Thermal Behavior and Phase Transition of Uric Acid and Its Dihydrate Form, the Common Biominerals Uricite and Tinnunculite

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Occurrence
2.2. Single-Crystal X-Ray Diffraction Study
2.3. High-Temperature Powder X-Ray Diffraction Study
2.4. Infrared Spectroscopy
3. Results and Discussion
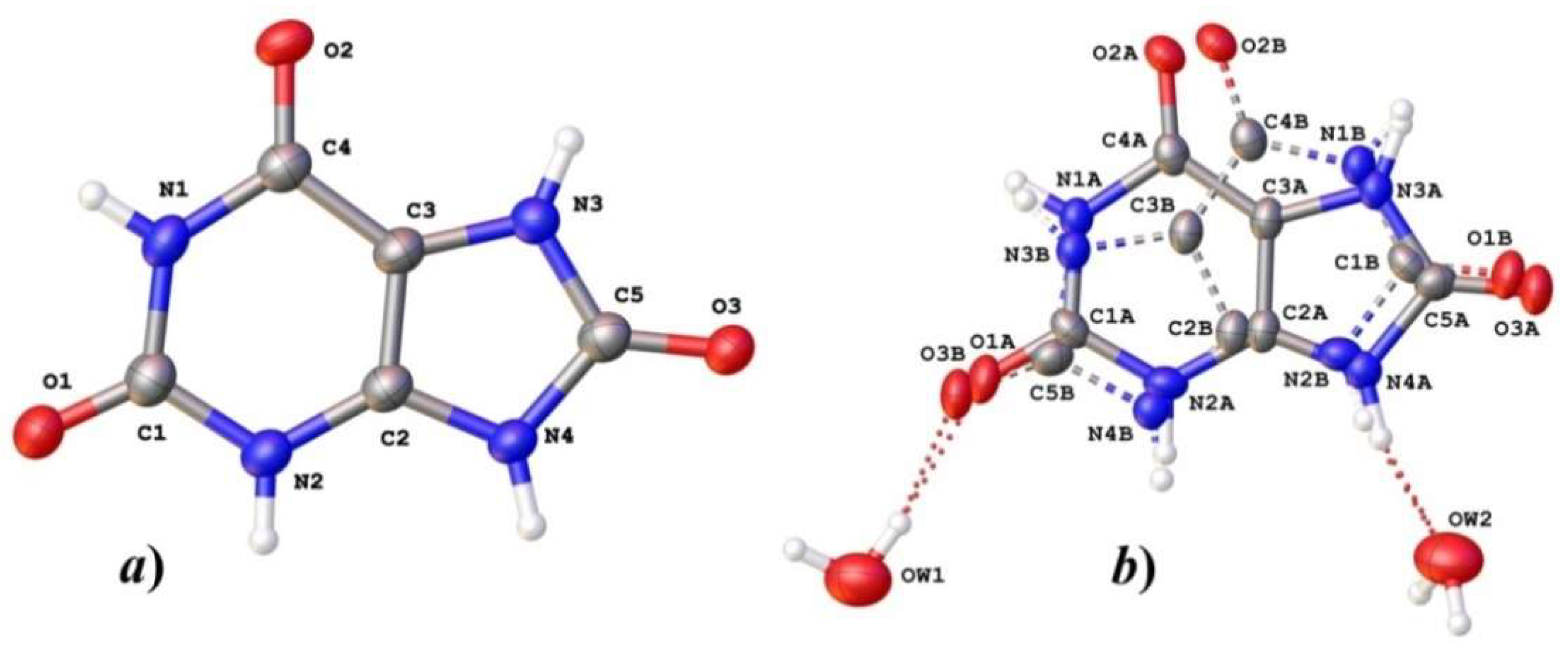
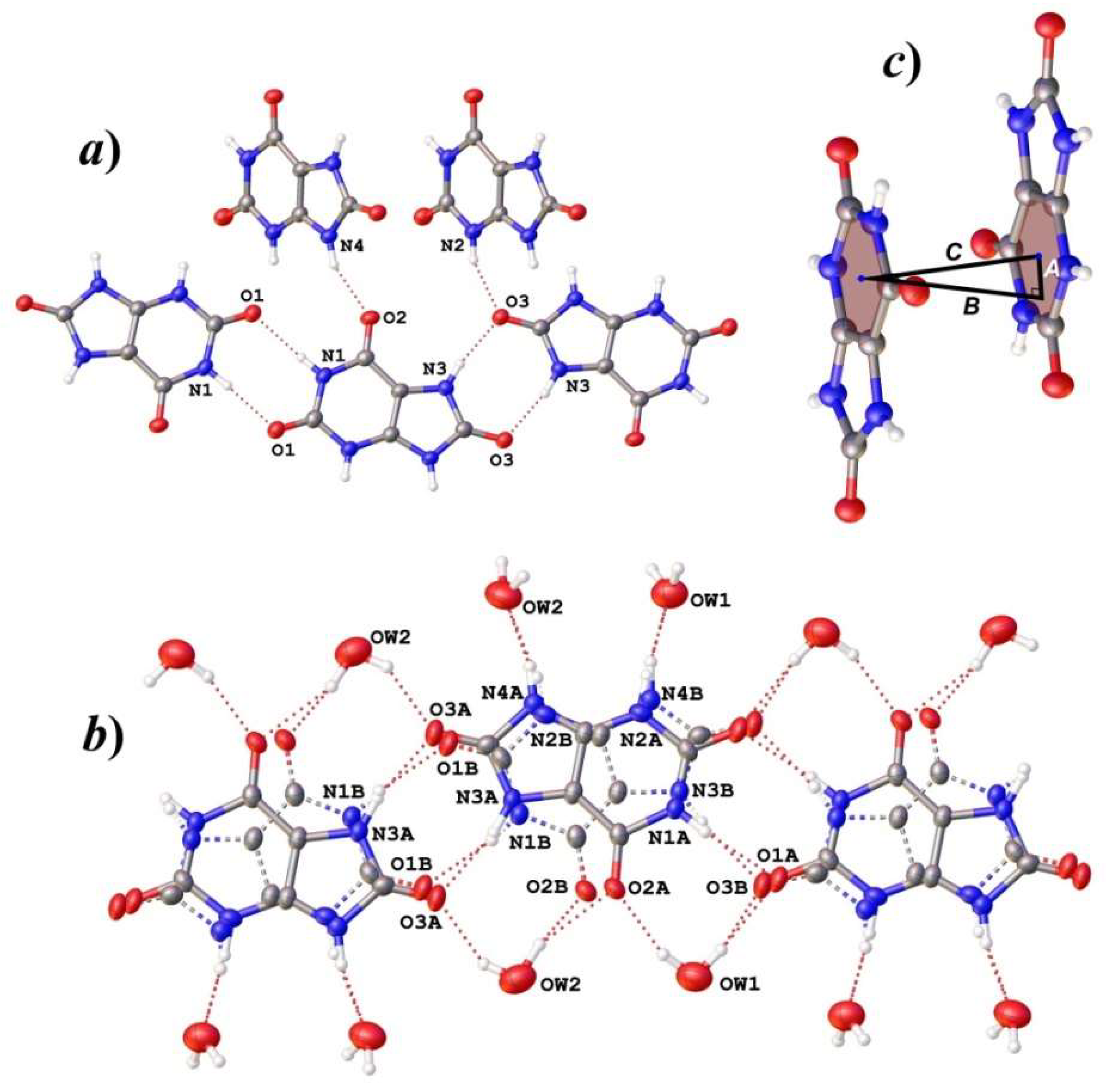
3.1. Structure Descriptions
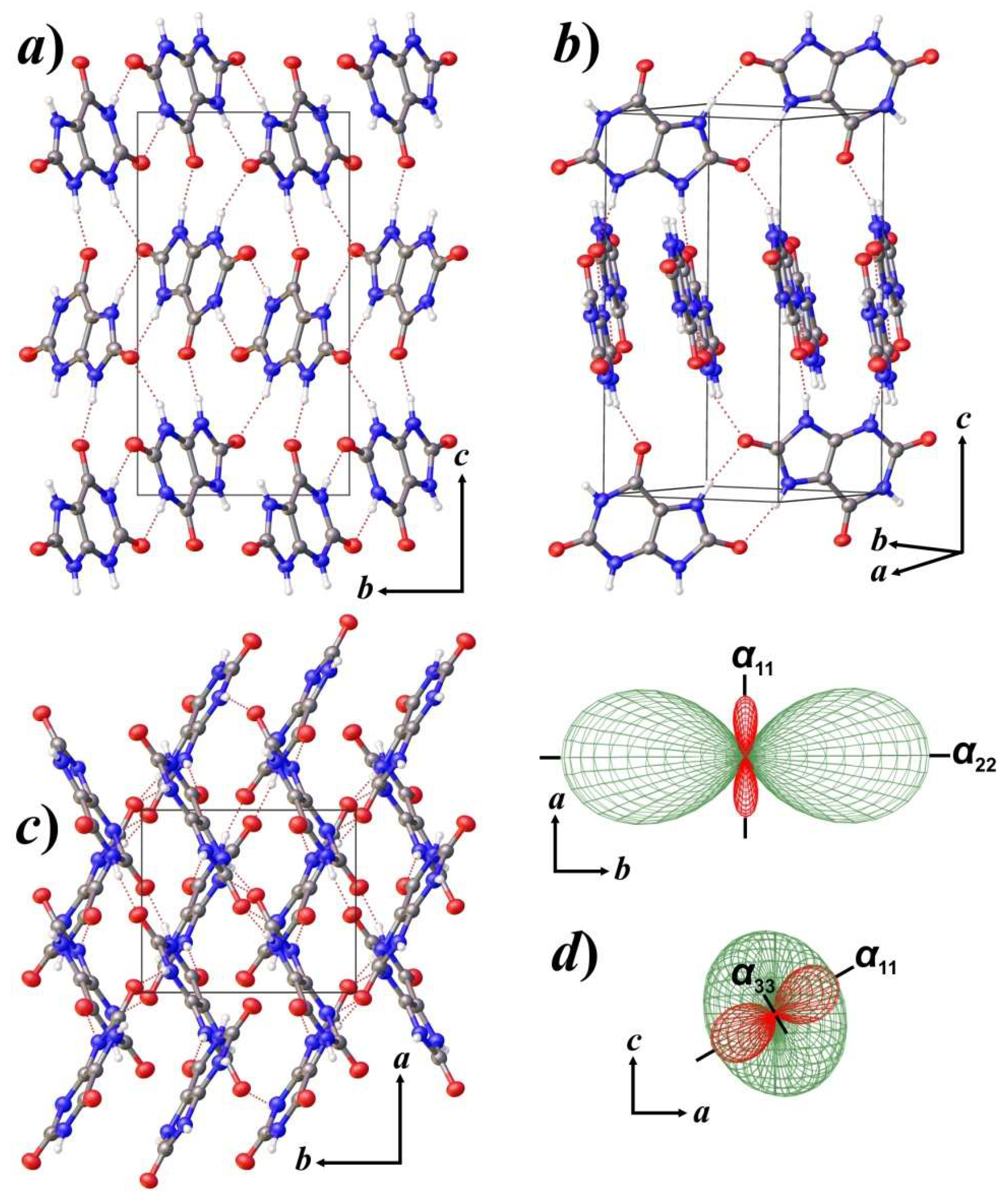
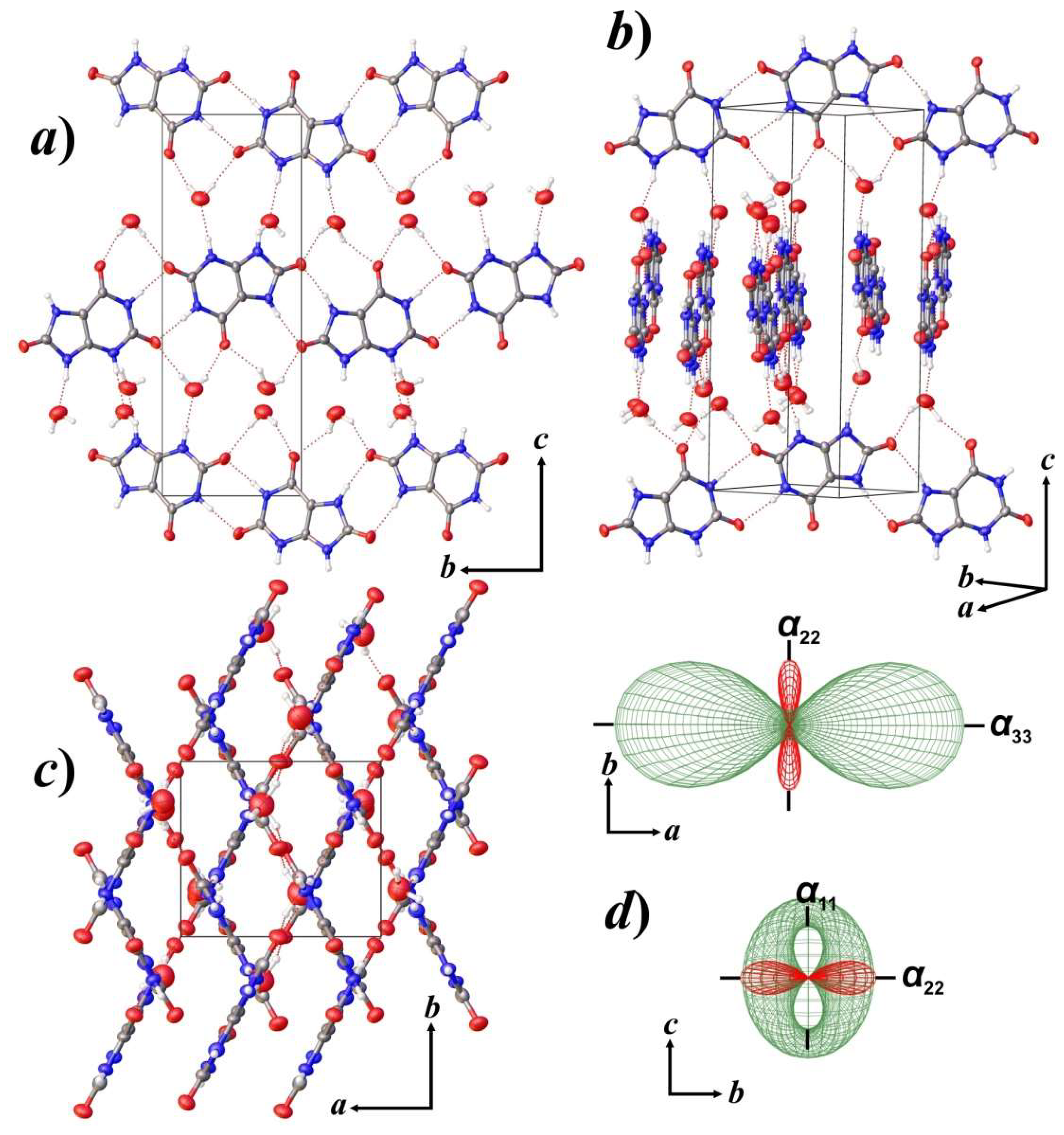
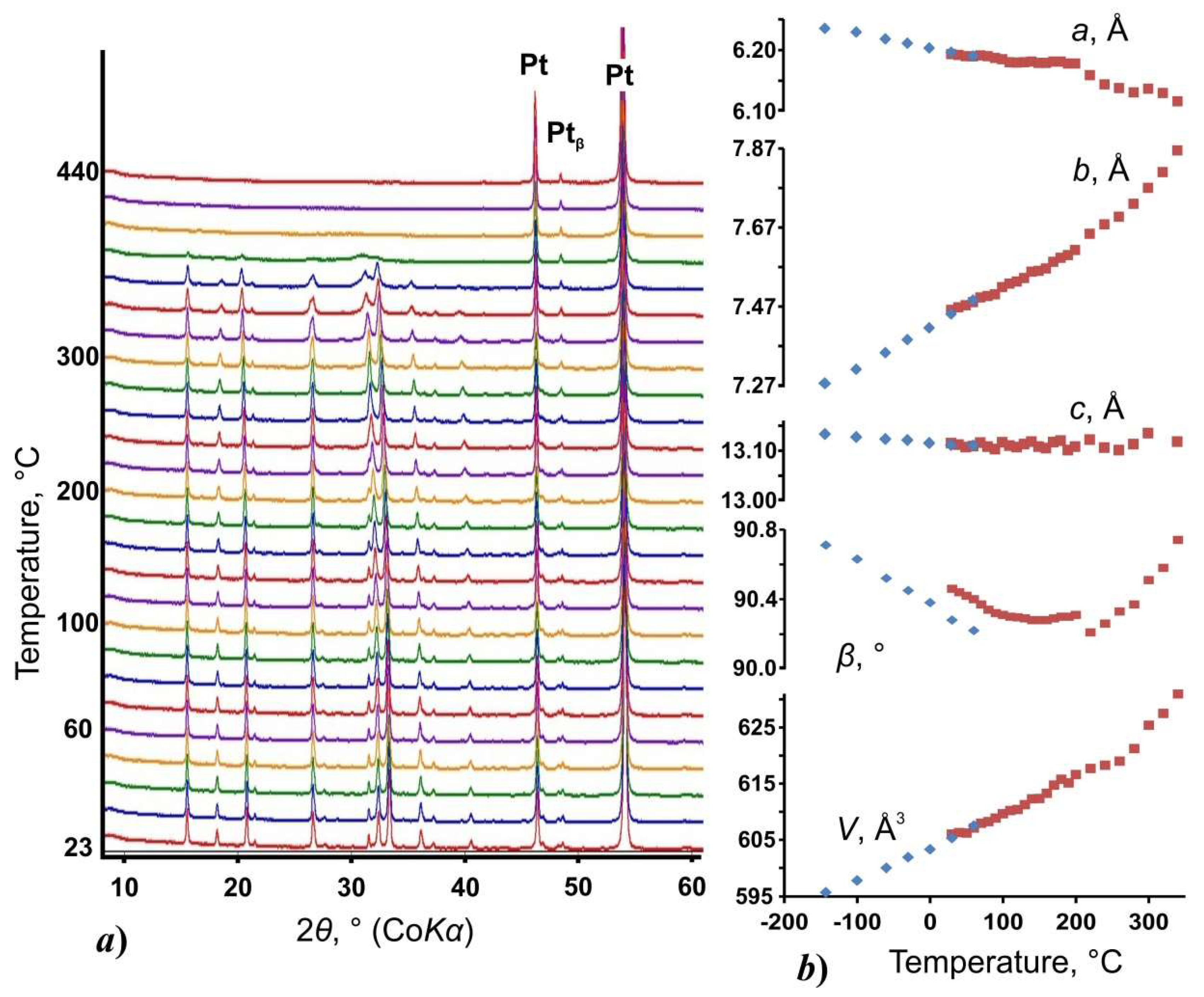
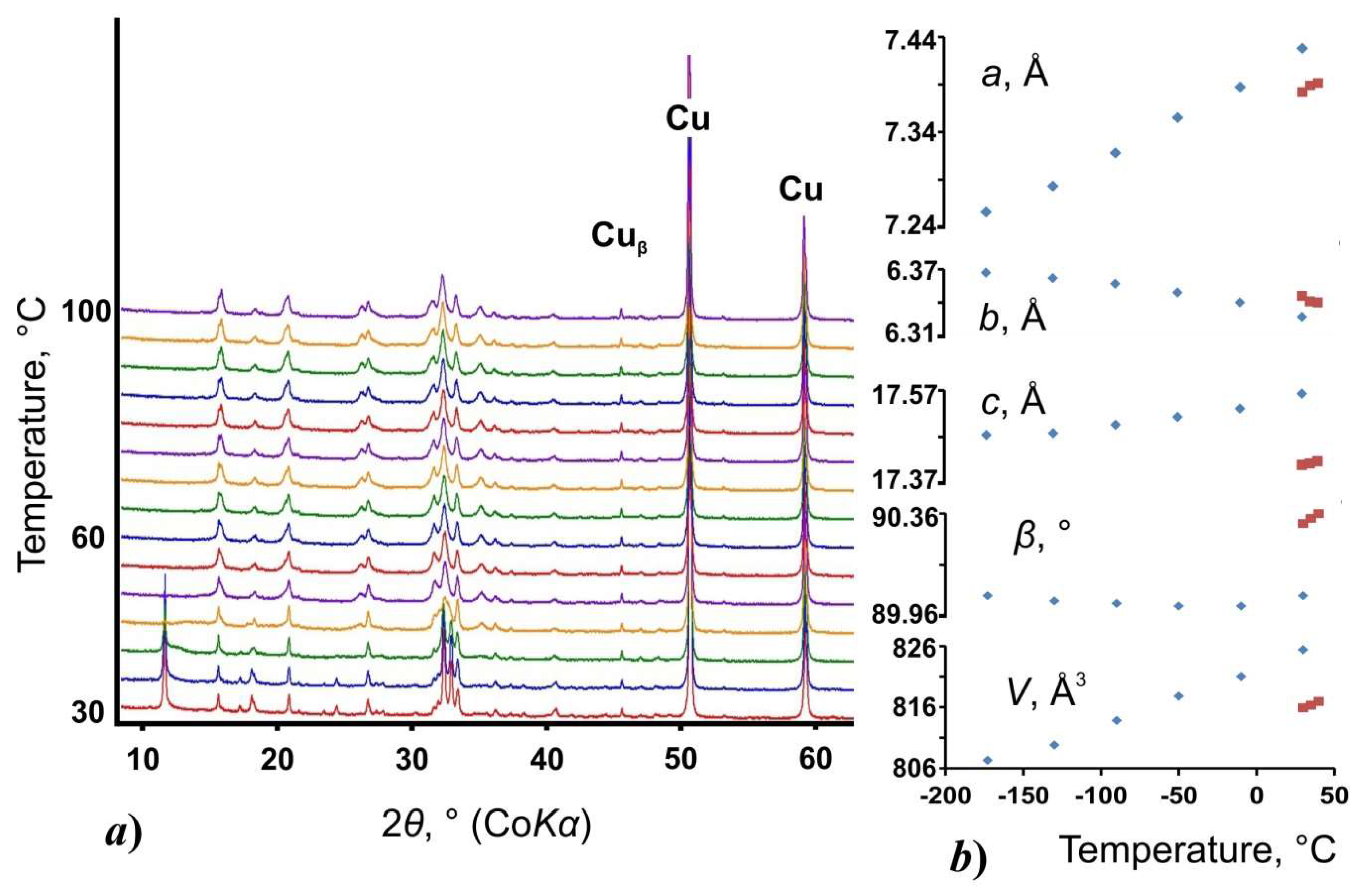
3.2. Thermal Behavior and Phase Transition
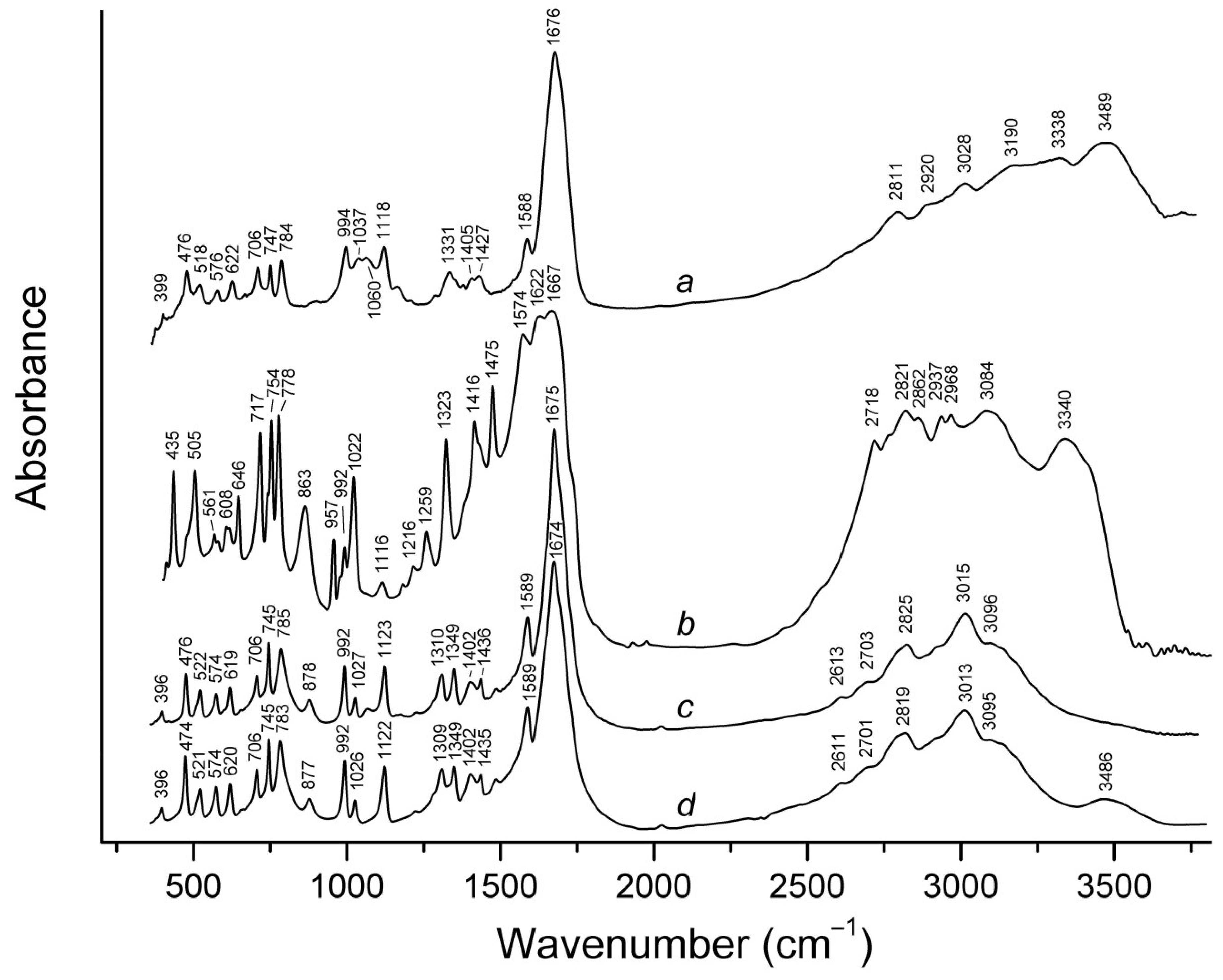
3.3. IR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kempe, S. Carbon in the rock cycle. In Scope 13, the Global Carbon Cycle; Bolin, B., Degens, E.T., Kempe, S., Ketner, P., Eds.; John Wiley and Sons: Chichester, UK, 1979; pp. 343–377. [Google Scholar]
- Hunt, J.M. Petroleum Geochemistry and Geology, 2nd ed.; Freeman: New York, NY, USA, 1995; 743p. [Google Scholar]
- Graustein, W.C.; Cromack, K.J.R.; Sollins, E. Calcium oxalate: Occurrence in soils and effect on nutrient and geochemical cycles. Science 1977, 23, 1252–1254. [Google Scholar] [CrossRef] [PubMed]
