3.1. Case 1
Firstly, we calculate the concentrations outside the cell. We assume that
is maintained at
= 1M, and that reaction 1 can occur freely, so that
is maintained in equilibrium with
; hence,
= 0.819 M.
is maintained at a fixed low concentration
= 0.01 M. If
were in equilibrium with
then its concentration would be
= 2.718 M. Thus, by fixing
to be much less than
, we ensure that the external environment is maintained out of equilibrium and that there is a driving force for the metabolic system inside the cell. The steady-state concentrations
and
are found by solution of the following differential equations using the fourth-order Runge-Kutta method until convergence is reached.
The rates
are given in
Table 1. As a measure of the metabolic rate, we use the rate of mass flow from food molecules (1 and 2) to biomolecules (3–5), which is
In the steady state, this rate is balanced by an equal rate of mass flow of biomolecules decaying to waste (Equation (5)):
In order to keep the concentrations of food molecules and waste fixed at the external concentrations, there must be a supply rate of food molecules and a removal rate of
by an environmental recycling or replenishment process. In the steady state, all four rates are equal (Equation (6)).
As this reaction system is bistable, the steady-state concentrations depend on the initial conditions. We consider two initial conditions, called low-concentration (LC) and high-concentration (HC) initial conditions. For the LC conditions, we start with M. For the HC conditions, we set the concentrations to , which is the concentration that would occur if the molecules were in equilibrium with and there were no decay to (i.e., if were zero).
Figure 2 shows the steady-state concentrations that arise as a function of
, starting from the two initial conditions. There is a transition point at
= 0.016. For
, there are two stable solutions—the HC solution with high concentrations of the three biomolecules and the LC solutions where the biomolecule concentrations are very small. Starting from HC initial conditions gives the HC solution, and starting from LC initial conditions gives the LC solution. For
