
Crystal Structure Prediction and Lattice Dynamical Calculations for the Rare Platinum-Group Mineral Zaccariniite (RhNiAs)


Abstract
:1. Introduction
2. Methods
3. Results and Discussion
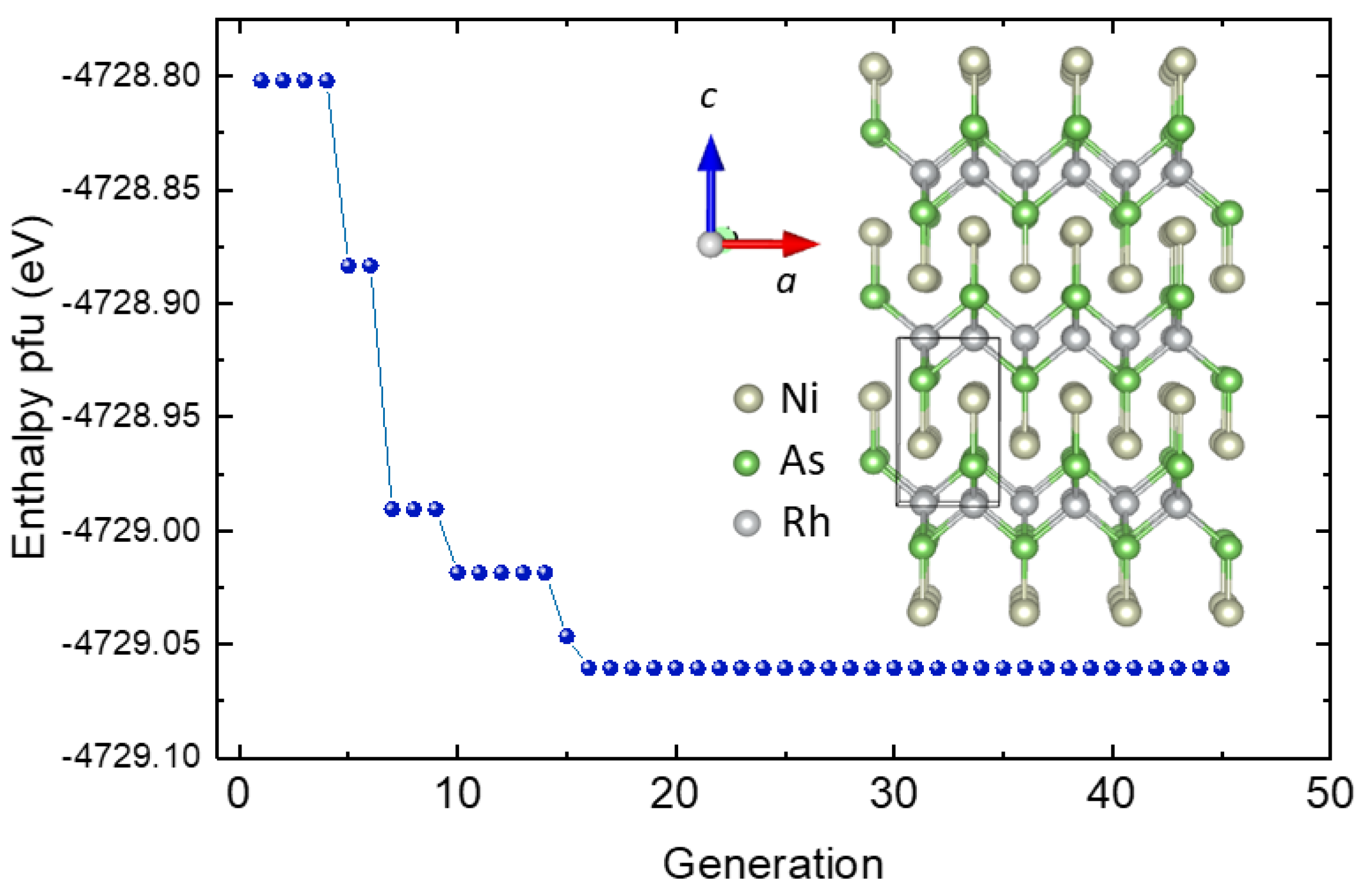
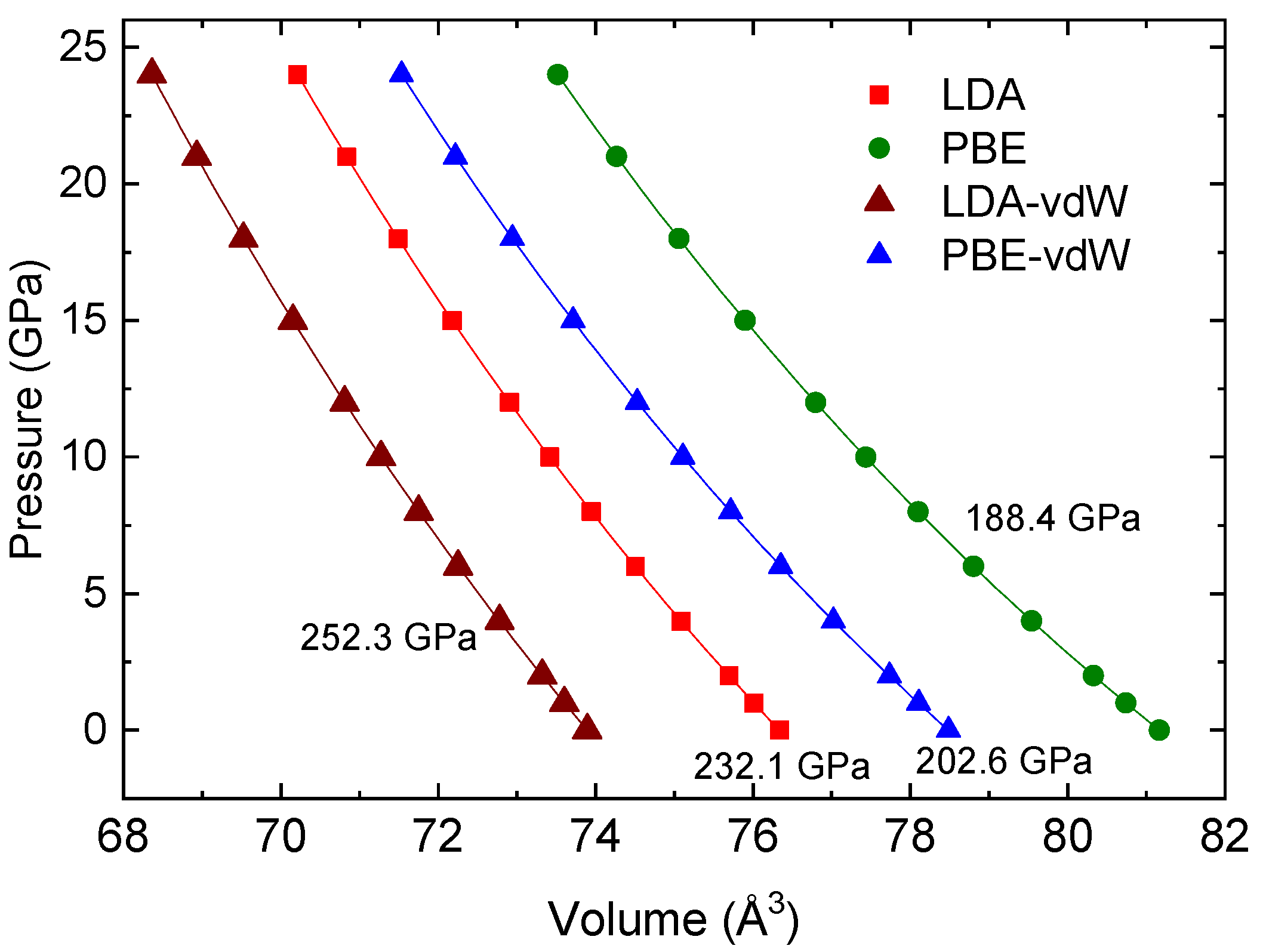
3.1. Crystal Structure Determination
3.2. Lattice Dynamical Calculations
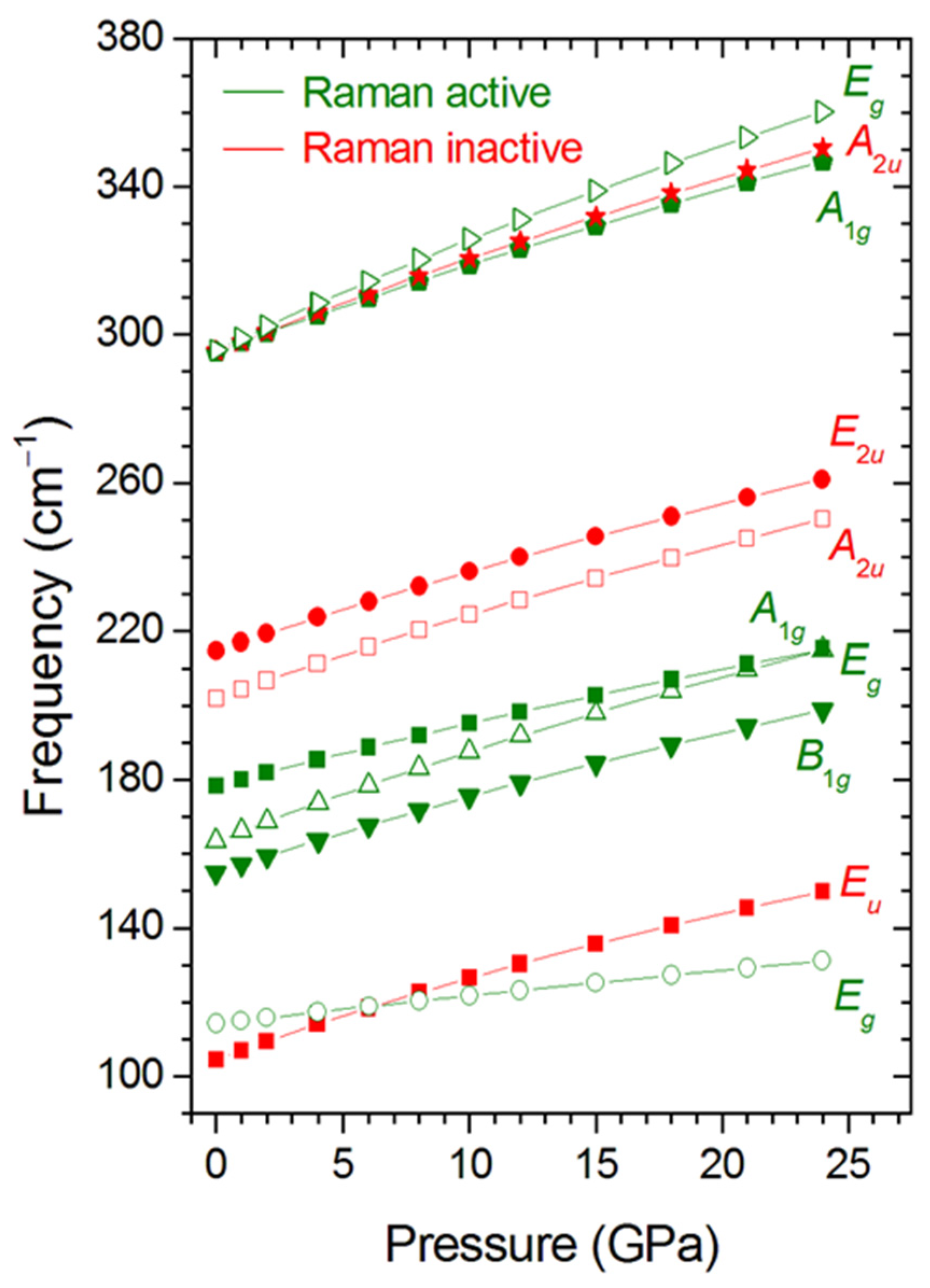
3.3. Zone-Center Phonons of RhNiAs as a Function of Pressure
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anhaeusser, C.R. The Geology, Geochemistry, Mineralogy and Mineral Beneficiation of Platinum-Group Elements.: Edited by Louis J. Cabri. CIM Special Volume 54, Canadian Institute of Mining, Metallurgy and Petroleum. Can. Mineral. 2003, 41, 544–546. [Google Scholar] [CrossRef]
- Vymazalová, A.; Laufek, F.; Drábek, M.; Cabral, A.R.; Haloda, J.; Sidorinová, T.; Lehmann, B.; Galbiatti, H.F.; Drahokoupil, J. Jacutingaite, Pt2HgSe3, a new platinum-group mineral species from the Cauê iron-ore deposit, itabira district, minas gerais, Brazil. Can. Mineral. 2012, 50, 431–440. [Google Scholar] [CrossRef]
- Vymazalová, A.; Laufek, F.; Drábek, M.; Stanley, C.J.; Baker, R.J.; Bermejo, R.; Garuti, G.; Thalhammer, O.; Proenza, J.A.; Longo, F. Zaccariniite, RhNiAs, a new platinum-group mineral from loma peguera, dominican republic. Can. Mineral. 2012, 50, 1321–1329. [Google Scholar] [CrossRef]
- Zaccarini, F.; Proenza, J.A.; Rudashevsky, N.S.; Cabri, L.J.; Garuti, G.; Rudashevsky, V.N.; Melgarejo, J.C.; Lewis, J.F.; Longo, F.; Bakker, R.J.; et al. The Loma Peguera ophiolitic chromitite (Central Dominican Republic): A source of new platinum group minerals (PGM) species. Neues Jahrb. für Mineral. Abh. 2009, 335–349. [Google Scholar] [CrossRef]