- Echigo, T.; Kimata, M. Crystal chemistry and genesis of organic minerals: A review of oxalate and polycyclic aromatic hydrocarbon minerals. Can. Mineral. 2010, 48, 1329–1358. [Google Scholar] [CrossRef]
- Izatulina, A.R.; Gurzhiy, V.V.; Krzhizhanovskaya, M.G.; Kuz’mina, M.A.; Leoni, M.; Frank-Kamenetskaya, O.V. Hydrated Calcium Oxalates: Crystal Structures, Thermal Stability and Phase Evolution. Cryst. Growth Des. 2018, 18, 5465–5478. [Google Scholar] [CrossRef]
- Grases, F.; Villacampa, A.I.; Costa-Bauza, A.; Sohnel, O. Uric acid calculi: Types, etiology and mechanisms of formation. Clin. Chim. Acta 2000, 302, 89–104. [Google Scholar] [CrossRef]
- Seregin, A.V.; Mulabaev, N.S.; Tolordava, E.R. Urolithiasis: Current Aspects of Etiology and Pathogenesis. Med. Bus. 2012, 4, 4–10. (In Russian) [Google Scholar]
- Klaproth, M.H. Beiträge zur Chemischen Kenntnis der Mineralkörper; Decker und Compagnie: Posen, Germany; Heinrich August Rottmann: Berlin/Leipzig, Germany, 1795; pp. 299–313. [Google Scholar]
- Bridge, P.J. Guanine and uricite, two new organic minerals from Peru and Western Australia. Min. Mag. 1974, 39, 889–890. [Google Scholar] [CrossRef]