, only the LC solution is stable, and this solution is reached starting from both initial conditions.
We now consider concentrations inside the cell. Initially, we consider the cell to be a small compartment of fixed volume which does not grow. Later, we will consider cells with membranes that can grow and divide. For, the fixed-volume compartment, the cell boundary is permeable to
and
, with inflow and outflow proportional to rate
and it is impermeable to
and
. Since
and
have variable concentration inside the cell, we need three additional differential equations for these molecules.
The Equations (1)–(3) for
and
are the same as outside the cell (although the concentrations will be different).
and
are calculated as before. Instead of the supply of food and removal of waste via environmental recycling, we now have diffusion of food into the cell and diffusion of waste out.
In the steady state, the mass flow rate at all the steps is equal.
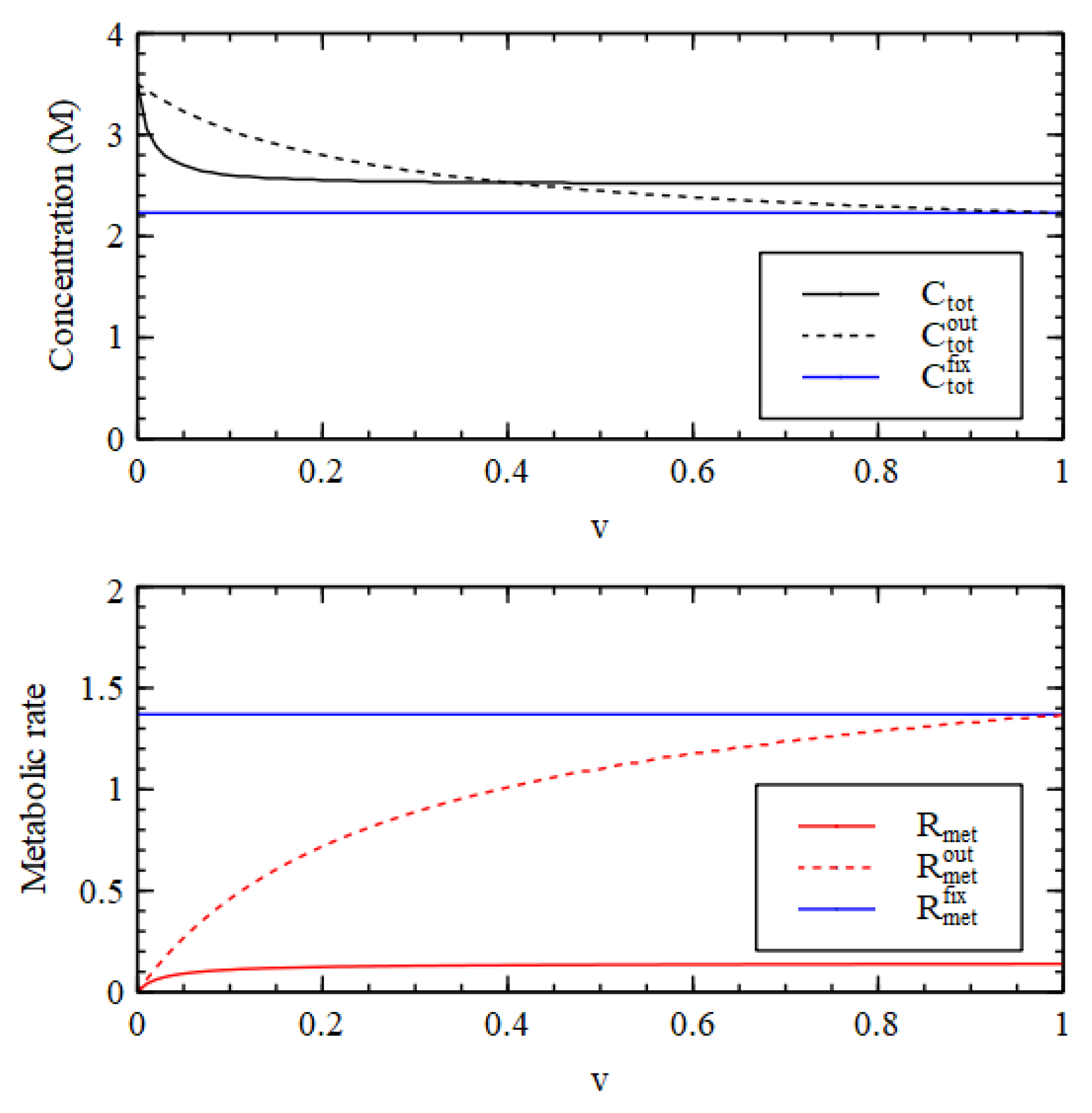
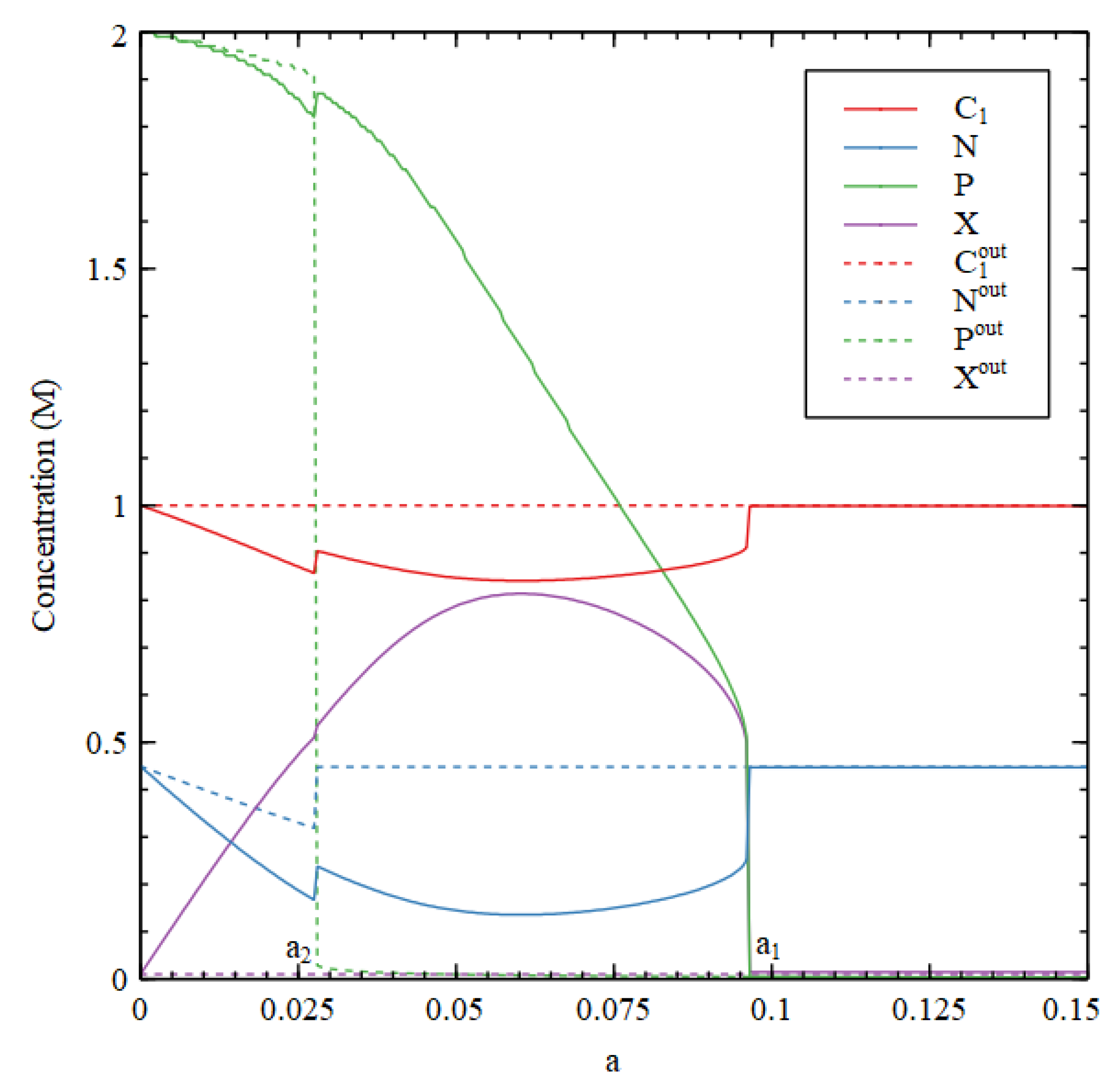
Figure 3 compares steady-state concentrations inside and outside the cell. We first determine the external concentrations, starting from the LC initial conditions. Then, keeping the external concentrations fixed in their steady state, we determine the steady state inside the cell by numerical solution of the differential equations for the inside, starting from the HC initial conditions. We want to show that the HC solution inside the cell can be stable when the outside is in the LC state.
There is an HC solution inside the cell that is stable up to a transition value
, which is slightly lower than the
in
Figure 2. It is apparent that the waste concentration
inside the cell is fairly high, whereas it is fixed at a low concentration outside. When the inside is in the HC state,
and
are lower inside than out because there has to be a net flow of food molecules inwards. The external concentrations
