1. Introduction
Economic processing of chalcopyrite ores through bioleaching, i.e., the mobilisation of metals from ore by microorganisms, has not been as successful as secondary copper sulphide leaching operations [
1]. This chalcopyrite “problem” needs to be solved, as it is the dominant copper mineral in many low-grade copper deposits. This has resulted in large quantities of low-grade waste material being stockpiled or discarded in mining operations, as they are not economic to process, though they do contain massive quantities of metals (i.e., copper) simply due to their combined volume [
1,
2,
3]. The highest copper recovery from chalcopyrite bioleaching has come from setups that tend to involve high energy costs due to heating or pre-processing, such as concentration, grinding or continuous stirring [
4,
5]. Because of these expensive overhead costs, it would be desirable to enhance bioleaching from chalcopyrite in a system with low operating costs, i.e., a leaching setup that requires minimal energy input over its lifetime, for instance ambient temperature heap (column) leaching systems that only require nutrient addition. By that logic, an ideal system would not entail any additional inputs; however, it is expected that certain costs associated with typical setups are unavoidable, such as agglomeration with acid or energy requirements for aeration and irrigation.
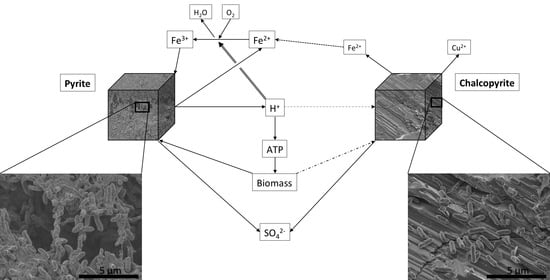
Leaching of sulphides occurs according to two distinct pathways, and by two different processes. The pathways are named according to the intermediate sulphur species formed, and the process names describe the oxidising agents. Pyrite, as an acid-nonsoluble sulphide can only be oxidised by ferric iron (Fe
3+), releasing thiosulphate (rxn. 1), whereas chalcopyrite, as an acid-soluble sulphide can be oxidised by Fe
3+ or by acid (H
+), resulting in polysulphide intermediates (rxn. 2). The chemistry is complex, thiosulphate can disproportionate (rxn. 3), and any remaining sulphur intermediates (e.g., from rxn. 2) can be subsequently oxidised further to elemental sulphur (rxn. 4) and/or to sulphate (rxns. 5–8) by sulphur oxidising bacteria. In these systems, ferrous iron (Fe
2+) is reoxidised to Fe
3+ by iron oxidisers (rxn. 9) to form an iron cycle (rxns. 9–12; Schippers and Sand [
6]). These reactions can take place as “contact” or “non-contact” leaching, whereby the former denotes attachment of cells to the substrate, potentially with extracellular polymeric substances (EPS) surrounding the cells and providing the reaction space, and the latter denotes planktonic cells with contact of the oxidants with the mineral surface through diffusion [
7,
8,
9].
Previous studies regarding the effect of additional ferric vs. ferrous iron in a leaching scenario have yielded contradictory results. Third, et al. [
10] observed that ferric iron inhibited chalcopyrite dissolution, which was attributed to an increased redox potential. In contrast, Bevilaqua et al. [
11] showed that the addition of ferrous iron improved copper solubilisation in an
A. ferrooxidans culture, concluding that ferric iron mediated chalcopyrite leaching (rxns. 9 and 11) was the dominant mechanism [
11].
Based on Bevilaqua et al. [
11], the addition of pyrite to the system should have an even greater bioleaching effect. While soluble iron contributes to the iron cycle, the bioleaching of pyrite would provide not only iron, but also acid, which should significantly contribute to chalcopyrite leaching efficiency even if ferric leach is the dominant process. Mehta and Murr [
12] showed that, in the presence of
A. ferrooxidans, the extraction of copper from chalcopyrite is higher in systems mixed with pyrite compared to pure chalcopyrite, and within the mixed systems, the highest extraction occurred with the highest pulp density.