- Chesnokov, B.V.; Kovalev, E.G.; Gorelov, P.N.; Kotlyarov, V.A.; Bushmakin, A.F.; Zhdanov, V.F.; Tinnunculite, C. 10H12N8O8, a new mineral, in Mineraly i mineral’noe syr’e gorno-promyshlennykh raionov Urala. In Minerals and Mineral Resources of Mining Districts of the Urals; UB of RAS: Sverdlovsk, Russia, 1989; pp. 20–24. [Google Scholar]
- Pekov, I.V.; Chukanov, N.V.; Yapaskurt, V.O.; Belakovskiy, D.I.; Lykova, I.S.; Zubkova, N.V.; Shcherbakova, E.P.; Britvin, S.N.; Chervonnyi, A.D.; Tinnunculite, C. 5H4N4O3·2H2O: Occurrences on the Kola Peninsula and Redefinition and Validation as a Mineral Species. Geol. Ore Depos. 2017, 59, 609–618. [Google Scholar] [CrossRef]
- Artioli, G.; Galli, E.; Ferrari, M. Uric acid dihydrate—A new mineral? Riv. Mineral. Ital. 1993, 4, 261–264. [Google Scholar]
- Kolitsch, U.; Raade, G. Uric acid dihydrate: Occurrences in Austria and Norway. Nor. Bergverksmus. Skr. 2011, 46, 25–29. [Google Scholar]
- Ringertz, H. Optical and crystallographic data of uric acid and its dihydrate. Acta Crystallogr. 1965, 19, 286. [Google Scholar] [CrossRef]
- Ringertz, H. The molecular and crystal structure of uric acid. Acta Crystallogr. 1966, 20, 397–403. [Google Scholar] [CrossRef]
- Artioli, G.; Masciocchi, N.; Galli, E. The elusive crystal structure of uric acid dihydrate: Implication for epitaxial growth during biomineralization. Acta Crystallogr. 1997, B53, 498–503. [Google Scholar] [CrossRef]