- Roqué Rosell, J.; Portillo Serra, J.; Aiglsperger, T.H.; Plana Ruiz, S.; Das, P.P.; Mendoza Gonzalvez, J.; Trifonov, T.; Proenza, J.A. Structural characterization and ab-initio resolution of natural occurring zaccariniite (RhNiAs) by means of Precession Electron Diffraction. Microchem. J. 2019, 148, 130–140. [Google Scholar] [CrossRef]
- Roqué Rosell, J.; Portillo Serra, J.; Aiglsperger, T.H.; Plana-Ruiz, S.; Pratim Das, P.; Mendoza Gonzalvez, J.; Trifonov, T.; Proenza, J.A. Crystallographic information data of natural occurring zaccariniite (RhNiAs) obtained by means of precession electron diffraction. Data Br. 2019, 25, 104346. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [Green Version]
- Lyakhov, A.O.; Oganov, A.R.; Stokes, H.T.; Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 2013, 184, 1172–1182. [Google Scholar] [CrossRef]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How Evolutionary Crystal Structure Prediction Works—And Why. Acc. Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ma, Y.; Oganov, A.R.; Wang, H.; Wang, H.; Xu, Y.; Cui, T.; Mao, H.K.; Zou, G. Superhard monoclinic polymorph of carbon. Phys. Rev. Lett. 2009, 102, 175506. [Google Scholar] [CrossRef] [Green Version]