It stands to reason that different chalcopyrite to pyrite ratios should result in different copper leaching rates, either through the production of ferric iron and acid as described above, or potentially by galvanic interaction. Galvanic interaction occurs with pyrite functioning as a cathode and chalcopyrite as an anode [
13], where pyrite is passivated and the copper and iron from chalcopyrite leached. This process forms elemental sulphur on the chalcopyrite surface that can subsequently be oxidised by bacteria to sulphuric acid, producing an overall acid-neutral dissolution of chalcopyrite. It is reasonable to expect that stable galvanic cells are more likely to form and affect leaching rates in systems composed mostly of the electrode materials (i.e., when leaching concentrates), as opposed to the low-grade setups studied here or in low grade, i.e., marginal stockpiles.
For this column leaching setup, pyrite and chalcopyrite were “diluted” in a quartz sand matrix to model low grade and subeconomic concentrations of copper as they would be present in some porphyry copper orebodies, waste piles or tailings. These materials are generally considered to be the primary targets for future bioleaching setups, as they represent currently unavailable or lost resources. The economic cut-off for copper extraction is dependent on the market; however, concentrations at which deposits have been considered economic begin at a range between 0.3 and 0.4% [
2,
14]. The only external influence on the leaching columns was the addition of nutrients at regular intervals, representing a setup with minimal operational costs. It is assumed that both the chemical reactivity of, and the attachment of bacteria to, the quartz surface is negligible, or in the case of attachment, easily quantified. This limited chemical reactivity removed gangue mineral effects from the system.
Combining iron and sulphur oxidising metabolisms has been shown to be important when bioleaching copper minerals.
Leptospirillum ferrooxidans, an iron oxidiser, has been shown to oxidise pyrite faster in combination with a sulphur oxidising species, such as
Acidithiobacillus thiooxidans, and faster than
Acidithiobacillus ferrooxidans (an iron and sulphur oxidiser) by itself [
15]. In a related study, Romo et al. [
16] found that the combination of
A. ferrooxidans and
A. thiooxidans resulted in the highest leaching rates from ore while Sand et al. [
17] found that the combination of all three species resulted in the best leaching behaviour for complex copper deposits. In all of these systems, it is commonly accepted that bioleaching along the polysulphide pathway, as is the case for chalcopyrite, requires both iron and sulphur oxidising species to avoid passivation by elemental sulphur [
6,
18].
The experimental work presented here is aimed at investigating how the leaching rate of chalcopyrite is affected both by the chalcopyrite content in the system and the chalcopyrite to pyrite ratio. It is aimed specifically at the interaction of these two minerals in the absence of any other chemically and biologically reactive gangue minerals [
19], i.e., removing the effect(s) of gangue minerals in order to quantify the effect of pyrite in enhancing chalcopyrite bioleaching.
2. Materials and Methods
2.1. Culturing and Substrates
A bacterial consortium consisting of
A. ferrooxidans (strain DSM 14882),
L. ferrooxidans (strain DSM 2705) and
A. thiooxidans (strain ATCC 19377) was used for the experiments. Stock cultures were grown in conical flasks in DSMZ 882 [
20] media at pH 1.5 and room temperature. All cultures were grown in the same basal medium to avoid a lag phase resulting from a change of media when inoculating the columns.
A. ferrooxidans and
L. ferrooxidans had FeSO
4·7H
2O [20 g/L] as an energy source, while
A. thiooxidans was maintained on elemental sulphur [10 g/L]. To harvest cells for use as inoculum in the experiment, sulphur grains > 11 μm were removed from the
A. thiooxidans culture by filtering through Whatman No. 1 filter paper. All three culture systems were individually filtered through 0.22 µm filters, washed three times on the filters using fresh basal media and suspended in fresh basal media. Cells were then counted in a Petroff-Hausser counting chamber using phase contrast light microscopy and combined with additional basal media to give final populations of 10
5 cells/mL of
A. thiooxidans and 10
6 cells/mL of
A. ferrooxidans and
L. ferrooxidans. Culture viability was confirmed using LIVE/DEAD™ BacLight™ staining. The population of
A. thiooxidans was lower than the iron oxidising cultures because of the removal of the > 11 μm sized sulphur particles and any associated cells [
21] by filtration.