and
are very close to zero and are not shown in
Figure 3.
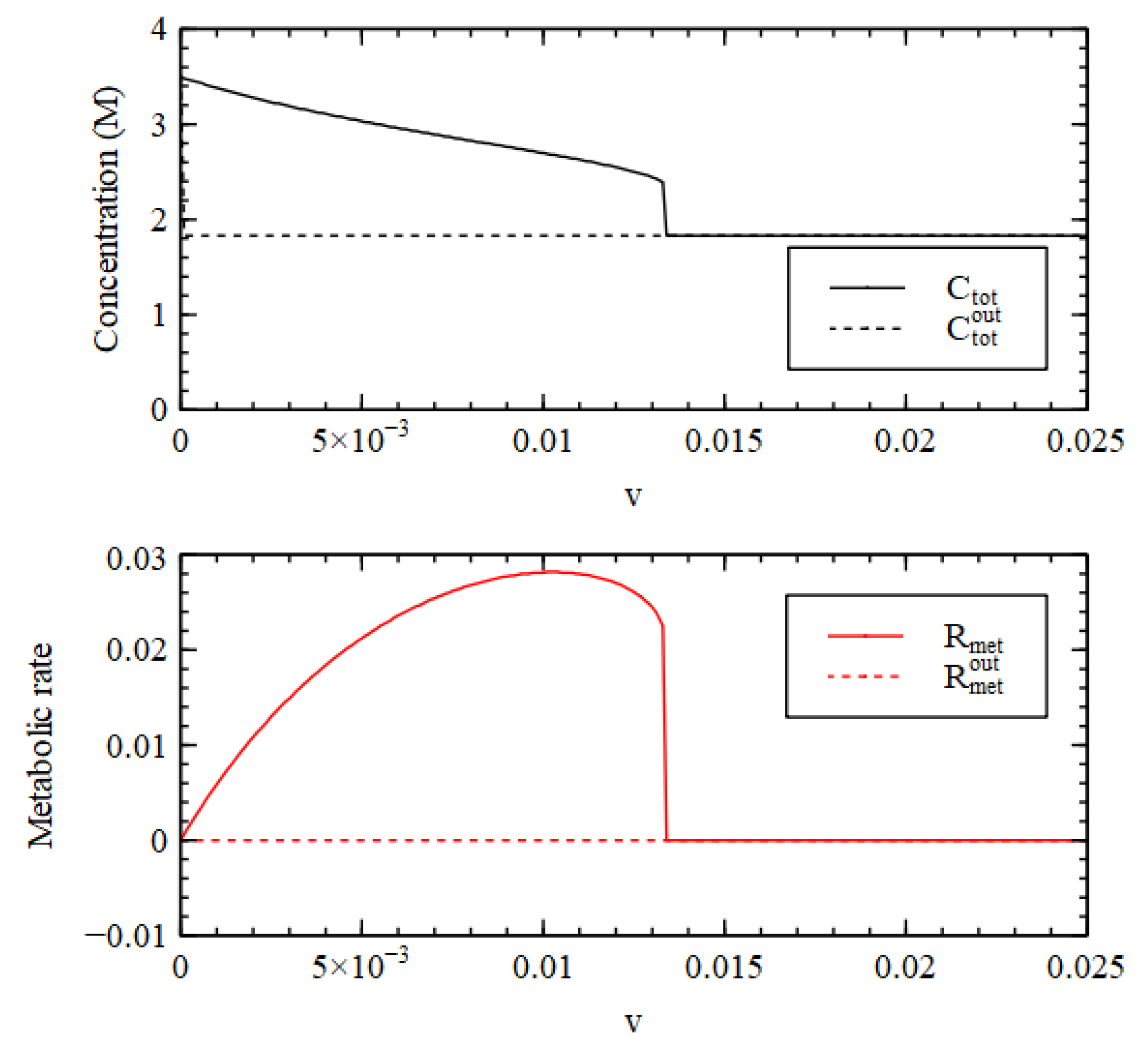
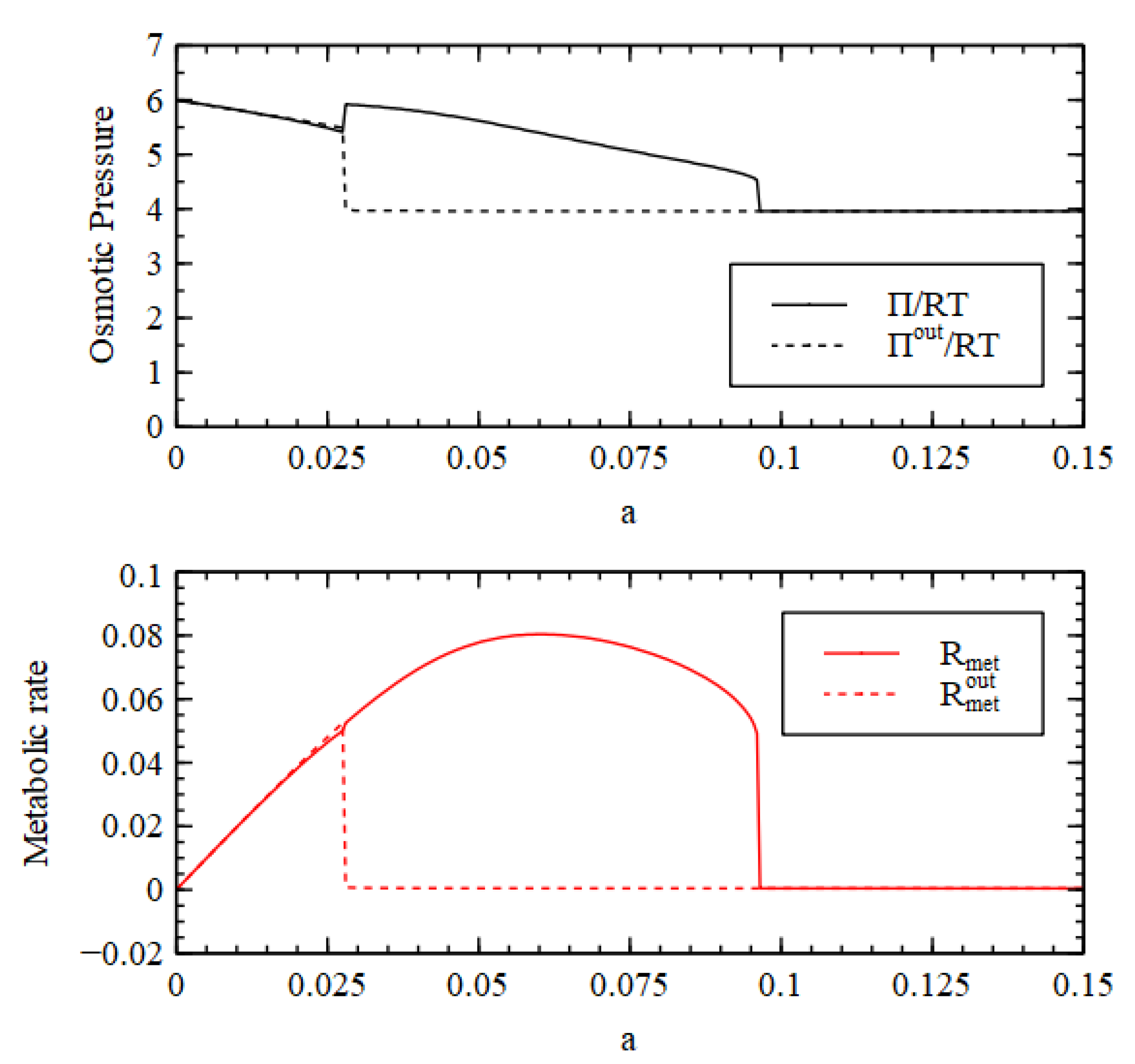
It is useful to look at the total concentration, shown in
Figure 4. For
, the total concentration is higher inside than out, hence there is a positive osmotic pressure inside the cell, as we require for Criterion 2. We will show in a later section of this paper that when this is the case, the osmotic pressure can drive growth and division of lipid vesicles. However, firstly, we want to establish under what conditions the reaction system can maintain a positive osmotic pressure in a compartment of fixed volume. For
, the total concentration inside collapses to the concentration outside, so the osmotic pressure is lost.
A similar result is seen in
Figure 4 for the metabolic rate
(Equation (4)). The external metabolic rate is very close to zero when the outside is in the LC state because the rate constants
and
are zero, and because the concentrations of the catalysts are very low, so the rates of the reactions in the catalytic processes are also very low. Thus, when
,
in the HC state inside the cell is much higher than
outside the cell, as we require for Criterion 3. Thus, all three criteria are satisfied for
, when the reaction system is bistable.
3.2. Case 2
We now consider Case 2, in which process P3 + 5 is permitted (by setting
= 1), and where processes P4 + 6 and P4 + 5 + 6 are prevented (by setting
= 0). This case still relies on an autocatalytic cycle to synthesize
, because the direct reaction 2 is not possible (
= 0). Case 2 is monostable, therefore it does not matter whether we start with LC or HC initial conditions.
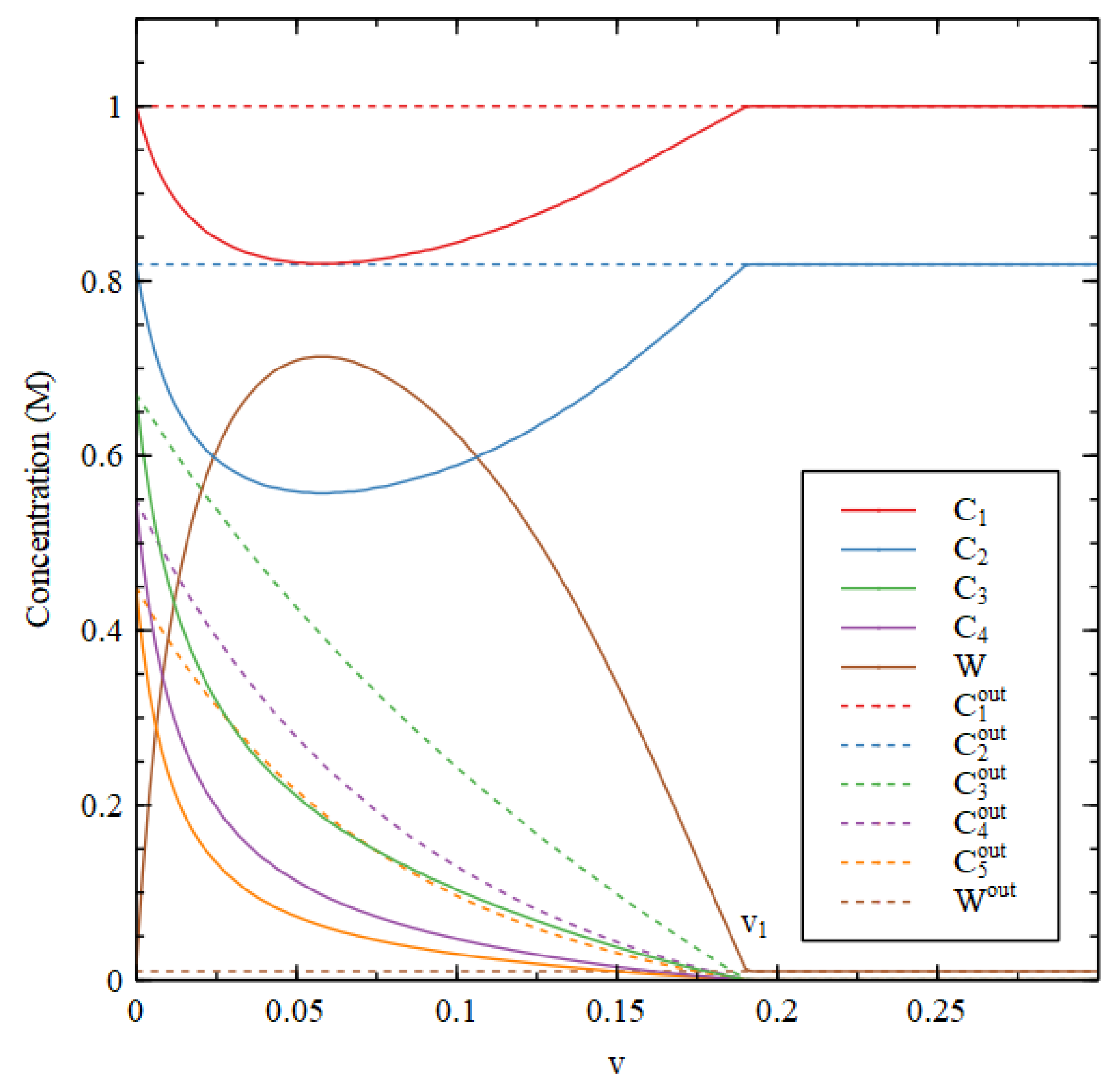
Figure 5 shows the stable state concentrations inside and out. There is a transition point at
= 0.192. For
, there is an HC solution with high
and
, whereas for
, these concentrations fall very close to zero and the concentrations
and
become equal to their external values.
