015, 51, 9567–9570. [Google Scholar] [CrossRef]
- Shin, W.S.; Han, J.; Verwilst, P.; Kumar, R.; Kim, J.H.; Kim, J.S. Cancer targeted enzymatic theranostic prodrug: Precise diagnosis and chemotherapy. Bioconjug. Chem. 2016, 27, 1419–1426. [Google Scholar] [CrossRef]
- Li, B.; Liu, P.; Yan, D.; Zeng, F.; Wu, S. A self-immolative and DT-diaphorase-activatable prodrug for drug-release tracking and therapy. J. Mater. Chem. B 2017, 5, 2635–2643. [Google Scholar] [CrossRef]
- Xu, S.; Yao, H.; Pei, L.; Hu, M.; Li, D.; Qiu, Y.; Wang, G.; Wu, L.; Yao, H.; Zhu, Z.; et al. Design, synthesis, and biological evaluation of NAD(P)H: Quinone oxidoreductase (NQO1)-targeted oridonin prodrugs possessing indolequinone moiety for hypoxia-selective activation. Eur. J. Med. Chem. 2017, 132, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Li, Z.; Wu, X.; Wu, Y.; You, Q.; Zhang, X. An NAD(P)H: Quinone oxidoreductase 1 responsive and self-immolative prodrug of 5-fluorouracil for safe and effective cancer therapy. Org. Lett. 2018, 20, 3635–3638. [Google Scholar] [CrossRef] [PubMed]
- Ross, D. Quinone reductases multitasking in the metabolic world. Drug Metab. Rev. 2004, 36, 639–654. [Google Scholar] [CrossRef]
- Liebler, D.C. The role of metabolismin the antioxidant function of vitamin E. Crit. Rev. Toxicol. 1993, 23, 147–169. [Google Scholar] [CrossRef]
- Bindoli, A.; Valente, M.; Cavallini, L. Inhibition of lipid peroxidation by alpha-tocopherolquinone and alpha-tocopherolhydroquinone. Biochem. Int. 1985, 10, 753–761. [Google Scholar] [PubMed]
- Kohar, I.; Baca, M.; Suarna, C.; Stocker, R.; Southwell-Keely, P.T. Is alpha-tocopherol a reservoir for alpha-tocopheryl hydroquinone? Free Radic. Biol. Med. 1995, 19, 197–207. [Google Scholar] [CrossRef]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Jia, Z.; Mahaney, J.E.; Ross, D.; Misra, H.P.; Trush, M.A.; Li, Y. The highly expressed and inducible endogenous NAD(P)H:quinone oxidoreductase 1 in cardiovascular cells acts as a potential superoxide scavenger. Cardiovasc. Toxicol. 2007, 7, 202–211. [Google Scholar] [CrossRef]
- Cao, Z.; Li, Y. The chemical inducibility of mouse cardiac antioxidants and phase 2 enzymes in vivo. Biochem. Biophys. Res. Commun. 2004, 317, 1080–1088. [Google Scholar] [CrossRef]
- Boothman, D.A.; Meyers, M.; Fukunaga, N.; Lee, S.W. Isolation of X-ray-inducible transcripts from radioresistant human melanoma cells. Proc. Natl. Acad. Sci. USA 1993, 90, 7200–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Lotem, J.; Cohen, B.; Sachs, L.; Shaul, Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc. Natl. Acad. Sci. USA 2001, 98, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.; Dehn, D.; Siegel, D.; Kepa, J.K.; Tang, L.J.; Pietenpol, J.A.; Ross, D. Interaction of human NAD(P)H:quinone oxidoreductase 1 (NQO1) with the tumor suppressor protein p53 in cells and cell-free systems. J. Biol. Chem. 2003, 278, 10368–10373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskander, K.; Gaikwad, A.; Paquet, M.; Long, D.J., 2nd; Brayton, C.; Barrios, R.; Jaiswal, A.K. Lower induction of p53 and decreased apoptosis in NQO1-null mice lead to increased sensitivity to chemical-induced skin carcinogenesis. Cancer Res. 2005, 65, 2054–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Bercovich, Z.; Tsvetkov, P.; Shaul, Y.; Kahana, C. 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol. Cell 2005, 17, 645–655. [Google Scholar] [CrossRef]