Chalcopyrite was obtained from the Salobo mine in Brazil, pyrite was purchased from Ward’s Science and quartz sand was purchased from Sigma. Chalcopyrite and pyrite were crushed and sieved to 150–300 µm; the quartz sand was graded to 50–70 mesh (212–300 µm) by the supplier. All materials were sonicated in deionised water to dislodge smaller particles, sterilised for 15 min using an autoclave and dried at 60°C. Chalcopyrite and pyrite were then analysed by XRD to confirm their purity. No additional phases were identified. All XRD data was gathered using a Bruker D8 Advance MKII, with a Cu source at 40 kV and 40 mA (spectra not shown).
2.2. Experimental Setup
Six distinct systems were set up: three at 0.1% Cu to represent subeconomic deposits and three at 0.5% Cu to represent a more contemporary economic cut-off grade. As the addition of pyrite has been shown to increase chalcopyrite dissolution by Mehta and Murr [
12], their chalcopyrite to pyrite ratio of 1:1 was used as a starting or reference point, with 1:2 and 2:1 ratio systems added to identify the influence of chalcopyrite:pyrite on copper extraction. The concentrations of copper in these systems were achieved by “diluting” chalcopyrite with sand to provide 35 g of solids for each column that would retain approximately 10.5 mL of culture medium.
Down flow leaching columns were set up in 20 mL syringes, lined at the bottom with sterile glass wool. The sulphide minerals and sand were weighed out and homogenised by stirring before adding the mixture to the columns. Inoculum was added until the column became “saturated”, but not draining, and the weight recorded. Columns were loosely covered with foil to allow for oxygen diffusion but limiting evaporation and incubated at approximately 20 °C. Abiotic controls were wetted with DSMZ 882 [
20] basal medium containing 0.2 g/L sodium benzoate as a biocide (modified after [
21,
22]) but otherwise identically treated. Columns were only fed on sampling days.
2.3. Sampling and Microscopy
Sampling took place every 10 days for 50 days, followed by additional sampling time points at 70 and 105 days. Sampling for chemistry consisted of flushing each column with 7.5 mL (approximately 75% of the liquid in the column, i.e., void volume; added all at once by pipette) of fresh media and collecting the effluent, as well as recording the weights of both media (input) and resulting sample (output) to determine dilution and evaporation and for measuring pH. The media used to flush the abiotic columns contained biocide as above. Chemical analyses were carried out in triplicate for each system at the Environmental Geochemistry Laboratory at the University of Queensland (UQ) using a Perkin Elmer Optima 8300DV with inductively coupled plasma optical emission spectrometer (ICP-OES) to measure Fe, Cu, P and S concentrations.
It was assumed that when flushing the columns with fresh media, the mass of liquid that would keep the column saturated was the same as the mass of the original inoculum. It was further assumed that the chemical composition of the solution in the columns was homogeneous and that the media added during sampling would displace the liquid in the column without mixing but that the two liquids would homogenise by diffusion between sampling points. Hence, for each time point, we determined the net change (e.g., total mass of dissolved copper present in the columns) from the previous sampling point, correcting for evaporation, and determined the “new” concentration of analysed elements after sampling, to use as the subsequent baseline for determining a change in aqueous chemistry for the next sampling point. Note that most of the iron concentrations at day 40 were above the calibrated range, and there was insufficient sample to repeat the analysis. Therefore, the Column type A (0.1% Cu 1:2 py:ccp) value, along with ratios of iron concentrations, e.g., column A:column B, from the previous sample time point was used to proportionately estimate these iron concentrations.