- Parkin, S.; Hope, H. Uric Acid Dihydrate Revisited. Acta Crystallogr. 1998, B54, 339–344. [Google Scholar] [CrossRef]
- Liu, F.; Hooks, D.E.; Nan, L.; Mara, N.A.; Swift, J.A. Mechanical Properties of Anhydrous and Hydrated Uric Acid Crystals. Chem. Mater. 2018, 30, 3798–3805. [Google Scholar] [CrossRef]
- Zellelow, A.Z.; Kim, K.-H.; Sours, R.E.; Swift, J.A. Solid-State Dehydration of Uric Acid Dihydrate. Cryst. Growth Des. 2010, 10, 418–425. [Google Scholar] [CrossRef]
- Schubert, G.; Gunter, R.; Jancke, H.; Kraus, W.; Patzelt, C. Uric acid monohydrate—A new urinary calculus phase. Urol. Res. 2005, 33, 231–238. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, version 1.171.38.46; Rigaku Oxford Diffraction: Oxford, UK, 2015.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Topas, version 4.2; General Profile and Structure Analysis Software for Powder Diffraction Data; Bruker AXS: Karlsruhe, Germany, 2009.
- Langreiter, T.; Kahlenberg, V. TEV—A Program for the Determination of the Thermal Expansion Tensor from Diffraction Data. Crystals 2015, 5, 143–153. [Google Scholar] [CrossRef]
- Filatov, S.K. Visokotemperaturnaia Kristallohimia. In High-Temperature Crystal Chemistry; Nedra: Leningrad, Russia, 1990. (In Russian) [Google Scholar]