- Hershkovitz Rokah, O.; Shpilberg, O.; Granot, G. NAD(P)H quinone oxidoreductase protects TAp63gamma from proteasomal degradation and regulates TAp63gamma-dependent growth arrest. PLoS ONE 2010, 5, e11401. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1 a, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.-J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1 a. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garate, M.; Wong, R.P.; Campos, E.I.; Wang, Y.; Li, G. NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of the tumour suppressor p33(ING1b). EMBO Rep. 2008, 9, 576–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alard, A.; Fabre, B.; Anesia, R.; Marboeuf, C.; Pierre, P.; Susini, C.; Bousquet, C.; Pyronnet, S. NAD(P)H quinone-oxydoreductase 1 protects eukaryotic translation initiation factor 4GI from degradation by the proteasome. Mol. Cell. Biol. 2010, 30, 1097–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanni, A.; Fiore, M.; De Salvia, R.; Cundari, E.; Ricordy, R.; Ceccarelli, R.; Degrassi, F. DNA damage and cytotoxicity induced by beta-lapachone: Relation to poly(ADPribose) polymerase inhibition. Mutat. Res. 1998, 401, 55–63. [Google Scholar] [CrossRef]
- Pink, J.J.; Wuerzberger-Davis, S.; Tagliarino, C.; Planchon, S.M.; Yang, X.; Froelich, C.J.; Boothman, D.A. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during beta-lapachone-mediated apoptosis. Exp. Cell Res. 2000, 255, 144–155. [Google Scholar] [CrossRef]
- Starcher, C.L.; Pay, S.L.; Singh, N.; Yeh, I.J.; Bhandare, S.B.; Su, X.; Huang, X.; Bey, E.A.; Motea, E.A.; Boothman, D.A. Targeting base excision repair in cancer: NQO1-bioactivatable drugs improve tumor selectivity and reduce treatment toxicity through radiosensitization of human cancer. Front. Oncol. 2020, 10, 1575. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S.; Bey, E.A.; Dong, Y.; Meng, J.; Patra, B.; Yan, J.; Xie, X.J.; Brekken, R.A.; Barnett, C.C.; Bornmann, W.G.; et al. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of beta-lapachone for pancreatic cancer therapy. Clin. Cancer Res. 2011, 17, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bey, E.A.; Reinicke, K.E.; Srougi, M.C.; Varnes, M.; Anderson, V.E.; Pink, J.J.; Li, L.S.; Patel, M.; Cao, L.; Moore, Z.; et al. Catalase abrogates beta-lapachone-induced PARP1 hyperactivation-directed programmed necrosis in NQO1-positive breast cancers. Mol. Cancer Ther. 2013, 12, 2110–2120. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, G.; Silvers, M.A.; Ilcheva, M.; Liu, Y.; Moore, Z.R.; Luo, X.; Gao, J.; Anderson, G.; Liu, L.; Sarode, V.; et al. Tumor-selective use of DNA base excision repair inhibition in pancreatic cancer using the NQO1 bioactivatable drug, beta-lapachone. Sci. Rep. 2015, 5, 17066. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.H.; Kim, D.W.; Jo, E.J.; Kim, Y.K.; Jo, Y.S.; Park, J.H.; Yoo, S.K.; Park, M.K.; Kwak, T.H.; Kho, Y.L.; et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes 2009, 58, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Hwang, J.H.; Noh, J.R.; Gang, G.T.; Kim, D.H.; Son, H.Y.; Kwak, T.H.; Shong, M.; Lee, I.K.; Lee, C.H. Activation of NAD(P)H:quinone oxidoreductase ameliorates spontaneous hypertension in an animal model via modulation of eNOS activity. Cardiovasc. Res. 2011, 91, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Park, A.H.; Lee, S.H.; Lee, S.H.; Kim, J.H.; Yang, S.J.; Yeom, Y.I.; Kwak, T.H.; Lee, D.; Lee, S.J.; et al. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice. PLoS ONE 2012, 7, e47122. [Google Scholar] [CrossRef]