In Case 2, biomolecules can survive at high concentration for much larger decay rates than in Case 1. Nevertheless, Case 2 is not a good model for a metabolism as it stands because all the molecules with the exception of
have a higher concentration outside than inside.
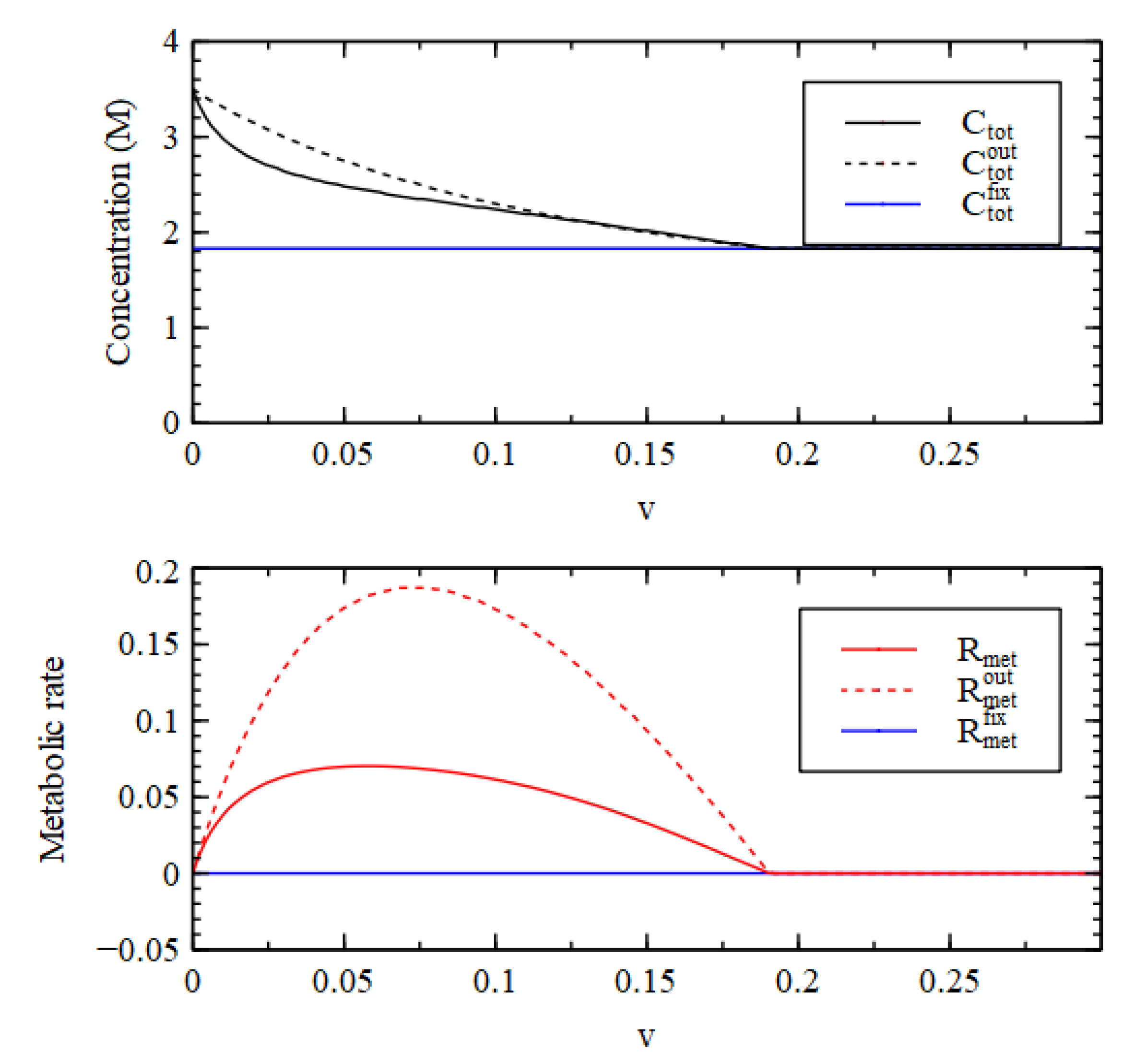
Figure 6 shows that the total concentration inside is less than outside and the metabolic rate inside is less than outside. Thus, Case 2 does not satisfy Criteria 2 and 3. This shows that the existence of an autocatalytic process in the reaction system (P3 + 5 in this case) and the presence of biomolecules that are maintained at a high concentration by this process is not sufficient for the reaction system to satisfy the requirements for a metabolism.
The reason for the failure of Case 2 is that there is no stable LC state on the outside. We could in principle begin with with zero concentration of outside, and we could argue that would never form because = 0. However, in a real case, there would always be some possibility of leakage of from the inside, either via a small rate of permeability through the membrane or due to occasional bursting of cells. There is also a very small rate of formation of from , via the reverse of reaction 7. Thus, we are bound to have a small initial concentration of these molecules by one means or another, and if the LC state is not stable, then the concentrations of and will rise to high values. This rules out Case 2 as a model for metabolism as it stands.
However, we can rescue Case 2 if we are prepared to make the assumption that the decay rate
of the biomolecules is larger on the outside than the inside. (We will discuss further below why this might be the case). We fixed the external decay rate to be
= 0.3, which is higher than
, and the decay rate
inside was allowed to vary as before. The concentrations inside the cell are the same as before, but
and
are now close to zero outside the cell because
.
Figure 6 shows that when
is fixed at a high rate, the total external concentration
and the external metabolic rate
are both lower than the internal values (
and
).
Thus, with the additional assumption that the decay rate is higher outside than in, Case 2 now satisfies the required criteria for a metabolism. It is possible to think of reasons why this might be true in a real case. The decay reactions 7–9 might be driven by UV light, and being inside the compartment might provide some protection from this, so would be larger outside than inside. Or possibly, the breakdown reactions could be driven by interaction with some additional molecule that is not permeable to the membrane, so the breakdown would occur outside but not inside. Although these reasons seem possible, they are somewhat ad hoc, in that an extra condition must be added that was not part of the definition of the reaction network. The actual degree of UV protection afforded by a lipid membrane may not be very large, and what impermeable molecule might be that would cause breakdown outside is unclear. Other reasons for the difference between inside and outside could be proposed if desired. However, if any of these mechanisms were true, we would have the rather odd situation that life would exist inside the cell because of the absence of an inhibiting effect that occurs outside the cell, rather than because of the presence of something constructive inside the cell. Life would be just waiting to pop up as soon as the inhibiting factor is removed! This does not fit with our intuition that the origin of life is an unlikely event that requires the coming together of favourable circumstances.