- Hazen, R.M.; Downs, R.T. (Eds.) Reviews in Mineralogy and Geochemistry: High-Temperature and High-Pressure Crystal Chemistry; Mineralogical Society of America: Washington, DC, USA, 2001; Volume 41, pp. 1–596. [Google Scholar]
- Filatov, S.K. General concept of increasing crystal symmetry with an increase in temperature. Crystallogr. Rep. 2011, 56, 953–961. [Google Scholar] [CrossRef]
- Filatov, S.K. Negative linear thermal expansion of oblique-angle (monoclinic and triclinic) crystals as a common case. Phys. Stat. Sol. (B) 2008, 245, 2490–2496. [Google Scholar] [CrossRef]
- Ostwald, W. Studien über die Bildung und Umwandlung fester Körper. Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar] [CrossRef]
- Golovanova, O.A.; Punin, Y.O.; Izatulina, A.R.; Yelnikov, V.Y.; Plotkina, Y.V. Structural-tex-tural features and ontogenetic regularities of renal stone formation. Vestn. St. Peterbg. Univ. Seriya Geol. I Geogr. 2009, 1, 26–34. (In Russian) [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | T (°C) | a (Å) | b (Å) | c (Å) | Β (°) | V (Å3) | R1 | CCDC |
|---|---|---|---|---|---|---|---|---|
| UA (s.g. P21/n) | −143 | 6.2352(5) | 7.2756(6) | 13.1328(11) | 90.710(8) | 595.72(8) | 0.055 | 1916030 |