- Oganov, A.R.; Chen, J.; Gatti, C.; Ma, Y.; Ma, Y.; Glass, C.W.; Liu, Z.; Yu, T.; Kurakevych, O.O.; Solozhenko, V.L. Ionic high-pressure form of elemental boron. Nature 2009, 457, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oganov, A.R.; Ma, Y.; Xu, Y.; Errea, I.; Bergara, A.; Lyakhov, A.O. Exotic behavior and crystal structures of calcium under pressure. Proc. Natl. Acad. Sci. USA 2010, 107, 7646–7651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Oganov, A.R.; Bergara, A.; Martinez-Canales, M.; Cui, T.; Iitaka, T.; Ma, Y.; Zou, G. Superconducting high pressure phase of germane. Phys. Rev. Lett. 2008, 101, 107002. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Oganov, A.R.; Goncharov, A.F.; Zhu, Q.; Boulfelfel, S.E.; Lyakhov, A.O.; Stavrou, E.; Somayazulu, M.; Prakapenka, V.B.; Konôpková, Z. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 2013, 342, 1502–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Oganov, A.R.; Lyakhov, A.O. Novel stable compounds in the Mg–O system under high pressure. Phys. Chem. Chem. Phys. 2013, 15, 7696–7700. [Google Scholar] [CrossRef] [PubMed]
- Zurek, E.; Hoffmann, R.; Ashcroft, N.W.; Oganov, A.R.; Lyakhov, A.O. A little bit of lithium does a lot for hydrogen. Proc. Natl. Acad. Sci. USA 2009, 106, 17640–17643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez-Insa, J. Reformulating table salt under pressure. Science 2013, 342, 1459–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum espresso: A modular and open-source software project for quantumsimulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; de Gironcoli, S.; Delugas, P.; Ruffino, F.F.; et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 2020, 152, 154105. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Molina-Sánchez, A.; Hummer, K.; Wirtz, L. Vibrational and optical properties of MoS2: From monolayer to bulk. Surf. Sci. Rep. 2015, 70, 554–586. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, J.; Woźniak, T.; Dybala, F.; Oliva, R.; Hernández, S.; Kudrawiec, R. High-pressure Raman scattering in bulk HfS2: Comparison of density functional theory methods in layered MS2 compounds (M = Hf, Mo) under compression. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Insa, J.; Woźniak, T.; Oliva, R.; Popescu, C.; Hernández, S.; López-Vidrier, J. Structural and High-Pressure Properties of Rheniite (ReS2) and (Re,Mo)S2. Minerals 2021, 11, 207. [Google Scholar] [CrossRef]
- Manjón, F.J.; Sans, J.Á.; Rodríguez-Hernández, P.; Muñoz, A. Combined Experimental and Theoretical Studies: Lattice-Dynamical Studies at High Pressures with the Help of Ab Initio Calculations. Minerals 2021, 11, 1283. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Lattice Parameters (Å) | Zero-Pressure Unit-Cell Volume (Å3) | Bulk Modulus (GPa) | |
|---|---|---|---|
| LDA | a = 3.54350 c = 6.07957 | 76.338 | 232.1 |
| LDA-vdW | a = 3.49898 c = 6.03559 | 73.893 | 252.3 |
| PBE | a = 3.61884 c = 6.19769 | 81.165 | 188.4 |
| PBE-vdW | a = 3.56793 c = 6.16537 | 78.486 | 202.6 |
| Experimental [3] | a = 3.5498 (1) c = 6.1573 (2) | 77.59 (1) | – |
| Structural data on zaccariniite (RhNiAs) obtained with PBE-vdW calculations | |
|---|---|
| Crystal data | |
| Space group | P4/nmm, No. 129 |
| Formula unit | RhNiAs, Z = 2 |
| Zero-pressure unit-cell parameters | a = 3.56793 Å |
| c = 6.16537 Å | |