An alternative way to rescue Case 2, rather than making
larger on the outside, would be to make the reactions of the autocatalytic processes (
) small or zero on the outside. This would obviously make the model work, because the
and
would remain low outside, but again it is ad hoc. It is not obvious why the chemistry should be different outside the cell from inside. Piedrafita et al. [
30] have studied two reaction networks which they call protometabolism 1 and 2—PM1 and PM2. PM2 is bistable, with similar properties to our Case 1, and PM1 is monostable, with similar properties to our Case 2. These authors did not look at the reactions outside the cell, and simply assumed that these reactions did not occur. This requires a reason why the reaction rates for the autocatalytic processes (analogous to our
) should be zero outside. This point was not considered by Piedrafita et al. [
30]. We consider this issue to be important, because if the rate constants are equal inside and out and if the system has only one stable solution with high biomolecule concentration, the reaction system will not satisfy the criteria for a metabolism.
3.3. Case 3
So far, we have shown that in bistable reaction systems, there is a natural explanation of why the difference in total concentration and metabolic rate between the inside and the outside is maintained, whereas in monostable systems, these differences are only maintained if there is an extra ad hoc reason why reaction rate constants are different on the inside and outside. We now take this argument one step further, by showing that if we do allow rate constants to be different inside and out, then we no longer need there to be any autocatalytic processes at all.
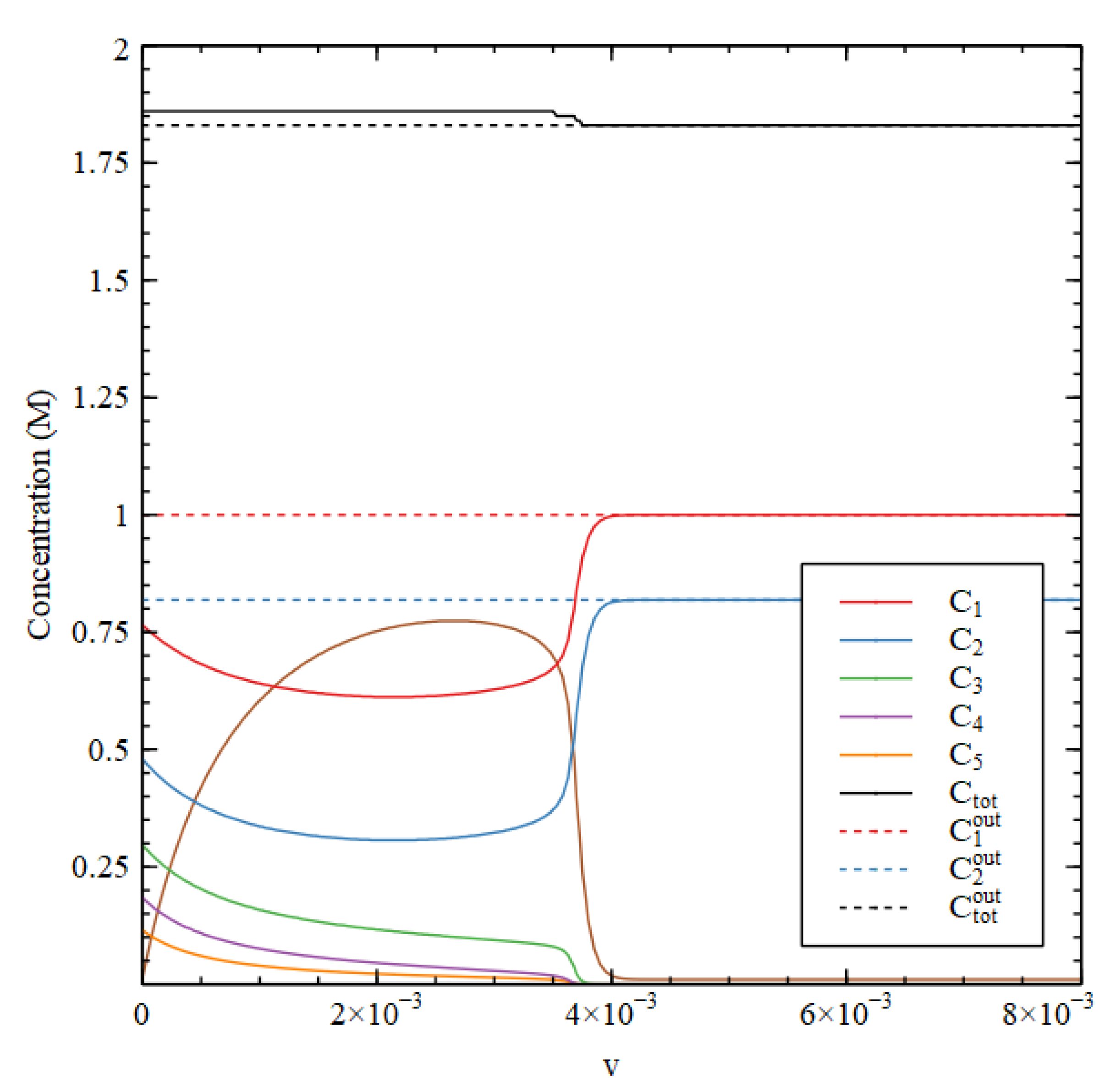
In Case 3, all the direct reactions 1–4 occur with
, and none of the autocatalytic processes occur, because
. In this case, there is only one solution. The concentrations
and
reach high values both inside and outside, that decrease slowly with
and there is no transtion point. The total concentration is lower inside than out at small
, but becomes higher inside for high
, mostly because of the accumulation of
(
Figure 7). However, the external metabolic rate is always much higher than the internal rate. It should be remembered that these metabolic rates are the rates of mass conversion from food molecules to biomolecules per unit volume. Given that the external environment has a much larger volume than the cell, this means huge amounts of food molecules must be supplied, which seems unreasonable.
Now we try to rescue Case 3 by fixing the external decay rate to be a higher rate than inside. If
is fixed at 1.0, then the total exterior concentration becomes
, shown in
Figure 7, which is now lower than the interior concentration
for the whole range of
. However, the external metabolic rate becomes
, which is very high. So this still does not satisfy Criterion 3.
We can force Case 3 to satisfy all the criteria, if we simply say that the synthesis reactions of the biomolecules do not occur outside the cell ( In this case only and exist outside the cell, and is fixed at zero, trivially, so Criterion 3 is also satisfied. Our point here is two-fold. Firstly, Case 3 seems to be too simple to be a good model for a metabolism, as it only works if we prohibit the direct synthesis reactions from occuring outside while allowing them to happen inside, which seems artificial. Secondly, however, if there really is a good reason why these reactions should be different inside and out, then this case shows that all three requirements for the existence of a metabolism can be satisfied with a reaction network having no autocatalytic processes at all.
3.4. Cell Growth and Division
So far, we considered only compartments of fixed volume. In this section, we will consider lipid vesicle compartments in which growth and division are possible. The mechanism for growth and division used here is very similar to that introduced in references [
28,
29,
30].
Experiments often use vesicles with a radius of approximately 50 nm [
31,
32]. We will therefore define a standard vesicle of this size, and measure sizes of growing vesicles relative to this standard size. The radius, suface area and volume of a standard vesicle are
,
, and
.
If a vesicle has a volume , then its surface area, if it were a sphere, would be . However, the actual surface area of the vesicle also depends on the amount of lipid in the membrane. If is the number of lipid molecules in the membrane, and is the surface area per lipid in a relaxed membrane, then the natural surface area, if it is a relaxed state, is , where the factor of 2 occurs because of the two sides of the lipid bilayer. The actual surface area is the larger of and . When the vesicle is an irregular, non-spherical shape because its surface area is larger than the minimum area needed to enclose a sphere of volume When , the vesicle is spherical, and the membrane is under tension, because its surface area is larger than the natural surface area of a relaxed membrane with lipid molecules.