| −100 | 6.2288(4) | 7.3113(5) | 13.1274(8) | 90.632(6) | 597.79(7) | 0.056 | 1916031 | |
| −60 | 6.2182(4) | 7.3530(5) | 13.1232(8) | 90.519(6) | 600.00(7) | 0.053 | 1916032 | |
| −30 | 6.2110(4) | 7.3857(5) | 13.1213(8) | 90.447(6) | 601.89(7) | 0.048 | 1916033 | |
| 0 | 6.2025(4) | 7.4163(5) | 13.1147(8) | 90.378(6) | 603.26(7) | 0.046 | 1916034 | |
| 30 | 6.1963(3) | 7.4512(4) | 13.1114(7) | 90.282(5) | 605.34(5) | 0.042 | 1916035 | |
| 60 | 6.1898(3) | 7.4857(4) | 13.1103(5) | 90.217(4) | 607.46(5) | 0.037 | 1916036 | |
| UAD (s.g. P21/c) | −173 | 7.2559(4) | 6.3669(4) | 17.4739(13) | 90.036(6) | 807.25(9) | 0.048 | 1916037 |
| −130 | 7.2830(3) | 6.3616(3) | 17.4781(9) | 90.020(4) | 809.79(7) | 0.047 | 1916038 | |
| −90 | 7.3175(3) | 6.3565(3) | 17.4962(8) | 90.010(4) | 813.82(6) | 0.047 | 1916039 | |
| −50 | 7.3553(4) | 6.3485(3) | 17.5125(9) | 90.005(5) | 817.75(7) | 0.050 | 1916040 | |
| −10 | 7.3872(4) | 6.3397(3) | 17.5312(9) | 90.004(5) | 821.03(7) | 0.048 | 1916041 | |
| 30 | 7.4283(8) | 6.3266(5) | 17.5627(16) | 90.044(10) | 825.37(13) | 0.094 | 1916042 |
| Sample | T (°C) | A | B | C | N1···O1 | N2···O3 | N3···O3 | N4···O2 |
| UA | −143 | 1.252(4) | 3.241(3) | 3.474(2) | 2.816(3) | 2.800(3) | 2.780(3) | 2.721(3) |