- Kim, H.J.; Oh, G.S.; Shen, A.; Lee, S.B.; Choe, S.K.; Kwon, K.B.; Lee, S.; Seo, K.S.; Kwak, T.H.; Park, R.; et al. Augmentation of NAD(+) by NQO1 attenuates cisplatin-mediated hearing impairment. Cell Death Dis. 2014, 5, e1292. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Dehn, D.D.; Bokatzian, S.S.; Quinn, K.; Backos, D.S.; Di Francesco, A.; Bernier, M.; Reisdorph, N.; de Cabo, R.; Ross, D. Redox modulation of NQO1. PLoS ONE 2018, 13, e0190717. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Kepa, J.K.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) localizes to the mitotic spindle in human cells. PLoS ONE 2012, 7, e44861. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Bersie, S.; Harris, P.; Di Francesco, A.; Armstrong, M.; Reisdorph, N.; Bernier, M.; de Cabo, R.; Fritz, K.; Ross, D. A redox-mediated conformational change in NQO1 controls binding to microtubules and alpha-tubulin acetylation. Redox. Biol. 2021, 39, 101840. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Wong, L.H.; Kalitsis, P.; Earle, E.; Shaffer, L.G.; Choo, K.H. Poly(ADPribose) polymerase 2 localizes to mammalian active centromeres and interacts with PARP-1, Cenpa, Cenpb and Bub 3, but not Cenpc. Hum. Mol. Genet. 2002, 11, 2319–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.; Coughlin, M.; Mitchison, T.J. Tankyrase-1 polymerization of poly(ADPribose) is required for spindle structure and function. Nat. Cell Biol. 2005, 7, 1133–1139. [Google Scholar] [CrossRef]
- Kang, H.J.; Song, H.Y.; Ahmed, M.A.; Guo, Y.; Zhang, M.; Chen, C.; Cristofanilli, M.; Horiuchi, D.; Vassilopoulos, A. NQO1 regulates mitotic progression and response to mitotic stress through modulating SIRT2 activity. Free Radic. Biol. Med. 2018, 126, 358–371. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [Green Version]
- Di Francesco, A.; Di Germanio, C.; Panda, A.C.; Huynh, P.; Peaden, R.; Navas-Enamorado, I.; Bastian, P.; Lehrmann, E.; Diaz-Ruiz, A.; Ross, D.; et al. Novel RNA-binding activity of NQO1 promotes SERPINA1 mRNA translation. Free Radic. Biol. Med. 2016, 99, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef]
- Chen, S.; Deng, P.S.; Bailey, J.M.; Swiderek, K.M. A two-domain structure for the two subunits of NAD(P)H:quinone acceptor oxidoreductase. Protein Sci. 1994, 3, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Danielson, L.; Ernster, L. Lack of relationship bewteen mitochondrial oxidative phosphorylation and the dicoumarol-sensitive flavoenzyme DT-diaphorase or vitamin K reductase. Nature 1962, 194, 155–157. [Google Scholar] [CrossRef]
- Winski, S.L.; Koutalos, Y.; Bentley, D.L.; Ross, D. Subcellular localization of NAD(P) H:quinone oxidoreductase 1 in human cancer cells. Cancer Res. 2002, 62, 1420–1424. [Google Scholar] [PubMed]
- Milkovic, L.; Tomljanovic, M.; Cipak Gasparovic, A.; Novak Kujundzic, R.; Simunic, D.; Konjevoda, P.; Mojzes, A.; Dakovic, N.; Zarkovic, N.; Gall Troselj, K. Nutritional stress in head and neck cancer originating cell lines: The sensitivity of the NRF2–NQO1 Axis. Cells 2019, 8, 1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, D.H.; Emerson, S.S.; Jo, D.G.; Mattson, M.P.; de Cabo, R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc. Natl. Acad. Sci. USA 2006, 103, 19908–19912. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Roe, A.L.; Vandale, S.E.; Bingham, E.; Oakley, G.G. NAD(P)H:quinone oxidoreductase (NQO1) polymorphism, exposure to benzene, and predisposition to disease: A HuGE review. Genet. Med. 2002, 4, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Traver, R.D.; Siegel, D.; Beall, H.D.; Phillips, R.M.; Gibson, N.W.; Franklin, W.A.; Ross, D. Characterization of a polymorphism in NAD(P)H: Quinone oxidoreductase (DT-diaphorase). Br. J. Cancer 1997, 75, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Gaedigk, A.; Tyndale, R.F.; Jurima-Romet, M.; Sellers, E.M.; Grant, D.M.; Leeder, J.S. NAD(P)H:quinone oxidoreductase: Polymorphisms and allele frequencies in Caucasian, Chinese and Canadian Native Indian and Inuit populations. Pharmacogenetics 1998, 8, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kiffmeyer, W.R.; Langer, E.; Davies, S.M.; Envall, J.; Robison, L.L.; Ross, J.A. Genetic polymorphisms in the Hmong population: Implications for cancer etiology and survival. Cancer 2004, 100, 411–417. [Google Scholar] [CrossRef]
- Chen, S.; Wu, K.; Zhang, D.; Sherman, M.; Knox, R.; Yang, C.S. Molecular characterization of binding of substrates and inhibitors to DTdiaphorase: Combined approach involving site-directed mutagenesis, inhibitorbinding analysis, and computer modeling. Mol. Pharmacol. 1999, 56, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. FAD binding overcomes defects in activity and stability displayed by cancer-associated variants of human NQO1. Biochim. Biophys. Acta 2014, 1842, 2163–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef]
- Kelsey, K.T.; Ross, D.; Traver, R.D.; Christiani, D.C.; Zuo, Z.F.; Spitz, M.R.; Wang, M.; Xu, X.; Lee, B.-K.; Schwartz, B.S.; et al. Ethnic variation in the prevalence of a common NAD(P)H quinone oxidoreductase polymorphism and its implications for anti-cancer chemotherapy. Br. J. Cancer 1997, 76, 852–854. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.S.; Han, Y.; Farabaugh, P.; Xia, H. Implication of alternative splicing for expression of a variant NAD(P)H:quinone oxidoreductase-1 with a single nucleotide polymorphism at 465C>T. Pharmacogenetics 2002, 12, 479–488. [Google Scholar] [CrossRef]
- Hu, L.T.; Stamberg, J.; Pan, S. The NAD(P)H:quinone oxidoreductase locus in human colon carcinoma HCT 116 cells resistant to mitomycin C. Cancer Res. 1996, 56, 5253–5259. [Google Scholar] [PubMed]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef]
- Lienhart, W.D.; Strandback, E.; Gudipati, V.; Koch, K.; Binter, A.; Uhl, M.K.; Rantasa, D.M.; Bourgeois, B.; Madl, T.; Zangger, K.; et al. Catalytic competence, structure and stability of the cancer-associated R139W variant of the human NAD(P)H:quinone oxidoreductase 1 (NQO1). FEBS J. 2017, 284, 233–1245. [Google Scholar] [CrossRef] [Green Version]
- Dovinova, I.; Kvandová, M.; Balis, P.; Gresova, L.; Majzunova, M.; Horakova, L.; Chan, J.Y.; Barancik, M. The role of Nrf2 and PPARgamma in the improvement of oxidative stress in hypertension and cardiovascular diseases. Physiol. Res. 2020, 69 (Suppl. S4), S541–S553. [Google Scholar] [CrossRef]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Chang, Y.C.; Whang, K.; Kim, C.H.; Lee, I.S. Role of NAD(P)H:quinone oxidoreductase 1 on tumor necrosis factor-alpha-induced migration of human vascular smooth muscle cells. Cardiovasc. Res. 2007, 76, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Jeoung, N.H.; Oh, C.J.; Choi, Y.K.; Lee, H.J.; Kim, H.J.; Kim, J.-Y.; Hwang, J.H.; Tadi, S.; Yim, Y.-H.; et al. Activation of NAD(P)H:quinone oxidoreductase 1 prevents arterial restenosis by suppressing vascular smooth muscle cell proliferation. Circ. Res. 2009, 104, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.J.; Kang, E.S.; Kim, H.J.; Kim, S.H.; Chun, S.W.; Ahn, C.W.; Cha, B.S.; Nam, M.; Lee, H.C. The C609T variant of NQO1 is associated with carotid artery plaques in patients with type 2 diabetes. Mol. Genet. Metab. 2009, 97, 85–90. [Google Scholar] [CrossRef]