The tension is proportional to the area strain
which is the relative increase in surface area above the natural area:
. This results in a pressure inside the vesicle proportional to
. If the concentrations of solutes inside and outside the vesicle are different, then there is an osmotic pressure difference
. For simplicity, we have assumed that all the solutes behave like ideal gases, so the osmotic pressure is directly proportional to the concentration. The balance of osmotic pressure and membrane tension results in the following equation for the growth of the volume.
For convenience, we have incorporated the factor into the definition of constants and . A factor of has been added, so is measured relative to the standard vesicle, and the constant for the membrane tension has units of concentration (molar). The volume increase is due to water passing through the membrane, therefor the rate scales in proportion to the surface area. A factor of has been added to give a volume scale appropriate to the standard size vesicle. The time scale for growth is determined by , which has units of time−1molar−1. In the examples below, we have and .
If there is a fixed amount of lipid in the membrane, and a positive osmotic pressure, the volume will grow until the osmotic pressure is balanced by the membrane tension. There is a maximum area strain
of approximately 0.1 that can be maintained by the membrane. A vesicle will burst and release some of its contents if
exceeds
[
33,
34]. Repeated cycles of swelling and bursting can be followed in experiments [
35]. However, if the osmotic pressure is not too large, bursting will not occur. In this paper, we assume that the membrane is strong enough to maintain the pressure generated by the internal metabolic reactions without bursting.
If lipid is available in the solution outside and inside the membrane, then transfer of lipid molecules into and out of the membrane is possible, so the membrane surface area
can grow at the same time the volume is increasing. Lipids are only sparingly soluble, and above a critical concentration
, lipid bilayers will spontaneously form, until the concentration falls to
. The number of lipid molecules on each surface of the bilayer is
. The rate of motion of lipid molecules into and out of the membrane on the two sides is
, where
and
are the lipid concentrations inside and outside the vesicle, and
and
are the rate constants for molecules entering and leaving the membrane. When the solution concentration is
, the membrane is in equilibrium with the solution. This means that
. We assume in what follows that the external lipid concentration is fixed at
and the internal concentration will also tend to
if the membrane is relaxed. A relaxed vesicle will not increase in membrane area if the solution on both sides of the membrane has concentration
. However, it is observed experimentally [
31] that transfer of lipids from relaxed vesicles to osmotically swollen vesicles occurs. This means that swollen vesicles gain lipids at solution concentrations where relaxed vesicles do not grow. To account for this, we assume that the rate of lipids entering the membrane increases when the membrane is under tension with input rate proportional to
, whereas the rate of lipids leaving the membrane remains unchanged when the membrane is under tension. The rate of change of the number of membrane lipids for a membrane under tension can therefore be written
. The rate of change of lipid surface area, assuming lipids flip between bilayers rapidly to keep equal numbers of molecules on both surfaces, is
times the rate of exchange of molecules. Hence, finally:
The rate constant is set to 1 in the examples below, which sets the timescale for membrane area growth comparable to that for vesicle volume growth.
Equation (14) says that the membrane area will grow either if the vesicle is under tension, or if the vesicle is relaxed but the internal concentration is higher than
. We therefore introduce further reactions into the system to allow for the possibility that the metabolism drives lipid synthesis inside the vesicle. A precursor molecule
is assumed to be present in the external environment, and combination of
with
yields the lipid
. However, this direct reaction does not occur, but it is driven in two steps by a catalytic process involving
, as shown in
Table 3. Thus, lipid synthesis will only occur when the metabolism is operating and maintaining a high concentration of
. We assume that the free energies of formation of the precursor
, the intermediate
and the final lipid
are
,
, and
respectively. The free energies of the reactions are given in
Table 3.
As the volume is changing with time, it is easier to deal with the number of molecules in the vesicle,
, and then to determine the concentrations as
, where
is in litres,
is in moles/litre and
is Avogadro’s number
. Equations (15)–(23) for the molecule numbers need to be solved concurrently with 13 and 14 for the volume and lipid surface area.
We have assumed that the membrane is permeable to the precursor , but not to the intermediate . The membrane is not permeable to the lipid itself, but can cross between the two sides by entering and leaving the membrane. In calculating the permeability terms above, we assumed that the constant in Equations (7)–(9) for the fixed-volume compartment applies to a compartment with volume and surface area . If the rate of concentration change in the standard vesicle is , the rate of change of the number of molecules is The rate of change in the vesicle of variable size increases in proportion to S, which gives a rate constant in the equations above.
We suppose that the critical lipid concentration is
M, which is appropriate for fatty acids, and the external concentration is fixed at
. The external concentrations of the precursor and intermediate are
M, and
M.
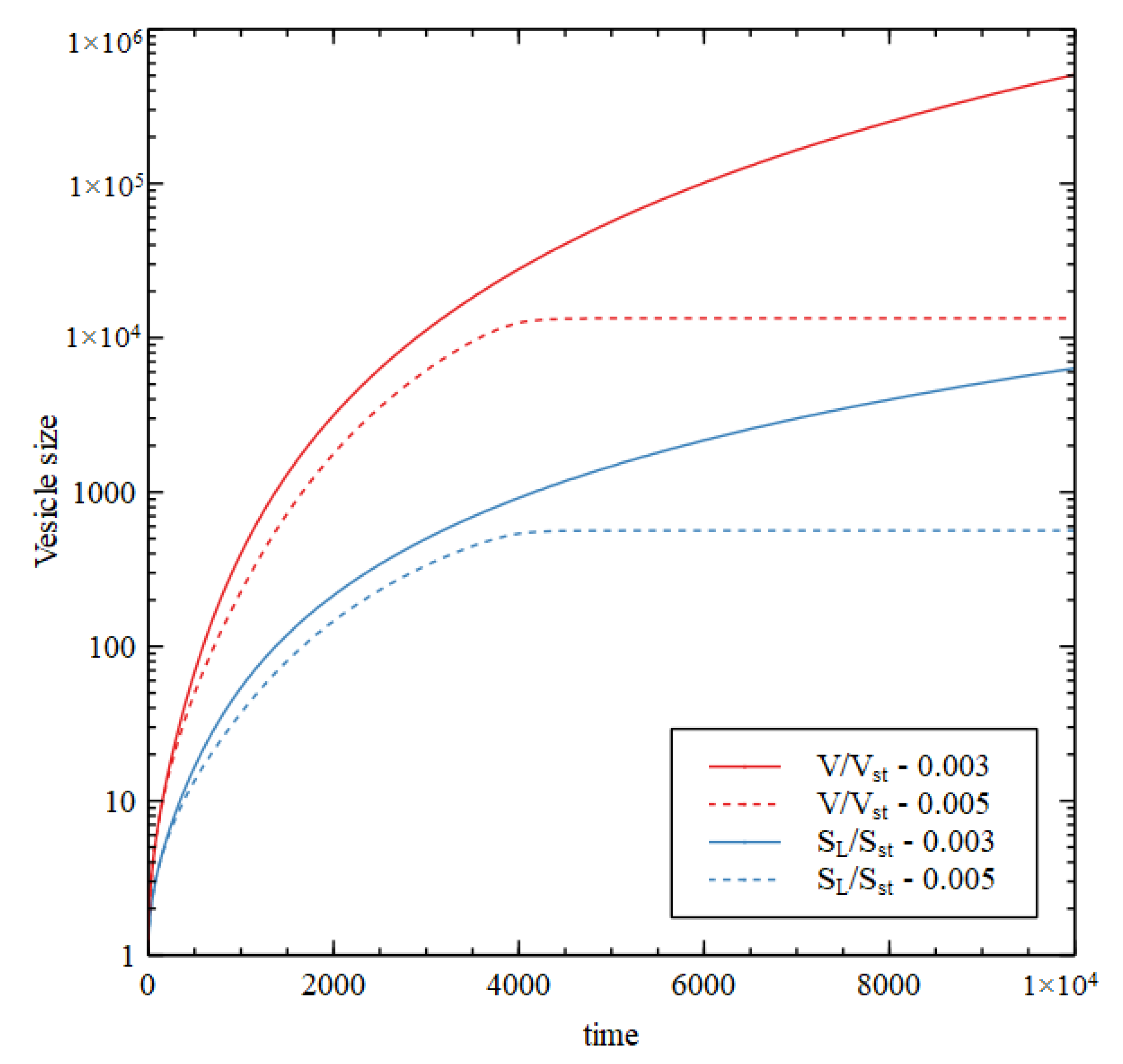
Figure 8 shows results using Case 1 reaction rates, assuming that
and
are present, but the rate constants for reaction 10 and 11 are
so there is no lipid synthesis by the metabolism. The inside begins with HC initial conditions while the outside is in the LC state. Therefore, there is a positive osmotic pressure that drives increase in volume. The membrane is then under tension (
, so
increases due to addition of lipid molecules from the outside. Continued growth is seen at
but at
the internal concentrations collapse to the external concentration after a time, so there is no further growth.