| −100 | 1.245(5) | 3.246(3) | 3.476(3) | 2.817(3) | 2.803(4) | 2.782(3) | 2.723(3) | |
| −60 | 1.231(4) | 3.254(3) | 3.479(2) | 2.819(3) | 2.806(3) | 2.783(3) | 2.725(3) | |
| −30 | 1.228(4) | 3.261(3) | 3.484(2) | 2.821(3) | 2.809(3) | 2.786(3) | 2.727(3) | |
| 0 | 1.224(4) | 3.265(3) | 3.487(2) | 2.824(3) | 2.810(3) | 2.790(3) | 2.729(3) | |
| 30 | 1.221(4) | 3.274(2) | 3.494(2) | 2.827(3) | 2.815(3) | 2.789(3) | 2.728(2) | |
| 60 | 1.218(3) | 3.280(2) | 3.499(2) | 2.829(2) | 2.818(2) | 2.793(2) | 2.730(2) | |
| T (°C) | a | b | c | N1A···O1A | N2A···OW1 | N3A···O3A | N4A···OW2 | |
| UAD | −173 | 1.619(11) | 3.242(7) | 3.624(6) | 2.832(12) | 2.832(7) | 2.836(14) | 2.566(8) |
| −130 | 1.629(12) | 3.240(8) | 3.627(6) | 2.808(16) | 2.816(8) | 2.858(18) | 2.555(7) | |
| −90 | 1.586(12) | 3.268(7) | 3.633(6) | 2.780(15) | 2.848(6) | 2.887(17) | 2.549(6) | |
| −50 | 1.611(11) | 3.273(7) | 3.648(6) | 2.837(12) | 2.844(6) | 2.879(13) | 2.559(7) | |
| −10 | 1.591(13) | 3.311(8) | 3.674(7) | 2.864(13) | 2.846(8) | 2.876(15) | 2.563(8) | |
| 30 | 1.48(4) | 3.24(3) | 3.57(2) | 2.71(4) | 2.84(2) | 2.94(4) | 2.51(2) |
| Sample | T (°C) | α11 | α22 | α33 | <α11a | <α22b | <α33c |
|---|---|---|---|---|---|---|---|
| UA | −100 | −45.7 | 129.0 | 2.0 | 29.60 | 0 | 28.98 |