- Martin, N.J.; Collier, A.C.; Bowen, L.D.; Pritsos, K.L.; Goodrich, G.G.; Arger, K.; Cutter, G.; Pritsos, C.A. Polymorphisms in the NQO1, GSTT and GSTM genes are associated with coronary heart disease and biomarkers of oxidative stress. Mutat. Res. 2009, 674, 93–100. [Google Scholar] [CrossRef]
- Sun, X.; Ji, Y.; Tahir, A.; Kang, J. Network pharmacology combined with transcriptional analysis to unveil the biological basis of astaxanthin in reducing the oxidative stress induced by diabetes mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 4281–4295. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, L.; Li, Q. Genetic polymorphisms of GSTT1, GSTM1, and NQO1 genes and diabetes mellitus risk in Chinese population. Biochem. Biophys. Res. Commun. 2006, 341, 310–313. [Google Scholar] [CrossRef]
- Kristiansen, O.P.; Larsen, Z.M.; Johannesen, J.; Nerup, J.; Mandrup-Poulsen, T.; Pociot, F. No linkage of P187S polymorphism in NAD(P)H: Quinone oxidoreductase (NQO1/DIA4) and type 1 diabetes in the Danish population. DIEGG and DSGD: Danish IDDM epidemiology and genetics group and the Danish study group of diabetes in childhood. Hum. Mutat. 1999, 14, 67–70. [Google Scholar] [CrossRef]
- Gaikwad, A.; Long, D.J., 2nd; Stringer, J.L.; Jaiswal, A.K. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J. Biol. Chem. 2001, 276, 22559–22564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palming, J.; Sjoholm, K.; Jernas, M.; Lystig, T.C.; Gummesson, A.; Romeo, S.; Lonn, L.; Lonn, M.; Carlsson, B.; Carlsson, L.M. The expression of NAD(P)H:quinone oxidoreductase 1 is high in human adipose tissue, reduced by weight loss, and correlates with adiposity, insulin sensitivity, and markers of liver dysfunction. J. Clin. Endocrinol. Metab. 2007, 92, 2346–2352. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Cao, W.; Oh, G.S.; Lee, S.; Shen, A.; Khadka, D.; Lee, S.-B.; Sharma, S.; Kim, S.Y.; Choe, S.-K.; et al. Augmentation of cellular NAD+ by NQO1 enzymatic action improves age-related hearing impairment. Aging Cell 2019, 18, e13016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SantaCruz, K.S.; Yazlovitskaya, E.; Collins, J.; Johnson, J.; DeCarli, C. Regional NAD (P)H:quinone oxidoreductase activity in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 63–69. [Google Scholar] [CrossRef]
- Wang, Y.; Santa-Cruz, K.; DeCarli, C.; Johnson, J.A. NAD(P)H:quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Aalzheimer’s disease. Neurobiol. Aging 2000, 21, 525–531. [Google Scholar] [CrossRef]
- Luo, J.; Li, S.; Qin, X.; Peng, Q.; Liu, Y.; Yang, S.; Qin, X.; Xiong, Y.; Zeng, Z. Association of the NQO1 C609T polymorphism with Alzheimer’s disease in Chinese populations: A meta-analysis. Int. J. Neurosci. 2016, 126, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Limon, A.; Alriquet, M.; Lang, W.H.; Calloni, G.; Wittig, I.; Vabulas, R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. USA 2016, 113, 12156–12161. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.L.; Varner, S.E.; Rao, A.S.; Grey, J.Y.; Thomas, S.; Cook, C.K.; Wasserman, M.A.; Medford, R.M.; Jaiswal, A.K.; Kunsch, C. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J. Biol. Chem. 2003, 278, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.Y.; Kim, S.H.; Choi, M.A.; Williams, D.R.; Lee, Y.H.; Kang, S.W.; Yadav, U.C.; Srivastava, S.K.; Jung, M.; Cho, J.W.; et al. Protective effects of magnesium lithospermate B against diabetic atherosclerosis via Nrf2-ARENQO1 transcriptional pathway. Atherosclerosis 2010, 211, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Isbir, C.S.; Ergen, A.; Tekeli, A.; Zeybek, U.; Gormus, U.; Arsan, S. The effect of NQO1 polymorphism on the inflammatory response in cardiopulmonary bypass. Cell Biochem. Funct. 2008, 26, 534–538. [Google Scholar] [CrossRef]