At 70 days, one column from each system was fixed in 2.5%(aq) glutaraldehyde. Samples were then taken from the top, middle and bottom of each column to enable identification of the presence of reaction gradients, i.e., an oxidising gradient indicated by the presence of iron oxides. Briefly, samples were processed through an ethanol dehydration series in 20% increments with three repeats at 100%, followed by critical point drying. Grains were then mounted for scanning electron microscopy (SEM) and carbon coated to 20 nm. The samples were imaged at the Centre for Microscopy and Microanalysis (CMM) at UQ on a JEOL JSM-7100 field emission SEM with an accelerating voltage of 1 kV and a working distance of approximately 8 mm, unless otherwise stated. Energy dispersive spectroscopy (EDS) was used to differentiate between the substrates in the system. This analysis was carried out at 15 kV and 10 mm working distance using a JEOL 129 eV resolution silicon drift detector fitted to the microscope.
3. Results
3.1. Chemistry
Cumulative leaching results for copper, iron and sulphur are shown in
Figure 1 (see also
Bostelmann_Southam_Chemistry_Data.xlsx; Supplementary Data). The cumulative release of copper from the leaching columns showed a constant increase in dissolved copper over time for all systems. Columns with higher pyrite to chalcopyrite ratios slightly exceeded the columns with lower ratios in terms of copper in solution; however, columns with the same initial copper concentrations reached similar leaching rates (i.e., slope). The two different concentrations of copper in the substrate resulted in two distinct populations of leaching curves. The abiotic controls showed similar leaching behaviour, with similar leach values at the first sampling point, but a continuous, albeit slight, decrease in leaching rate over the following time points.
The abiotic iron release also plotted as two “groupings” distinguishable by their copper content, with the higher-pyrite columns plotting slightly higher for each grouping. The abiotic iron release was mostly linear, with only a slight decrease towards the last sampling time point. In contrast, the cumulative release of iron in the biologically active columns was linear and tiered according to the iron content in the systems. For the systems containing only chalcopyrite, the iron release varied according to their iron content but had lower total extraction rates than the system with the lowest pyrite concentration. Sulphur release in the active columns followed a very similar pattern, while sulphur concentrations in the abiotic systems (i.e., sulphate from the culture medium) continuously decreased.
Fe:S ratios were based on solution analysis and calculated from the change in molar concentration of each element from one sampling time point to the next. For example, if between time A and time B Fe increased by 20 mmol and S increased by 40 mmol, the resulting Fe:S ratio would be 0.5. The Fe:S ratio at 10 days was enriched in iron due to carryover of iron within the inoculum. After this time point, the systems had an average molar Fe:S ratio of 0.52 ± 0.06. The abiotic controls gave negative ratios corresponding to sulphur adsorption within the columns, which presumably occurred in the biotic systems as well, explaining the slight enrichment in iron. Negative ratios result from one element increasing and the other decreasing in concentration. The Fe:S ratios resulting from chalcopyrite dissolution (i.e., in the chalcopyrite only systems) varied markedly over the course of the experiment and with copper concentration, increasing from 0.26 to 0.54 and from 0.37 to 1.11 in the 0.1% and 0.5% Cu systems, respectively, suggesting that sulphur retention was occurring in the columns, though copper recovery did not plateau, indicating that passivation was not taking place. Subtracting the effect of chalcopyrite, the pyrite contribution to iron and sulphur in solution had an average ratio of 0.54 ± 0.05, indicating that (along with the lack of any secondary mineral precipitates, below) the congruent dissolution of pyrite dominates the leachate chemistry.
The pH data was corrected for evaporation and dilution, and the total consumption (or release) of protons between each sampling point calculated (shown in
Figure 2). For the first 50 days, the columns buffered to pH ca. 1.7–1.8 after each feeding with media at pH 1.5, indicating constant acid consumption. After this, acid consumption plateaued, and in the case of the 0.5% Cu systems, they became acid producing.