| −50 | −48.3 | 139.7 | 2.8 | 29.17 | 0 | 28.67 | |
| 0 | −51.0 | 150.1 | 3.6 | 28.76 | 0 | 28.39 | |
| 50 | −53.6 | 160.3 | 4.4 | 28.36 | 0 | 28.12 | |
| UAD | −100 | 19.8 | −24.7 | 110.8 | 88.41 | 0 | 88.40 |
| −50 | 28.8 | −34.7 | 120.7 | 88.04 | 0 | 88.04 | |
| 0 | 37.7 | −44.8 | 130.5 | 87.66 | 0 | 87.68 | |
| 30 | 43.1 | −50.9 | 136.2 | 87.43 | 0 | 87.47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izatulina, A.R.; Gurzhiy, V.V.; Krzhizhanovskaya, M.G.; Chukanov, N.V.; Panikorovskii, T.L. Thermal Behavior and Phase Transition of Uric Acid and Its Dihydrate Form, the Common Biominerals Uricite and Tinnunculite. Minerals 2019, 9, 373. https://doi.org/10.3390/min9060373
Izatulina AR, Gurzhiy VV, Krzhizhanovskaya MG, Chukanov NV, Panikorovskii TL. Thermal Behavior and Phase Transition of Uric Acid and Its Dihydrate Form, the Common Biominerals Uricite and Tinnunculite. Minerals. 2019; 9(6):373. https://doi.org/10.3390/min9060373
Chicago/Turabian StyleIzatulina, Alina R., Vladislav V. Gurzhiy, Maria G. Krzhizhanovskaya, Nikita V. Chukanov, and Taras L. Panikorovskii. 2019. "Thermal Behavior and Phase Transition of Uric Acid and Its Dihydrate Form, the Common Biominerals Uricite and Tinnunculite" Minerals 9, no. 6: 373. https://doi.org/10.3390/min9060373