- Shyu, H.Y.; Fong, C.S.; Fu, Y.P.; Shieh, J.C.; Yin, J.H.; Chang, C.Y.; Wang, H.-W.; Cheng, C.-W. Genotype polymorphisms of GGCX, NQO1, and VKORC1 genes associated with risk susceptibility in patients with large-artery atherosclerotic stroke. Clin. Chem. Acta 2010, 411, 840–845. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Kensler, T.W. New player on an old field; the keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr. Diabetes Rev. 2013, 9, 137–145. [Google Scholar] [CrossRef]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [Green Version]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, Y.; Uruno, A.; Chartoumpekis, D.V.; Kensler, T.W.; Yamamoto, M. Nrf2 represses the onset of type 1 diabetes in non-obese diabetic mice. J. Endocrinol. 2019, 240, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell Biol. 2014, 34, 3305–3320. [Google Scholar] [PubMed] [Green Version]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental nonalcoholic steatohepatitis and liver fibrosis are ameliorated by pharmacologic activation of Nrf2 (NF-E2 p45-related factor 2). Cell Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, S.; Jiang, X.; Wang, Y.H.; Li, F.; Wang, Y.G.; Zheng, Y.; Cai, L. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev. Endocr. Metab. Disord. 2015, 16, 35–45. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; McCullough, P.A.; Sowers, J.R. The role of oxidative stress in the metabolic syndrome. Rev. Cardiovasc. Med. 2011, 12, 21–29. [Google Scholar]
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Novel insights in the metabolic syndromeinduced oxidative stress and inflammation-mediated atherosclerosis. Curr. Cardiol. Rev. 2018, 14, 4–14. [Google Scholar] [CrossRef]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef] [Green Version]
- De Cabo, R.; Cabello, R.; Rios, M.; Lopez-Lluch, G.; Ingram, D.K.; Lane, M.A.; Navas, P. Calorie restriction attenuates age-related alterations in the plasma membrane antioxidant system in rat liver. Exp. Gerontol. 2004, 39, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H.; Hernandez, J.O.; Mattson, M.P.; de Cabo, R. The plasma membrane redox system in aging. Ageing Res. Rev. 2006, 5, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, A.; Lanasa, M.; Garcia, J.; Mora, H.; Fan, F.; Martin-Montalvo, A.; Di Francesco, A.; Calvo-Rubio, M.; Salvador-Pascual, A.; Aon, M.A.; et al. Overexpression of CYB5R3 and NQO1, two NAD(+) -producing enzymes, mimics aspects of caloric restriction. Aging Cell 2018, 17, e12767. [Google Scholar] [CrossRef]
- Diaz-Ruiz, A.; Di Francesco, A.; Carboneau, B.A.; Levan, S.R.; Pearson, K.J.; Price, N.L.; Ward, T.M.; Bernier, M.; de Cabo, R.; Mercken, E.M. Benefits of caloric restriction in longevity and chemical-induced tumorigenesis are transmitted independent of NQO1. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 74, 155–162. [Google Scholar] [CrossRef]
- Raina, A.K.; Templeton, D.J.; Deak, J.C.; Perry, G.; Smith, M.A. Quinone reductase (NQO1), a sensitive redox indicator, is increased in Alzheimer’s disease. Redox Rep. 1999, 4, 23–27. [Google Scholar] [CrossRef]