Cell counts in sampled liquor were within the same order of magnitude as the original inoculum (ca. 106 bacteria/mL) at all sampling time points, so with every sampling, approximately 75% of the ‘original’ planktonic bacteria were washed out, with the planktonic culture subsequently recovering. Cell numbers on chalcopyrite and sand were approx. 105–106 cells/mm2 for regions with attached cells, although there were proportionately more empty areas (overall, possessing ca. 2 × 103 cells/mm2). Therefore, for the chalcopyrite system, cells were roughly distributed between planktonic, chalcopyrite-bound and sand-bound surfaces. In contrast, biofilm cell numbers dominated the pyrite-containing systems, with a conservative estimate of biomass corresponding to ca. 107 total planktonic numbers (106 cells/mL in 10 mL of column liquid volume) and 108–109 cells as biofilms on pyrite grains (assuming a monolayer of cells covering the entire surface of each grain). The abiotic controls were negative, i.e., no cells were evident in phase contrast light microscopy with a detection limit of 104 cells/mL. There were no phenotypic colour changes, i.e., rust coloured iron oxides or blue-green copper sulphates/phosphates, observed in any of the columns.
3.2. Microscopy
All samples, regardless of the pyrite content of the column or where the sample was collected, showed the same colonisation characteristics. The samples for microscopy were separated into “sulphides” and “sand” before imaging. In the “sulphide minerals” group, some grains were completely encased in biofilm (
Figure 3A–D) while others contained only individual or small clusters of cells scattered across the surface (
Figure 3E,F). EDS analysis showed that all grains that were covered in biofilm were pyrite and all other grains were chalcopyrite. The sand grains had only individual cells attached (
Figure 3G,H). Additional images of pyrite (
Figure S1), chalcopyrite (
Figure S2) and quartz (
Figure S3) are given in
supplementary materials. All imaged cells were rod-shaped. None of the systems had evidence of secondary mineralisation. The abiotic controls had no cells on any of the imaged grains.
4. Discussion
4.1. Leaching Rates
The general leaching rate of copper was proportional to the copper content of the columns. However, the biooxidation of iron and sulphur from pyrite did not result in a concomitant increase in the recovery of copper from chalcopyrite. Assuming that the columns reached a steady leaching rate and using the final analysis time point to calculate the time necessary to bioleach the chalcopyrite in the columns to completion, the columns containing 0.1% Cu would take an average of 42.6 (±11.8) years [abiotic 192 (±6) years], whereas the 0.5% Cu columns would require 40.4 (±1.8) years [abiotic 184 (±14) years]. These values also suggest no inhibitory effect of the achieved copper concentrations, as the leaching rate of the higher concentration columns is proportionally higher than that of the lower concentration columns. In fact, this proportionality suggests that copper release in a system such as those used in this trial is simply a direct function of the surface area of copper containing minerals, and the reaction may be a purely chemical interaction of the mineral with the surrounding liquid. In this case, accelerating the process would be achievable only by grain size reduction, which may increase processing costs and affect the financial viability of the process. Over the course of the experiment, the 0.1% Cu and 0.5% Cu columns released copper at similar degree relative to the surface area of chalcopyrite, e.g., 0.81 (± 0.08) µg/mm2 when assuming cubes of chalcopyrite with a grain size of 300 µm. As the surface area of chalcopyrite is only an estimate, this leaching calculation is to be taken as an approximation.