Figure 9 shows the concentrations inside the cell at the end of the simulation run (time 10000). There is a transition point at
. For
the osmotic pressure difference is maintained by the metabolism, and the volume and lipid surface area grow indefinitely. If
the osmotic pressure difference is not maintained and the vesicle stops growing. For
, all three criteria for a metabolism are satisfied while the vesicle is growing continuously.
When lipid synthesis occurs inside the vesicle, we need to consider the possibility of vesicle division. As has been shown previously [
27,
28,
29,
30], a spherical vesicle cannot divide without losing contents because the membrane area needed to enclose a sphere of volume
is less than the membrane area needed to enclose two spheres of volume
. Thus, if a vesicle is growing with a membrane under tension, as in
Figure 8, then it cannot divide. For division to occur we need the membrane to be relaxed, i.e., the vesicle must be non-spherical, with a lipid surface area
greater than the minimal surface area required for the sphere of the current volume
. After division, the minimal surface area for the two smaller spheres is
. Thus, division of the original vesicle can occur only if
To achieve vesicle division at the same time as the osmotic pressure is driving volume increase, we need the surface area to grow proportionately faster than the volume. This means that the lipid concentration must be higher than
, so that there is a net addition of molecules to the relaxed membrane. The exterior concentration can be temporarily higher than
if there is a sudden addition of lipid to the external medium. It is known that sudden addition of lipid micelles to vesicles causes vesicle division [
32,
36,
37]. On the other hand, if the external concentration remains at
, the internal concentration can become higher than
if there is lipid synthesis inside the vesicle.
We now consider the case of internal lipid synthesis driven by the metabolism. We consider Case 1 with
, which is able to support continued vesicle growth when there is no internal lipid synthesis (as in
Figure 8). We then add the lipid synthesis reactions 10 and 11, with rate constant
. We begin with one vesicle of standard size with internal concentrations equal to the HC initial conditions. Whenever the lipid surface area becomes large enough to satisfy the division condition,
the vesicle is divided into two half-sized vesicles, and we continue to follow the growth of one of these.
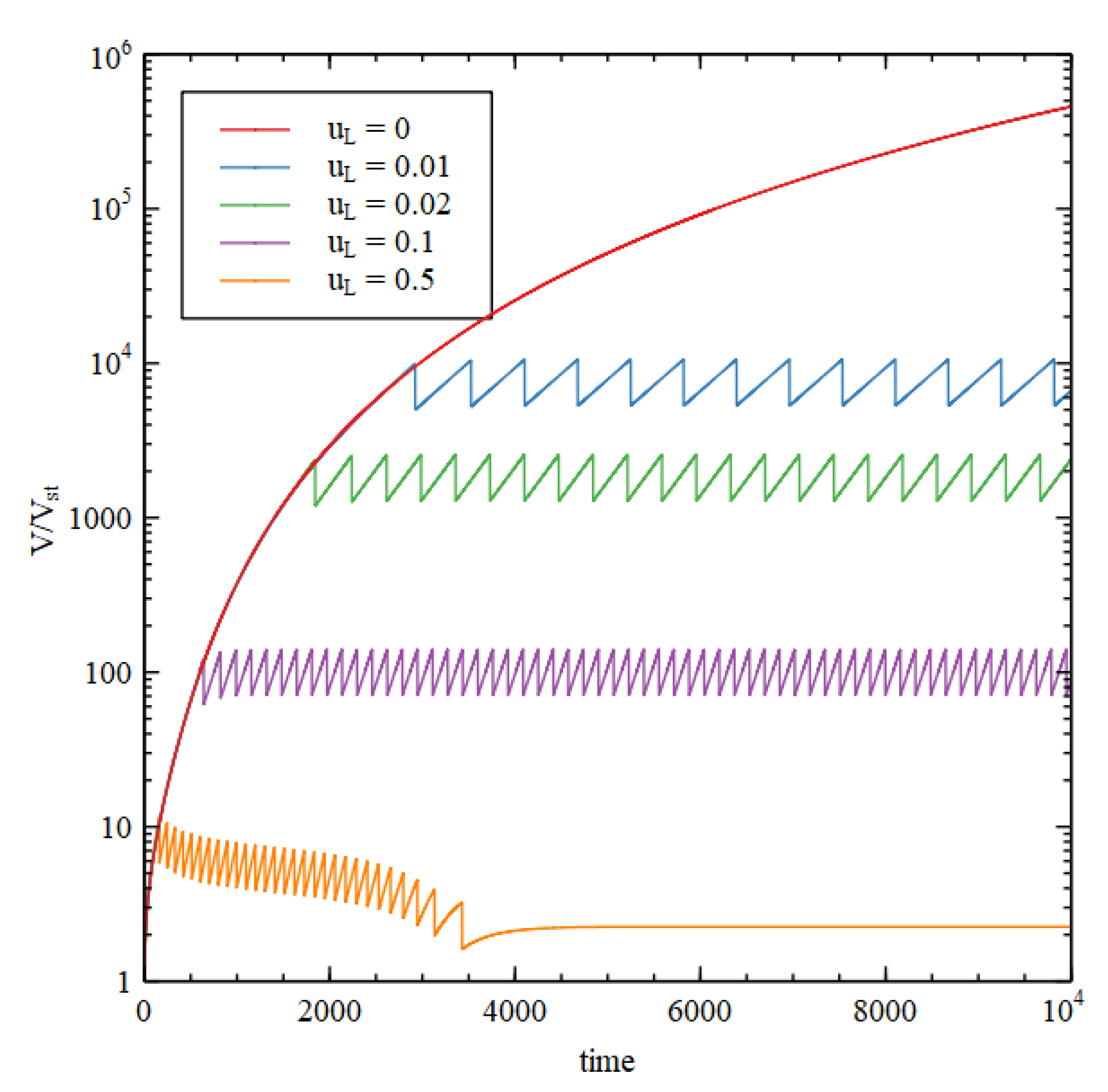
Figure 10 shows the volume as a function of time for five values of
. For
, we have continued growth without division, as before. For
, we have repeated growth and division. The time required for division and the size at which division occurs decrease with increasing lipid synthesis rate. Interestingly, it is possible for lipid synthesis to be too fast. For the highest
considered, the vesicle gets smaller on each division, because the division condition is reached before the size has doubled. Smaller vesicles have a larger surface area to volume ratio, so when the vesicle gets smaller, the permeability terms become larger relative to the internal reaction terms, and the difference between the internal and external concentrations cannot be maintained. Hence, growth and division stop after a few cycles.