| Atomic positions | |
| Ni | Wyckoff position: 2a |
| x = ¾, y = ¼, z = 0 | |
| Rh | Wyckoff position: 2c |
| x = ¼, y = ¼, z = 0.3612 | |
| As | Wyckoff position: 2c |
| x = ¼, y = ¼, z = 0.7475 | |
| Equation of state (3rd order Birch-Murnaghan) | |
| Zero-pressure unit-cell volume | 78.485 (1) Å3 |
| Zero-pressure bulk modulus | B0 = 202.6 (1) GPa |
| First derivative of the bulk modulus | B’ = 5.26 (1) |
| Symmetry (Activity) | Frequency ω0 (cm−1) | a (cm−1 GPa−1) | b (cm−1 GPa−2) |
|---|---|---|---|
| Eu (IR) | 104.62 | 2.41 | −0.0222 |
| Eg (Raman) | 114.19 | 0.79 | −0.0036 |
| B1g (Raman) | 154.96 | 2.21 | −0.0162 |
| Eg (Raman) | 163.72 | 2.56 | −0.0178 |
| A1g (Raman) | 178.37 | 1.89 | −0.0110 |
| A2u (IR) | 202.11 | 2.38 | −0.0161 |
| Eu (IR) | 214.77 | 2.27 | −0.0146 |
| A2u (IR) | 295.48 | 2.67 | −0.0161 |
| A1g (Raman) | 295.48 | 2.48 | −0.0145 |
| Eg (Raman) | 295.94 | 3.19 | −0.0218 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibáñez-Insa, J. Crystal Structure Prediction and Lattice Dynamical Calculations for the Rare Platinum-Group Mineral Zaccariniite (RhNiAs). Minerals 2022, 12, 98. https://doi.org/10.3390/min12010098
Ibáñez-Insa J. Crystal Structure Prediction and Lattice Dynamical Calculations for the Rare Platinum-Group Mineral Zaccariniite (RhNiAs). Minerals. 2022; 12(1):98. https://doi.org/10.3390/min12010098
Chicago/Turabian StyleIbáñez-Insa, Jordi. 2022. "Crystal Structure Prediction and Lattice Dynamical Calculations for the Rare Platinum-Group Mineral Zaccariniite (RhNiAs)" Minerals 12, no. 1: 98. https://doi.org/10.3390/min12010098