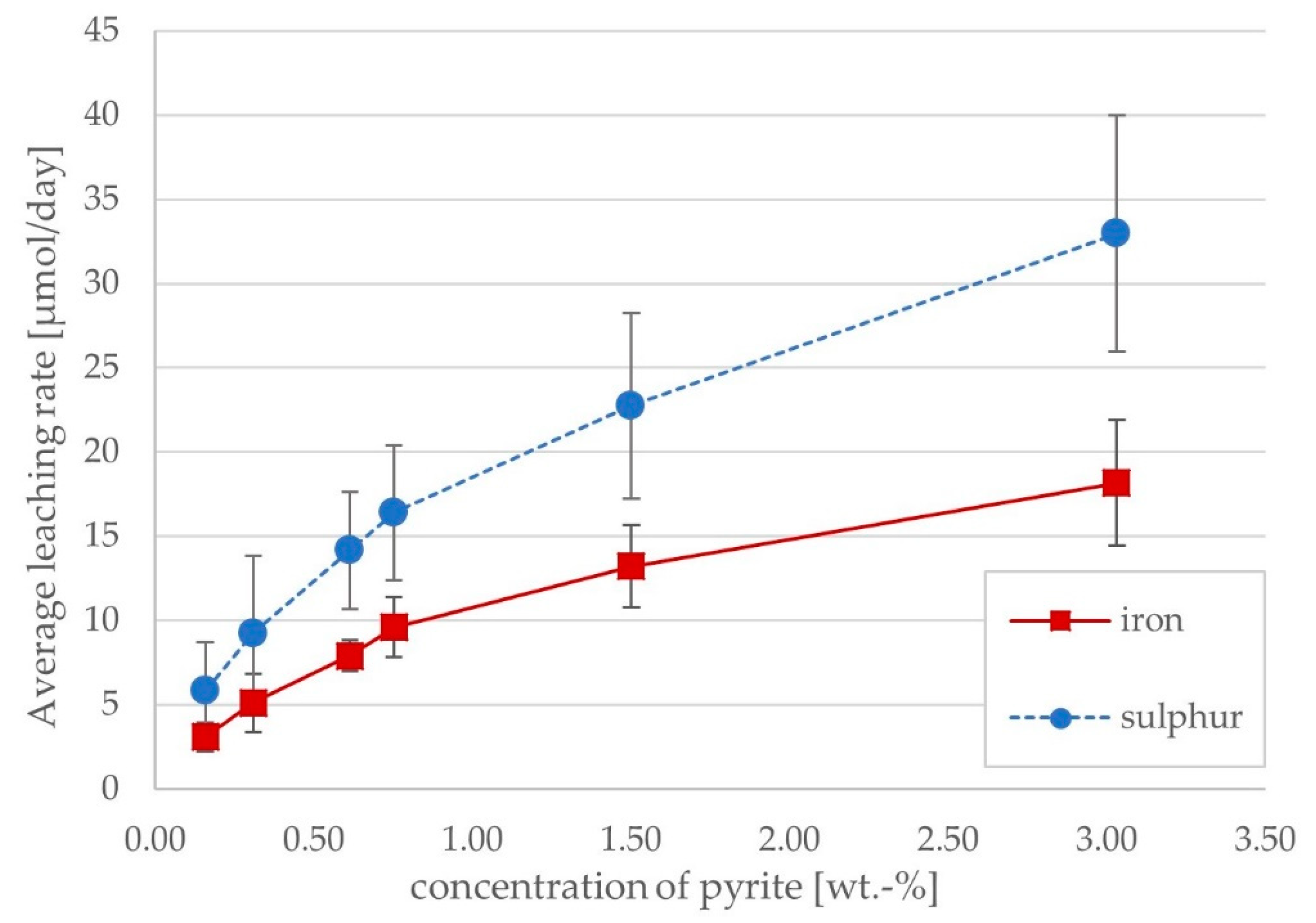
The relationship between daily iron and sulphur release and the concentration of pyrite in the system was expected to follow a linear trend, given that the leaching reactions are restricted to the surface of the mineral grains. While the leaching rates of iron and sulphur were proportional to pyrite content, the rates decreased at higher pyrite contents (
Figure 4), suggesting that there may be some limiting condition, e.g., phosphate availability, at higher pyrite contents. The similarity of iron and sulphur leaching rates and their correlation with pyrite content indicate that the bulk of dissolved iron and sulphur stem from pyrite dissolution. The absence of a gap or disconnect between the populations (i.e., 0.1% and 0.5% Cu) indicates that the copper in solution had no inhibitory effect at the concentrations achieved. This is as expected, as inhibitory levels of copper have been reported to be several times to several orders of magnitude higher than those achieved in the columns [
23,
24,
25].
4.2. Leaching Mechanism
The iron and acid generated from the oxidation of pyrite should have enhanced the leaching of chalcopyrite through increased iron concentrations in solution and therefore a higher degree of ferric leaching (rxn. 11), as well as acid dissolution (rxn. 2). The systems do show higher concentrations of iron in solution as a result of higher pyrite contents, but not a significant, discernible effect on copper concentration. Therefore, there does not appear to be significant ferric iron leaching of chalcopyrite. Rather, the dominant process of low-grade chalcopyrite dissolution at the given conditions is by acid leach or proton attack.