In the case where continued growth occurs without division, as the growth is driven by autocatalytic reactions, we might expect that volume would grow exponentially with time. However, this is not so, because the rate of growth is limited by the rate of supply of food molecules. The vesicle settles into a state of steady growth where the internal concentrations are approximately constant in time and there is an approximately constant osmotic pressure. The membrane is only slightly swollen, and the tension term in equation 13 is small in comparison to the osmotic pressure. In this case the rate of volume increase is roughly proportional to the surface area. The rate of import of food molecules is also proportional to surface area, as is required if the internal concentration is constant. We thus have
, hence
increases in proportion to
, not exponentially with time. However, in the case where repeated cell division occurs, the time required for each division is constant. The number of vesicles doubles at each division; hence, the number of vesicles increases exponentially with time. The time per division is only a constant when there is balanced growth of the membrane and the vesicle contents, i.e., the time for doubling of the vesicle volume is equal to the time for doubling of the membrane area (more details in [
28]). These doubling times are dependent on vesicle size, and it can be seen that the vesicle naturally tends to a size when they are equal. In this self-reproducing state, each subsequent generation has the same size and composition as the previous one.
In this section, we have considered the Case 1 reaction network coupled to lipid synthesis and shown that it can drive vesicle growth and division. Any of the other models could also be coupled to lipid synthesis in the same way, so that in all the cases where the criteria for a metabolism are satisfied in a fixed-volume compartment, the addition of internal lipid synthesis will lead to growth and division in a lipid vesicle.
3.5. RNA Synthesis
The assumption of metabolism-first theories is that it is possible to have a self-sustaining small-molecule metabolic system without the presence of information-carrying polymers such as RNA. The previous examples have all focused on this case, showing that it is possible in theory, provided the reactions system satisfies the three criteria. If a small-molecule metabolism did get going in a protocell, it could later begin to synthesize nucleotides by some process that is coupled to the metabolism (in the way that the lipid synthesis was coupled to metabolism in the previous section). Polymerization of nucleotides could then arise in a protocell that already had an established metabolism.
However, there is still little experimental evidence for a self-sustaining small-molecule metabolism, so it is still possible to argue for replication-first. In this section, we make the point that template-directed synthesis of RNA is inherently autocatalytic because RNA strands are required to synthesize more strands. This means that the RNA polymerization process itself can produce a reaction system that satisfies the criteria for a metabolism. RNA World theories obviously require a means of synthesis of nucleotides before polymerization of nucleotides into RNA strands can occur. However, there is no requirement that the reaction pathways that generate nucleotides should be autocatalytic or that they should satisfy the criteria for a metabolism. This section will show a clear example where nucleotides are synthesized by a direct reaction pathway that is not autocatalytic and which does not by itself satisfy the metabolism criteria. Polymerization then occurs autocatalytically, and the metabolism requirements are satisfied due to the polymerization reactions, not the small-molecule reactions.
In this section, we identify molecule
as a nucleotide, and we call it
hereafter. Reactions 1–4 represent steps in the pathway synthesizing nucleotides. We assume that
= 1 for all these direct reactions, and that the small-molecule autocatalytic processes do not occur (as in Case 3). We already showed above that Case 3 does not satisfy the metabolism criteria, unless we set
to be zero for the formation reactions outside the cell. Here, on the contrary, reactions 1–4 occur freely both inside and out. So the nucleotide-synthesis pathway does not constitute a metabolism by itself, and cannot support cell growth and division. We then suppose that nucleotides can polymerize by a reaction
, where
represents a nucleotide that is part of an RNA polymer. Polymerization will generate many different sequences of many different lengths, and incorporation of all these things is beyond the scope of the present model. However, the simple
reaction captures the essence of the process. The polymerization rate is
where
and
are rates of spontaneous polymerization (independent of current strand concentration), non-enzymatic template-directed synthesis (proportional to template concentration
), and ribozyme-catalyzed synthesis (proportional to both template and catalyst concentration—hence
). All three synthesis processes are proportional to nucleotide concentration
, and they are limited by some saturation of resources or space when
reaches a maximum concentration
. Inclusion of
is necessary to prevent a runaway increase in polymer concentration. Cleavage reactions convert polymeric nucleotides back to single nucleotides
. We assume that the nucleotide
produced by cleavage is not the same as the nucleotide
prior to polymerization. For example,
could be an activated nucleotide, whereas
is not. We assume
cannot repolymerize, hence it is a waste molecule. The cleavage reaction occurs at rate
. We ignore the other waste molecule
, because it is not important for the point we are making in this section. The decay reactions 7–9 do not occur in this example. Treating
as a waste molecule is the least favourable case for establishing polymerization, in comparison to cases where cleavage directly reforms
, or where recycling of
back to
is possible.
When
is high and
and
are both low, RNA polymerization is bistable. In earlier papers [
38,
39,
40,
41] we have shown that this bistability results in two states that we called living (high polymer concentration with RNA synthesis dominated by the catalyzed process), and non-living (low polymer concentration with RNA synthesis dominated by the spontaneous process). These earlier papers focused on RNA replication without considering metabolism. Here, we make a link to metabolism.
Figure 11 and
Figure 12 consider a reaction network for RNA synthesis consisting of reactions 1–4 plus polymerization and cleavage reactions. We set
,
,
, and
, so that the reaction system is bistable and RNA synthesis is predominantly autocatalytic. We consider a fixed-volume compartment. Molecules
and
are maintained at constant concentration outside, as before. Molecules
and
are all formed outside as well as inside, and are considered as food molecules. The biomolecule is
. The waste molecule is
, which is fixed at a low concentration
= 0.01 M outside.
Figure 11 shows the stationary concentrations as a function of cleavage rate. Outside the cell there is an HC and an LC solution, as with the Case 1 small-molecule network, but ‘low’ and ‘high’ refer to the concentration of
in this example because
is the molecule whose presence denotes the living state. There are two transition values in
. For
, only the LC concentration is stable. For
, both states are stable and the state reached depends on the initial conditions. For
, only the HC state is stable. We start the outside in LC initial conditions and the inside in HC initial conditions. Thus, for
, the inside goes to the HC state while outside goes to the LC state. For
, both inside and outside go to the LC state; for
, both inside and outside go to the HC state. Note that
are also present in this example, but for clarity, they are not shown in
Figure 11.
The metabolic rate is the polymerization rate in this case
. We also assume that the single nucleotides are ionized, so they make a contribution to the osmotic pressure which is twice the number of molecules. Under the ideal gas approximation, a polymer contributes only once per chain rather than once per monomer, so the contribution per polymeric nucleotide
is
(1 for the ion and
for the polymer). We set the polymer length to be
. The resulting osmotic pressure is
There are theories giving the osmotic pressure of nucleic acids that are much more acurate than the ideal gas theory [
42], but these are more complicated than we require here. The values of these osmotic coefficients do not affect the conclusions of this paper. The main point is that, even though the osmotic coefficient for
is less than that for
, since
is impermeable, the conversion of
to
creates a positive osmotic pressure difference that can drive cell growth.
Figure 12 shows that both the osmotic pressure and the metabolic rate are higher inside than out when
. Thus, all three criteria for a metabolism are satisfied by the autocatalytic process of RNA polymerization, even though they are not satisfied by the small-molecule reactions that generate the nucleotides. The origin of metabolism occurs only because of the origin of replication in this case.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}