- Chhetri, J.; King, A.E.; Gueven, N. Alzheimer’s disease and NQO1: Is there a link? Curr. Alzheimer Res. 2018, 15, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [Green Version]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Vatolin, S.; Radivoyevitch, T.; Maciejewski, J.P. New drugs for pharmacological extension of replicative life span in normal and progeroid cells. NPJ Aging Mech. Dis. 2019, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Pardo, M.; Qiu, X.; Zimmermann, R.; Rudich, Y. Particulate Matter Toxicity Is Nrf2 and Mitochondria Dependent: The Roles of Metals and Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2020, 33, 1110–1120. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Morales Vasconsuelo, A.B.; Chiaramello, M.; Ferreira, V.F.; Macedo Oliveira, M.; Baptista Ferreira, S.; Rivarola, V.A.; Rumie Vittar, N.B. NQO1 induction mediated by photodynamic therapy synergizes with β-Lapachone-halogenated derivative against melanoma. Biomed. Pharmacother. 2018, 108, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Digby, E.M.; Sadovski, O.; Beharry, A.A. An Activatable Photosensitizer Targeting Human NAD(P)H:Quinone Oxidoreductase 1. Chemistry 2020, 26, 2713–2718. [Google Scholar] [CrossRef] [PubMed]
- Li, W.P.; Yen, C.J.; Wu, B.S.; Wong, T.W. Recent Advances in Photodynamic Therapy for Deep-Seated Tumors with the Aid of Nanomedicine. Biomedicines 2021, 9, 69. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Author | Published (Year) | Role of NQO1 | Reference |
|---|---|---|---|
| Atherosclerosis and Cardiovascular Diseases | |||
| Lee, et al. | 2007 | Reduced TNF-α-induced migration of human VSMCs | [131] |
| Kim, et al. | 2009 | Prevented arterial restenosis | [132] |
| Han, et al. | 2009 | C609T variant was associated with carotid artery plaques in T2DM patients | [133] |
| Isbir, et al. | 2008 | C609T variant was related to higher IL-6 levels | [133] |
| Martin, et al. | 2009 | Lack of activity of NQO1 was associated with elevation of CHD and CRP | [134] |
| Insulin Resistance and Diabetes Mellitus | |||
| Sun, et al. | 2020 | Astaxanthin reduced oxidative stress and insulin resistance | [135] |
| Wang, et al. | 2006 | C609T variant was not associated with DM in Chinese subjects | [136] |
| Kristiansen, et al. | 1999 | C609T variant was not related to DM in Danish subjects | [137] |
| Gaikward, et al. | 2001 | NQO1-null mice were insulin resistant | [138] |
| Metabolic Syndrome | |||
| Hwang, et al. | 2009 | β-Lapachone showed improvement of metabolic syndrome | [101] |
| Gaikward, et al. | 2001 | NQO1-null mice exhibited higher NAD(P)H/NAD(P)+, higher TG level, and lower abdominal fat | [138] |
| Palming, et al. | 2007 | NQO1 was correlated with adiposity and liver dysfunction | [139] |
| Aging | |||
| Lee, et al. | 2012 | β-Lapachone prevented health decline in aged mice | [100] |
| Kim, et al. | 2019 | β-Lapachone improved age-related hearing impairment | [140] |
| Alzheimer’s Disease | |||
| SantaCruz, et al. | 2004 | NQO1 was located adjacent to senile plaques | [141] |
| Wang, et al. | 2000 | NQO1 activity was increased in AD | [142] |
| Luo, et al. | 2016 | C609T variant was associated with risk of AD in Chinese subjects | [143] |
| Martinez-Limon, et al. | 2016 | NQO1 could bind together with β-amyloid fibrils | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.-S.; Ham, W.; Kim, J. Roles of NAD(P)H:quinone Oxidoreductase 1 in Diverse Diseases. Life 2021, 11, 1301. https://doi.org/10.3390/life11121301
Lee W-S, Ham W, Kim J. Roles of NAD(P)H:quinone Oxidoreductase 1 in Diverse Diseases. Life. 2021; 11(12):1301. https://doi.org/10.3390/life11121301
Chicago/Turabian StyleLee, Wang-Soo, Woojin Ham, and Jaetaek Kim. 2021. "Roles of NAD(P)H:quinone Oxidoreductase 1 in Diverse Diseases" Life 11, no. 12: 1301. https://doi.org/10.3390/life11121301