If acid leach is the dominant process in the bioleaching of low-grade chalcopyrite systems, any buffering of pH would likely decrease the efficacy of this process. Net chalcopyrite leaching is acid neutral (rxn. 14) but can be acid consuming if the iron moiety is further oxidised (rxn. 9) and acid producing if the iron is hydrolysed (rxn. 12). Since pyrite can only be dissolved by ferric leaching and the pyrite grains observed are heavily colonised and show dissolution features, an iron oxidation cycle is present in the system. This cycle would automatically include all iron in the system, and the presence of an active iron cycle at low pH is consistent with the lack of iron mineral precipitates or colouration that can be expected from hydrolysis. Therefore, if pyrite (rxn. 13) and chalcopyrite leach at the same rate (i.e., mol/unit time), the sum reaction (rxns. 9 + 13 + 14) would be acid neutral (rxn. 15), but as pyrite leaching exceeded that of chalcopyrite, the net reaction would be acid producing.
4.3. Bacterial Activity
A purely chemical interaction of the solution with the minerals could imply a lack of activity of the utilised inoculum. However, several factors are evidence of a healthy bacterial consortium. The number of planktonic cells was determined at every sampling time point and showed that planktonic cell numbers in the column “recovered” after every dilution to approximately the same quantity of cells. Though the method did not determine if or how the population changed over time, as a whole, cell numbers grew throughout the experiment. The morphologically distinct
L. ferrooxidans was notably absent from all imaged surfaces, indicating a change in consortium composition. It may have been outperformed by the other species in terms of attachment and subsequently washed out. Alternatively, it may have initially attached more successfully (i.e., faster), only to be obscured by the growth of the
Acidithiobacillus-dominated biofilm on the pyrite grains. The encrustation of pyrite grains is generally accepted to be the norm with pyrite bioleaching [
9,
26,
27], further supporting an active culture. The reduced attachment of cells to sand and chalcopyrite is consistent with previous observations and does not detract from the conclusion that the consortium was thriving. It is not clear whether the reduced attachment to chalcopyrite is a result of preferential colonisation of pyrite or if the surface properties of both chalcopyrite and the bacteria could be a limiting factor. The data obtained in the columns does not allow one to determine if different cells contribute to different processes (e.g., biofilm formation causing pyrite dissolution along with planktonic cells causing acid production and chalcopyrite leaching).
The chemical data for iron and sulphur concentrations in the inoculated columns further supports the interpretation that the bacteria are responsible for the chemistry of the system. The molar ratios of these elements should be 1:2 in the columns if either pyrite or chalcopyrite is leached. This is the case both for the systems as a whole and for the contribution of pyrite to iron and sulphur concentrations in solution, which, along with the colonisation data, demonstrates that pyrite is, in fact, being actively leached. In contrast, in the abiotic columns, iron concentrations increased, but sulphur concentrations decreased from one sampling point to the next, indicating a different mechanism to the inoculated columns. Additionally, the iron leaching rates from the abiotic columns plot as two groups distinguishable by copper content (
Figure 1), which indicates that the iron present in the abiotic systems is likely to have originated from chalcopyrite dissolution (rather than pyrite), which would occur abiotically through acid dissolution of chalcopyrite, consistent with the acid pH of the systems.
4.4. Secondary Mineralisation Considerations
The copper to iron ratio should be 1:1 in all abiotic systems as well as the inoculated systems containing only chalcopyrite but iron release consistently exceeds that of copper, indicating that either iron is preferentially released in this acid leach reaction or copper is later removed from solution. Preferential release of iron would likely result in a copper-enriched surface layer, which, based on the size of chalcopyrite grains in this study should be several 100s of nm to µm scale in thickness (Initially, the total volume of chalcopyrite in each system was determined based on chalcopyrite quantity and density. From this, the available surface area was calculated using the grain sizes given in the methods section and assuming a cube shape. The difference between Cu and Fe in solution was taken as the amount of Cu in the “enriched surface layer”. Coupled with the density of various copper sulphides, a total volume of “enriched surface layer” was calculated. The thickness of this layer follows from its volume spread over the surface area of chalcopyrite). This would be noticeable both in EDS analysis and possibly also optically, neither of which was the case. No secondary copper precipitates were identified using SEM, but their formation is still possible; however, a more likely explanation for the discrepancy in leaching values is that “planktonic” precipitates would have washed out during sample processing, and any colloidal precipitates coating the overall mineral grains in the system could have been obscured by the carbon coating, as even the most “generous” calculated thickness amounts to less than 20 nm (i.e., assuming the smallest possible surface area for the leached substrate and hydrated precipitates with low density). For the abiotic columns, the reduction in sulphur concentration over time likewise indicates possible sulphur mineral precipitation mechanisms additional to those described above, which would be similarly unlikely to be identified with the methods used here.
During the biooxidation of pyrite, the thiosulphate intermediate, formed during the first oxidation step (rxn. 1; [
6]), will disproportionate under acidic conditions to form elemental sulphur and sulphite (rxn. 3; [
28]). The precipitation of elemental sulphur (i.e., the removal of up to half the available sulphur from solution) would noticeably skew the Fe:S ratio. As this is not the case (
Figure 2), the only way to maintain that ratio is either for iron to be removed from solution at the same rate (considered unlikely, as no precipitates were found), or the elemental sulphur must be oxidised to a soluble chemical species and finally sulphate. As chalcopyrite is acid soluble, its oxidation follows the polysulphide pathway. These higher polysulphides will decompose at acidic pH (rxn. 4) with concomitant precipitation of elemental sulphur [
6,
29], which has been implicated as a cause for chalcopyrite surface passivation. Elemental sulphur was not observed in the systems analysed, either as particles, colloids or surface layers. It is possible that it was present and lost during leaching or during processing; (a) particles or colloids may have been flushed out of the column during sampling, or (b) they may have remained in the column during sampling, but unintentionally discarded during processing (dehydration) for SEM. However, if elemental sulphur forms as a result of thiosulphate disproportionation and is then oxidised, any sulphur formed from the polysulphide pathway of chalcopyrite oxidation would also be oxidised, and therefore solubilised.
4.5. pH Buffering and Biomass Formation
In the examined systems, different pyrite contents resulted in different iron and sulphur concentrations in solution, which should have resulted in differing acid generation or consumption patterns, and therefore should have affected the dissolution rates of chalcopyrite and consequently copper concentrations in solution. However, this was not the case. The measured pH of the samples did not correlate with iron or sulphur concentrations; pH values are more or less consistent across all systems, indicating significant buffering capacity proportional to the amount of acidity produced. The increase of pH in the columns after each sampling point is unlikely to be solely due to acid dissolution of chalcopyrite, as acid consumption significantly exceeds copper release. Therefore, the pH of the system even with a quartz matrix is effectively buffered.
Measured iron or sulphur concentrations in the columns can be taken as a proxy for the amount of pyrite oxidised, and consequently, the total amount of acidity produced. The synthesis of ATP (adenosine triphosphate), required for the formation of biomass through the Calvin cycle, consumes protons, thereby buffering the system. Based on the total acidity consumed in the columns (i.e., protons from pyrite oxidation plus measured pH increase) it is possible to determine the amount of ATP synthesised, and, in combination with the biomass yield of
A. ferrooxidans, determine the total mass of carbon fixed in biomass [
30,
31]. From this, it is possible to determine the corresponding quantity of bacterial cells produced, based on the proportion of carbon in biomass and the size and weight of
A. ferrooxidans relative to
E. coli [
32,
33,
34]. The calculated cell numbers based on the above method are in agreement with the cell number estimate for the bacterial biofilms encasing the pyrite grains. Therefore, any acidity produced through the biooxidation of pyrite was consumed in order to synthesise biomass and was consequently not available for chalcopyrite dissolution.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



